
Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases


Abstract
1. TTR Structure and Major Functions
2. Structural Stability, Cleavage, and Amyloidogenesis of TTR
3. Unaggregated TTR Is a Redox-Sensing Factor
4. TTR and ECM Remodeling
5. Link between TTR and Ca2+
6. Involvement of TTR in Biomineralization
7. Calcium-Containing Protein–Mineral Nanoparticles
8. TTR Involvement in Noncardiac Disorders
9. TTR Contribution to Cardiovascular Disease and Vascular Calcification
10. TTR and Wound Healing (Fibrinolysis vs. Thrombosis)
11. TTR Interconnection with Inflammation
12. TTR Role in Atherosclerosis
13. TTR Regulation of Angiogenesis
14. TTR at the Crossroads of Vascular Calcification and Bone Mineralization
15. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACS | Acute coronary syndrome |
| RAGE | Advanced glycation end products |
| ATTRwt | Aggregated wild type TTR |
| AD | Alzheimer’s disease |
| Aβ | Amyloid β |
| APP | Amyloid β precursor protein |
| ApoA1 | Apolipoprotein A1 |
| ATTR CA | ATTR cardiac amyloidosis |
| CAVD | Calcific aortic valve disease |
| CNPs | Calcifying nanoparticles |
| CA | Cardiac amyloidosis |
| CVD | Cardiovascular disease |
| CSF | Cerebrospinal fluid |
| CAD | Coronary artery disease |
| DR | Diabetic retinopathy |
| ECM | Extracellular matrix |
| FAP | Familial amyloidotic polyneuropathy |
| HF | Heart failure |
| HFpEF | Heart failure with preserved ejection fraction |
| HDL | High density lipoprotein |
| HMW | High molecular weight |
| HCy | Homocysteine |
| hRECs | Human retinal endothelial cells |
| AL | Immunoglobulin light chain amyloid |
| JIA | Juvenile idiopathic arthritis |
| lncRNA | Long noncoding RNA |
| MMP | Matrix metalloproteinases |
| TFAM | Mitochondrial transcription factor A |
| MMD | Moya-moya disease |
| MPO | Myeloperoxidase |
| NPY | Neuropeptide Y |
| OA | Osteoarthritis |
| PILP | Polymer-induced liquid phase |
| ROS | Reactive oxygen species |
| RA | Rheumatoid arthritis |
| RBP | Retinol-binding protein |
| SSA | Senile systemic amyloidosis |
| SAA | Serum amyloid A |
| TUDCA | Tauroursodeoxycholic acid |
| tPA | Tissue-type plasminogen activator |
| T2DM | Type 2 diabetes mellitus |
| UPR | Unfolded protein response |
| PAM | Peptidylglycine α-amidating monooxygenase |
| VC | Vascular calcification |
| VSMCs | Vascular smooth muscle cells |
References
- Liz, M.A.; Mar, F.M.; Franquinho, F.; Sousa, M.M. Aboard transthyretin: From transport to cleavage. IUBMB Life 2010, 62, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Reixach, N. Transthyretin: The servant of many masters. Cell. Mol. Life Sci. 2009, 66, 3095–3101. [Google Scholar] [CrossRef]
- Richardson, S.J. Evolutionary changes to transthyretin: Evolution of transthyretin biosynthesis. FEBS J. 2009, 276, 5342–5356. [Google Scholar] [CrossRef] [PubMed]
- Aleshire, S.L.; Bradley, C.A.; Richardson, L.D.; Parl, F.F. Localization of human prealbumin in choroid plexus epithelium. J. Histochem. Cytochem. 1983, 31, 608–612. [Google Scholar] [CrossRef]
- Bailey, M.J.; Coon, S.L.; Carter, D.A.; Humphries, A.; Kim, J.-S.; Shi, Q.; Gaildrat, P.; Morin, F.; Ganguly, S.; Hogenesch, J.B.; et al. Night/Day Changes in Pineal Expression of >600 Genes. J. Biol. Chem. 2009, 284, 7606–7622. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, T.; Martone, R.; Dwork, A.J.; Schon, E.A.; Herbert, J. The retinal pigment epithelium is the unique site of transthyretin synthesis in the rat eye. Investig. Ophthalmol. Vis. Sci. 1990, 31, 497–501. [Google Scholar]
- Refai, E.; Dekki, N.; Yang, S.-N.; Imreh, G.; Cabrera, O.; Yu, L.; Yang, G.; Norgren, S.; Rossner, S.M.; Inverardi, L.; et al. Transthyretin constitutes a functional component in pancreatic -cell stimulus-secretion coupling. Proc. Natl. Acad. Sci. USA 2005, 102, 17020–17025. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Jono, H.; Misumi, Y.; Senokuchi, T.; Guo, J.; Ueda, M.; Shinriki, S.; Tasaki, M.; Shono, M.; Obayashi, K.; et al. Novel function of transthyretin in pancreatic alpha cells. FEBS Lett. 2012, 586, 4215–4222. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, S.; Maeda, S.; Shimada, K. Structure and Expression of the Mouse Prealbumin Gene. J. Biochem. 1986, 100, 49–58. [Google Scholar] [CrossRef]
- Costa, R.H.; Grayson, D.R.; Darnell, J.E. Multiple hepatocyte-enriched nuclear factors function in the regulation of transthyretin and alpha 1-antitrypsin genes. Mol. Cell. Biol. 1989, 9, 1415–1425. [Google Scholar] [CrossRef]
- Wang, X.; Cattaneo, F.; Ryno, L.; Hulleman, J.; Reixach, N.; Buxbaum, J.N. The Systemic Amyloid Precursor Transthyretin (TTR) Behaves as a Neuronal Stress Protein Regulated by HSF1 in SH-SY5Y Human Neuroblastoma Cells and APP23 Alzheimer’s Disease Model Mice. J. Neurosci. 2014, 34, 7253–7265. [Google Scholar] [CrossRef]
- Oka, S.; Leon, J.; Sakumi, K.; Ide, T.; Kang, N.; LaFerla, F.M.; Nakabeppu, Y. Human mitochondrial transcriptional factor A breaks the mitochondria-mediated vicious cycle in Alzheimer’s disease. Sci. Rep. 2016, 6, 37889. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-W.; Lee, M.H.; Choi, J.-O.; Park, H.-Y.; Jung, S.-C. Tissue-specific activation of mitogen-activated protein kinases for expression of transthyretin by phenylalanine and its metabolite, phenylpyruvic acid. Exp. Mol. Med. 2010, 42, 105–115. [Google Scholar] [CrossRef]
- Martinho, A.; Gonsalves, J.; Costa, M.; Santos, C.R. Stress and glucocorticoids increase transthyretin expression in rat choroid plexus via mineralocorticoid and glucocorticoid receptorse. J. Mol. Neurosci. 2012, 48, 1–13. [Google Scholar] [CrossRef]
- Blake, C.C.F.; Burridge, J.M.; Oatley, S.J. X-Ray Analysis of Thyroid Hormone Binding to Prealbumin. Biochem. Soc. Trans. 1978, 6, 1114–1118. [Google Scholar] [CrossRef]
- Ferguson, R.F.; Edelhoch, H.; Saroff, H.; Robins, J.A.; Cahnmann, H.J. Negative cooperativity in the binding of thyroxine to human serum prealbumin. Biochemistry 1875, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Hagen, G.A.; Elliott, W.J. Transport of Thyroid Hormones in Serum and Cerebrospinal Fluid. J. Clin. Endocrinol. Metab. 1973, 37, 415–422. [Google Scholar] [CrossRef]
- Rabah, S.A.; Gowan, I.L.; Pagnin, M.; Osman, N.; Richardson, S.J. Thyroid Hormone Distributor Proteins During Development in Vertebrates. Front. Endocrinol. 2019, 10, 506. [Google Scholar] [CrossRef]
- Benvenga, S.; Bartalena, L.; Antonelli, A.; Calzi, L.L.; Di Pasquale, G.; Trimarchi, F.; Pinchera, A. Radioimmunoassay for human thyroxine-binding prealbumin. Ann. Clin. Lab. Sci. 1986, 16, 231–240. [Google Scholar] [PubMed]
- Weisner, B.; Roethig, H.-J. The Concentration of Prealbumin in Cerebrospinal Fluid (CSF), Indicator of CSF Circulation Disorders. Eur. Neurol. 1983, 22, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Gião, T.; Saavedra, J.; Cotrina, E.; Quintana, J.; Llop, J.; Arsequell, G.; Cardoso, I. Undiscovered Roles for Transthyretin: From a Transporter Protein to a New Therapeutic Target for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2075. [Google Scholar] [CrossRef]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis. Int. J. Mol. Sci. 2019, 20, 1287. [Google Scholar] [CrossRef] [PubMed]
- Pullakhandam, R.; Srinivas, P.; Nair, M.K.; Reddy, G.B. Binding and stabilization of transthyretin by curcumin. Arch. Biochem. Biophys. 2009, 485, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Choi, S.; Buxbaum, J.N.; Kelly, J.W.; Price, J.L.; Reixach, N. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem. Biophys. Res. Commun. 2011, 410, 707–713. [Google Scholar] [CrossRef]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef]
- Sousa, M.; Du Yan, S.; Stern, D.; Saraiva, M.J. Interaction of the Receptor for Advanced Glycation End Products (RAGE) with Transthyretin Triggers Nuclear Transcription Factor kB (NF-kB) Activation. Lab. Investig. 2000, 80, 1101–1110. [Google Scholar] [CrossRef]
- Smeland, S.; Kolset, S.O.; Lyon, M.; Norum, K.R.; Blomhoff, R. Binding of perlecan to transthyretin qin vitro. Biochem. J. 1997, 326, 829–836. [Google Scholar] [CrossRef]
- Sousa, M.; Berglund, L.; Saraiva, M.J. Transthyretin in high density lipoproteins: Association with apolipoprotein A-I. J. Lipid Res. 2000, 41, 58–65. [Google Scholar] [CrossRef]
- Gomes, J.; Nogueira, R.S.; Vieira, M.; Santos, S.D.; Ferraz-Nogueira, J.P.; Relvas, J.B.; Saraiva, M.J. Transthyretin provides trophic support via megalin by promoting neurite outgrowth and neuroprotection in cerebral ischemia. Cell Death Differ. 2016, 23, 1749–1764. [Google Scholar] [CrossRef] [PubMed]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994, 91, 8368–8372. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, Y.; Sanders, C.R.; Buxbaum, J.N.; Diego, S.; Squibb, B.; Development, B.P.; Medicine, E.; Torrey, N.; Road, P.; et al. Transthyretin Suppresses Amyloid-β secretion by Interfering with Processing of The Amyloid-β Precursor Protein. J. Alzheimers Dis. 2016, 52, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.D.; Lambertsen, K.L.; Clausen, B.H.; Akinc, A.; Alvarez, R.; Finsen, B.; Saraiva, M.J. CSF transthyretin neuroprotection in a mouse model of brain ischemia. J. Neurochem. 2010, 115, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Kalkunte, S.S.; Neubeck, S.; Norris, W.E.; Cheng, S.-B.; Kostadinov, S.; Hoang, D.V.; Ahmed, A.; von Eggeling, F.; Shaikh, Z.; Padbury, J.; et al. Transthyretin Is Dysregulated in Preeclampsia, and Its Native Form Prevents the Onset of Disease in a Preclinical Mouse Model. Am. J. Pathol. 2013, 183, 1425–1436. [Google Scholar] [CrossRef]
- Mangrolia, P.; Murphy, R.M. Retinol-Binding Protein Interferes with Transthyretin-Mediated β-Amyloid Aggregation Inhibition. Biochemistry 2018, 57, 5029–5040. [Google Scholar] [CrossRef] [PubMed]
- Fleming, C.E.; Saraiva, M.J.; Sousa, M.M. Transthyretin enhances nerve regeneration. J. Neurochem. 2007, 103, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Conti, S.; Mannini, B.; Li, X.; Buxbaum, J.N.; Tiribilli, B.; Chiti, F.; Cecchi, C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 2302–2314. [Google Scholar] [CrossRef]
- Liz, M.A.; Coelho, T.; Bellotti, V.; Fernandez-Arias, M.I.; Mallaina, P.; Obici, L. A Narrative Review of the Role of Transthyretin in Health and Disease. Neurol. Ther. 2020, 9, 395–402. [Google Scholar] [CrossRef]
- Sousa, J.; Marques, F.; Dias-Ferreira, E.; Cerqueira, J.; Sousa, N.; Palha, J.A. Transthyretin influences spatial reference memory. Neurobiol. Learn. Mem. 2007, 88, 381–385. [Google Scholar] [CrossRef]
- Vieira-Coelho, M.A.; Saraiva, M.J. Transthyretin regulates hippocampal 14-3-3ζ protein levels. FEBS Lett. 2013, 587, 1482–1488. [Google Scholar] [CrossRef]
- Ingenbleek, Y.; Bernstein, L.H. Plasma Transthyretin as a Biomarker of Lean Body Mass and Catabolic States. Adv. Nutr. 2015, 6, 572–580. [Google Scholar] [CrossRef]
- Liz, M.A.; Faro, C.J.; Saraiva, M.J.; Sousa, M.M. Transthyretin, a new cryptic protease. J. Biol. Chem. 2004, 279, 21431–21438. [Google Scholar] [CrossRef] [PubMed]
- Liz, M.A.; Leite, S.C.; Juliano, L.; Saraiva, M.J.; Damas, A.M.; Bur, D.; Sousa, M.M. Transthyretin is a metallopeptidase with an inducible active site. Biochem. J. 2012, 443, 769–778. [Google Scholar] [CrossRef]
- Costa, A.R.; Silva, F.; Saraiva, M.J.; Cardoso, I. Transthyretin Protects against A-Beta Peptide Toxicity by Proteolytic Cleavage of the Peptide: A Mechanism Sensitive to the Kunitz Protease Inhibitor. PLoS ONE 2008, 3, e2899. [Google Scholar] [CrossRef] [PubMed]
- Stevens, F.J. Possible evolutionary links between immunoglobulin light chains and other proteins involved in amyloidosis. Amyloid 2008, 15, 96–107. [Google Scholar] [CrossRef]
- Hörnberg, A.; Eneqvist, T.; Olofsson, A.; Lundgren, E.; Sauer-Eriksson, E. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J. Mol. Biol. 2000, 302, 649–669. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.D.F.-; Palmieri, L.; Valory, M.; Silva, J.; Lashuel, H.; Kelly, J.W.; Foguel, D. Hydration and Packing are Crucial to Amyloidogenesis as Revealed by Pressure Studies on Transthyretin Variants that Either Protect or Worsen Amyloid Disease. J. Mol. Biol. 2003, 328, 963–974. [Google Scholar] [CrossRef]
- Wieczorek, E.; Kędracka-Krok, S.; Bystranowska, D.; Ptak, M.; Wiak, K.; Wygralak, Z.; Jankowska, U.; Ożyhar, A. Destabilisation of the structure of transthyretin is driven by Ca2+. Int. J. Biol. Macromol. 2021, 166, 409–423. [Google Scholar] [CrossRef]
- Lim, K.H.; Dyson, H.J.; Kelly, J.W.; Wright, P.E. Localized Structural Fluctuations Promote Amyloidogenic Conformations in Transthyretin. J. Mol. Biol. 2013, 425, 977–988. [Google Scholar] [CrossRef]
- Das, J.K.; Mall, S.S.; Bej, A.; Mukherjee, S. Conformational Flexibility Tunes the Propensity of Transthyretin to Form Fibrils Through Non-Native Intermediate States. Angew. Chem. Int. Ed. 2014, 53, 12781–12784. [Google Scholar] [CrossRef]
- Yee, A.W.; Aldeghi, M.; Blakeley, M.P.; Ostermann, A.; Mas, P.J.; Moulin, M.; De Sanctis, D.; Bowler, M.W.; Mueller-Dieckmann, C.; Mitchell, E.P.; et al. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019, 10, 925. [Google Scholar] [CrossRef]
- Brito, R.; Damas, A.; Saraiva, M. Amyloid Formation by Transthyretin: From Protein Stability to Protein Aggregation. Curr. Med. Chem. Immunol. Endocr. Metab. Agents 2003, 3, 349–360. [Google Scholar] [CrossRef]
- Ribeiro, C.A.; Saraiva, M.J.; Cardoso, I. Stability of the Transthyretin Molecule as a Key Factor in the Interaction with A-Beta Peptide—Relevance in Alzheimer’s Disease. PLoS ONE 2012, 7, e45368. [Google Scholar] [CrossRef]
- Palmieri, L.D.C.; Lima, L.M.T.; Freire, J.B.; Bleicher, L.; Polikarpov, I.; Almeida, F.C.; Foguel, D. Novel Zn2+-binding Sites in Human Transthyretin: Implications for amyloidogenesis and retinol-binding protein recognition. J. Biol. Chem. 2010, 285, 31731–31741. [Google Scholar] [CrossRef]
- Ciccone, L.; Tonali, N.; Shepard, W.; Nencetti, S.; Orlandini, E. Physiological Metals Can Induce Conformational Changes in Transthyretin Structure: Neuroprotection or Misfolding Induction? Crystals 2021, 11, 354. [Google Scholar] [CrossRef]
- Zhang, Q.; Kelly, J.W. Cys10 Mixed Disulfides Make Transthyretin More Amyloidogenic under Mildly Acidic Conditions. Biochemistry 2003, 42, 8756–8761. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Buxbaum, J.N.; Reixach, N. Age-Related Oxidative Modifications of Transthyretin Modulate Its Amyloidogenicity. Biochemistry 2013, 52, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Mizuguchi, M.; Kouno, T.; Shinohara, Y.; Aizawa, T.; Demura, M.; Mori, Y.; Shinoda, H.; Kawano, K. Destabilization of transthyretin by pathogenic mutations in the DE loop. Proteins Struct. Funct. Genet. 2007, 66, 716–725. [Google Scholar] [CrossRef]
- Foumthuim, C.J.D.; Corazza, A.; Berni, R.; Esposito, G.; Fogolari, F. Dynamics and Thermodynamics of Transthyretin Association from Molecular Dynamics Simulations. BioMed Res. Int. 2018, 2018, 7480749. [Google Scholar] [CrossRef]
- Shnyrov, V.L.; Villar, E.; Zhadan, G.G.; Sanchez-Ruiz, J.M.; Quintas, A.; Saraiva, M.J.M.; Brito, R. Comparative calorimetric study of non-amyloidogenic and amyloidogenic variants of the homotetrameric protein transthyretin. Biophys. Chem. 2000, 88, 61–67. [Google Scholar] [CrossRef]
- Foss, T.R.; Wiseman, L.; Kelly, J.W. The Pathway by Which the Tetrameric Protein Transthyretin Dissociates. Biochemistry 2005, 44, 15525–15533. [Google Scholar] [CrossRef]
- Pires, R.H.; Karsai, A.; Saraiva, M.J.; Damas, A.M.; Kellermayer, M.S.Z. Distinct Annular Oligomers Captured along the Assembly and Disassembly Pathways of Transthyretin Amyloid Protofibrils. PLoS ONE 2012, 7, e44992. [Google Scholar] [CrossRef]
- Quintas, A.; Vaz, D.B.D.M.C.; Cardoso, I.; Saraiva, M.J.M.; Brito, R. Tetramer Dissociation and Monomer Partial Unfolding Precedes Protofibril Formation in Amyloidogenic Transthyretin Variants. J. Biol. Chem. 2001, 276, 27207–27213. [Google Scholar] [CrossRef]
- Dasari, A.K.R.; Hughes, R.M.; Wi, S.; Hung, I.; Gan, Z.; Kelly, J.W.; Lim, K.H. Transthyretin Aggregation Pathway toward the Formation of Distinct Cytotoxic Oligomers. Sci. Rep. 2019, 9, 33. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Transthyretin Amyloidosis: Update on the Clinical Spectrum, Pathogenesis, and Disease-Modifying Therapies. Neurol. Ther. 2020, 9, 317–333. [Google Scholar] [CrossRef]
- Chuang, E.; Hori, A.; Hesketh, C.D.; Shorter, J. Amyloid assembly and disassembly. J. Cell Sci. 2018, 131, jcs189928. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Jamerlan, A.; Shim, K.H.; An, S.S.A. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. Int. J. Mol. Sci. 2019, 20, 2982. [Google Scholar] [CrossRef] [PubMed]
- Bergström, J.; Gustavsson, Å.; Hellman, U.; Sletten, K.; Murphy, C.L.; Weiss, D.T.; Solomon, A.; Olofsson, B.-O.; Westermark, P. Amyloid deposits in transthyretin-derived amyloidosis: Cleaved transthyretin is associated with distinct amyloid morphology. J. Pathol. 2005, 206, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Bergström, J.; Solomon, A.; Murphy, C.; Sletten, K. Transthyretin-derived senile systemic amyloidosis: Clinicopathologic and structural considerations. Amyloid 2003, 10, 48–54. [Google Scholar] [CrossRef]
- Marcoux, J.; Mangione, P.P.; Porcari, R.; Degiacomi, M.; Verona, G.; Taylor, G.W.; Giorgetti, S.; Raimondi, S.; Cianferani, S.; Benesch, J.; et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 2015, 7, 1337–1349. [Google Scholar] [CrossRef]
- Raimondi, S.; Mangione, P.P.; Verona, G.; Canetti, D.; Nocerino, P.; Marchese, L.; Piccarducci, R.; Mondani, V.; Faravelli, G.; Taylor, G.W.; et al. Comparative study of the stabilities of synthetic in vitro and natural ex vivo transthyretin amyloid fibrils. J. Biol. Chem. 2020, 295, 11379–11387. [Google Scholar] [CrossRef]
- Bateman, D.A.; Tycko, R.; Wickner, R.B. Experimentally Derived Structural Constraints for Amyloid Fibrils of Wild-Type Transthyretin. Biophys. J. 2011, 101, 2485–2492. [Google Scholar] [CrossRef][Green Version]
- Poulsen, K.; Mc Bahl, J.; Simonsen, A.H.; Hasselbalch, S.G.; Heegaard, N.H. Distinct transthyretin oxidation isoform profile in spinal fluid from patients with Alzheimer’s disease and mild cognitive impairment. Clin. Proteom. 2014, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Gales, L.; Saraiva, M.J.; Damas, A.M. Structural basis for the protective role of sulfite against transthyretin amyloid formation. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2007, 1774, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.D.; Minamino, N.; Takao, T. Free Thiol of Transthyretin in Human Plasma Most Accessible to Modification/Oxidation. Anal. Chem. 2015, 87, 10785–10791. [Google Scholar] [CrossRef]
- Sharma, M.; Khan, S.; Rahman, S.; Singh, L.R. The Extracellular Protein, Transthyretin Is an Oxidative Stress Biomarker. Front. Physiol. 2019, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Garai, K.; Posey, A.E.; Li, X.; Buxbaum, J.N.; Pappu, R.V. Inhibition of amyloid beta fibril formation by monomeric human transthyretin. Protein Sci. 2018, 27, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Stankovic-Valentin, N.; Drzewicka, K.; König, C.; Schiebel, E.; Melchior, F. Redox regulation of SUMO enzymes is required for ATM activity and survival in oxidative stress. EMBO J. 2016, 35, 1312–1329. [Google Scholar] [CrossRef]
- Gupta, M.K.; McLendon, P.M.; Gulick, J.; James, J.; Khalili, K.; Robbins, J. UBC9-Mediated Sumoylation Favorably Impacts Cardiac Function in Compromised Hearts. Circ. Res. 2016, 118, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Wada, H.; Abe, Y.; Niikura, T. Alteration of global protein SUMOylation in neurons and astrocytes in response to Alzheimer’s disease-associated insults. Biochem. Biophys. Res. Commun. 2018, 500, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, E.; Kędracka–Krok, S.; Sołtys, K.; Jankowska, U.; Hołubowicz, R.; Seliga, J.; Ożyhar, A. Is Transthyretin a Regulator of Ubc9 SUMOylation? PLoS ONE 2016, 11, e0160536. [Google Scholar] [CrossRef]
- Ahmad, Y.; Sharma, N.; Garg, I.; Ahmad, M.F.; Sharma, M.; Bhargava, K. An Insight into the Changes in Human Plasma Proteome on Adaptation to Hypobaric Hypoxia. PLoS ONE 2013, 8, e67548. [Google Scholar] [CrossRef]
- Domoto, H.; Iwaya, K.; Ikomi, F.; Matsuo, H.; Tadano, Y.; Fujii, S.; Tachi, K.; Itoh, Y.; Sato, M.; Inoue, K.; et al. Up-Regulation of Antioxidant Proteins in the Plasma Proteome during Saturation Diving: Unique Coincidence under Hypobaric Hypoxia. PLoS ONE 2016, 11, e0163804. [Google Scholar] [CrossRef]
- Sartiani, L.; Bucciantini, M.; Spinelli, V.; Leri, M.; Natalello, A.; Nosi, D.; Doglia, S.M.; Relini, A.; Penco, A.; Giorgetti, S.; et al. Biochemical and Electrophysiological Modification of Amyloid Transthyretin on Cardiomyocytes. Biophys. J. 2016, 111, 2024–2038. [Google Scholar] [CrossRef]
- Teixeira, P.F.; Cerca, F.; Santos, S.D.; Saraiva, M.J. Endoplasmic Reticulum Stress Associated with Extracellular Aggregates. J. Biol. Chem. 2006, 281, 21998–22003. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanaka, K.; Essick, E.E.; Doros, G.; Tanriverdi, K.; Connors, L.H.; Seldin, D.C.; Sam, F. Circulating Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Cardiac Amyloidosis. J. Am. Heart Assoc. 2013, 2, e005868. [Google Scholar] [CrossRef] [PubMed]
- Fong, V.H.; Vieira, A. Cytotoxic Effects of Transthyretin Aggregates in an Epidermoid Cell Line. Pathobiology 2017, 84, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.; Iovanna, J.L.; Mak, T.W. DJ-1/PARK7 is an important mediator of hypoxia-induced cellular responses. Proc. Natl. Acad. Sci. USA 2009, 106, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Koide-Yoshida, S.; Niki, T.; Ueda, M.; Himeno, S.; Taira, T.; Iguchi-Ariga, S.M.; Ando, Y.; Ariga, H. DJ-1 degrades transthyretin and an inactive form of DJ-1 is secreted in familial amyloidotic polyneuropathy. Int. J. Mol. Med. 2007, 19, 885–893. [Google Scholar] [CrossRef]
- Sousa, M.M.; Amaral, J.; Guimarães, A.; Saraiva, M.J. Upregulation of the Extracellular Matrix Remodeling Genes, Biglycan, Neutrophil Gelatinase-Associated Lipocalin and Matrix Metalloproteinase-9 in Familial Amyloid Polyneuropathy. Amyloid Amyloidosis 2004, 20, 320–322. [Google Scholar] [CrossRef]
- Martins, D.; Moreira, J.; Gonçalves, N.P.; Saraiva, M.J. MMP-14 overexpression correlates with the neurodegenerative process in familial amyloidotic polyneuropathy. Dis. Model. Mech. 2017, 10, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Dubrey, S.W.; Cha, K.; Skinner, M.; LaValley, M.; Falk, R.H. Familial and primary (AL) cardiac amyloidosis: Echocardiographically similar diseases with distinctly different clinical outcomes. Heart 1997, 78, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [PubMed]
- Moore, K.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Gong, L.; Zhu, L.; Wang, S.; Zhang, Z. Transthyretin regulates the migration and invasion of JEG-3 cells. Oncol. Lett. 2016, 13, 1242–1246. [Google Scholar] [CrossRef]
- Zhu, L.; Baczyk, R.; Lye, S.J.; Zhang, Z. Preeclampsia is associated with low placental transthyretin levels. Taiwan J. Obstet. Gynecol. 2016, 55, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-B.; Nakashima, A.; Sharma, S. Understanding Pre-Eclampsia Using Alzheimer’s Etiology: An Intriguing Viewpoint. Am. J. Reprod. Immunol. 2015, 75, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Solomon, J.P.; Reixach, N.; Kelly, J.W. Sulfated Glycosaminoglycans Accelerate Transthyretin Amyloidogenesis by Quaternary Structural Conversion. Biochemistry 2010, 50, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Mechler, A.; Martin, L.L.; Aguilar, M.-I.; Small, D.H. Cholesterol and anionic phospholipids increase the binding of amyloidogenic transthyretin to lipid membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2008, 1778, 198–205. [Google Scholar] [CrossRef]
- Hou, X.; Richardson, S.; Aguilar, M.-I.; Small, D.H. Binding of Amyloidogenic Transthyretin to the Plasma Membrane Alters Membrane Fluidity and Induces Neurotoxicity. Biochemistry 2005, 44, 11618–11627. [Google Scholar] [CrossRef]
- Poltash, M.L.; Shirzadeh, M.; McCabe, J.W.; Moghadamchargari, Z.; Laganowsky, A.; Russell, D.H. New insights into the metal-induced oxidative degradation pathways of transthyretin. Chem. Commun. 2019, 55, 4091–4094. [Google Scholar] [CrossRef]
- Fleming, C.E.; Mar, F.M.; Franquinho, F.; Saraiva, M.J.; Sousa, M.M. Transthyretin Internalization by Sensory Neurons Is Megalin Mediated and Necessary for Its Neuritogenic Activity. J. Neurosci. 2009, 29, 3220–3232. [Google Scholar] [CrossRef]
- Hou, X.; Parkington, H.C.; Coleman, H.A.; Mechler, A.; Martin, L.; Aguilar, M.-I.; Small, D.H. Transthyretin oligomers induce calcium influx via voltage-gated calcium channels. J. Neurochem. 2007, 100, 446–457. [Google Scholar] [CrossRef]
- Gasperini, R.J.; Hou, X.; Parkington, H.; Coleman, H.; Klaver, D.W.; Vincent, A.J.; Foa, L.C.; Small, D.H. TRPM8 and Nav1.8 sodium channels are required for transthyretin-induced calcium influx in growth cones of small-diameter TrkA-positive sensory neurons. Mol. Neurodegener. 2011, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.J.; Bradwell, A. Identification of the serum binding proteins for iron, zinc, cadmium, nickel, and calcium. Clin. Chem. 1983, 29, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.C.; Smith, W.O.; Wulff, B. Calcium-45 binding by human prealbumin. J. Appl. Physiol. 1959, 14, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Masri, A.; Bukhari, S.; Eisele, Y.S.; Soman, P. Molecular Imaging of Cardiac Amyloidosis. J. Nucl. Med. 2020, 61, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Rose-Martel, M.; Smiley, S.; Hincke, M.T. Novel identification of matrix proteins involved in calcitic biomineralization. J. Proteom. 2015, 116, 81–96. [Google Scholar] [CrossRef]
- Wieczorek, E.; Chitruń, A.; Ożyhar, A. Destabilised human transthyretin shapes the morphology of calcium carbonate crystals. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 313–324. [Google Scholar] [CrossRef]
- Mori, Y.; Urushida, Y.; Nakano, M.; Uchiyama, S.; Kamino, K. Calcite-specific coupling protein in barnacle underwater cement. FEBS J. 2007, 274, 6436–6446. [Google Scholar] [CrossRef]
- So, C.R.; Liu, J.; Fears, K.P.; Leary, D.H.; Golden, J.P.; Wahl, K.J. Self-Assembly of Protein Nanofibrils Orchestrates Calcite Step Movement through Selective Nonchiral Interactions. ACS Nano 2015, 9, 5782–5791. [Google Scholar] [CrossRef]
- Mohanram, H.; Kumar, A.; Verma, C.S.; Pervushin, K.; Miserez, A. Three-dimensional structure of Megabalanus rosa Cement Protein 20 revealed by multi-dimensional NMR and molecular dynamics simulations. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190198. [Google Scholar] [CrossRef]
- Sullan, R.M.A.; Gunari, N.; Tanur, A.E.; Chan, Y.; Dickinson, G.H.; Orihuela, B.; Rittschof, D.; Walker, G.C. Nanoscale structures and mechanics of barnacle cement. Biofouling 2009, 25, 263–275. [Google Scholar] [CrossRef]
- Evans, J.S. “Liquid-like” biomineralization protein assemblies: A key to the regulation of non-classical nucleation. CrystEngComm 2013, 15, 8388–8394. [Google Scholar] [CrossRef]
- Rani, R.S.; Saharay, M. Molecular dynamics simulation of protein-mediated biomineralization of amorphous calcium carbonate. RSC Adv. 2019, 9, 1653–1663. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Tan, H.; Chen, G.; Guo, L.; Yang, L. Analysis of the Mineral Composition of the Human Calcified Cartilage Zone. Int. J. Med Sci. 2012, 9, 353–360. [Google Scholar] [CrossRef]
- Kajander, E.O.; Çiftçioglu, N. Nanobacteria: An alternative mechanism for pathogenic intra- and extracellular calcification and stone formation. Proc. Natl. Acad. Sci. USA 1998, 95, 8274–8279. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.; Young, J.D.-E. Purported nanobacteria in human blood as calcium carbonate nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 5549–5554. [Google Scholar] [CrossRef] [PubMed]
- Jahnen-Dechent, W.; Büscher, A.; Köppert, S.; Heiss, A.; Kuro-o, M.; Smith, E.R. Mud in the blood: The role of protein-mineral complexes and extracellular vesicles in biomineralisation and calcification. J. Struct. Biol. 2020, 212, 107577. [Google Scholar] [CrossRef]
- Çiftçioğlu, N.; McKay, D.S. Pathological Calcification and Replicating Calcifying- Nanoparticles: General Approach and Correlation. Pediatr. Res. 2010, 67, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Yuzhalin, A.; Borisov, V.V.; Velikanova, E.A.; Frolov, A.V.; Sakharova, V.M.; Brusina, E.B.; Golovkin, A. Calcifying nanoparticles: One face of distinct entities? Front. Microbiol. 2014, 5, 214. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Hewitson, T.; Hanssen, E.; Holt, S.G. Biochemical transformation of calciprotein particles in uraemia. Bone 2018, 110, 355–367. [Google Scholar] [CrossRef]
- Martel, J.; Young, D.; Young, A.; Wu, C.-Y.; Chen, C.-D.; Yu, J.-S.; Young, J.D. Comprehensive proteomic analysis of mineral nanoparticles derived from human body fluids and analyzed by liquid chromatography–tandem mass spectrometry. Anal. Biochem. 2011, 418, 111–125. [Google Scholar] [CrossRef]
- Simon, J.; Kuhn, G.; Fichter, M.; Gehring, S.; Landfester, K.; Mailänder, V. Unraveling the In Vivo Protein Corona. Cells 2021, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, J.E. A study on the mechanism of protein adsorption to TiO2. Biomaterials 1991, 12, 593–596. [Google Scholar] [CrossRef]
- Heller, D.; Helmerhorst, E.; Oppenheim, F. Saliva and Serum Protein Exchange at the Tooth Enamel Surface. J. Dent. Res. 2017, 96, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Reixach, N.; Matsuzaki, T.; Alvarez-Garcia, O.; Olmer, M.; Iwamoto, Y.; Buxbaum, J.N.; Lotz, M.K. Transthyretin Deposition in Articular Cartilage: A Novel Mechanism in the Pathogenesis of Osteoarthritis. Arthritis Rheumatol. 2015, 67, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Siepen, F.A.D.; Hein, S.; Prestel, S.; Baumgärtner, C.; Schönland, S.; Hegenbart, U.; Röcken, C.; Katus, H.A.; Kristen, A.V. Carpal tunnel syndrome and spinal canal stenosis: Harbingers of transthyretin amyloid cardiomyopathy? Clin. Res. Cardiol. 2019, 108, 1324–1330. [Google Scholar] [CrossRef]
- Westermark, P.; Westermark, G.T.; Suhr, O.; Berg, S. Transthyretin-derived amyloidosis: Probably a common cause of lumbar spinal stenosis. Upsala J. Med. Sci. 2014, 119, 223–228. [Google Scholar] [CrossRef]
- Lee, J.; Mun, S.; Kim, D.; Lee, Y.; Sheen, D.; Ihm, C.; Lee, S.H.; Kang, H. Proteomics Analysis for Verification of Rheumatoid Arthritis Biomarker Candidates Using Multiple Reaction Monitoring. Proteom. Clin. Appl. 2019, 13, e1800011. [Google Scholar] [CrossRef]
- Ni, M.; Wei, W.; Feng, Q.; Sun, X.G.; Wang, Y.C.; Gu, Y.J.; Zheng, F. Transthyretin as a potential serological marker for the diagnosis of patients with early rheumatoid arthritis. Clin. Exp. Rheumatol. 2013, 31, 394–399. [Google Scholar]
- Edilova, M.I.; Akram, A.; Abdul-Sater, A.A. Innate immunity drives pathogenesis of rheumatoid arthritis. Biomed. J. 2020, 44, 172–182. [Google Scholar] [CrossRef]
- Clement, C.C.; Moncrieffe, H.; Lele, A.; Janow, G.; Becerra, A.; Bauli, F.; Saad, F.A.; Perino, G.; Montagna, C.; Cobelli, N.; et al. Autoimmune response to transthyretin in juvenile idiopathic arthritis. JCI Insight 2016, 1, e85633. [Google Scholar] [CrossRef]
- Takinami, Y.; Yoshimatsu, S.; Uchiumi, T.; Toyosaki-Maeda, T.; Morita, A.; Ishihara, T.; Yamane, S.; Fukuda, I.; Okamoto, H.; Numata, Y.; et al. Identification of Potential Prognostic Markers for Knee Osteoarthritis by Serum Proteomic Analysis. Biomark. Insights 2013, 8, BMI-S11966. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Akasaki, Y.; Olmer, M.; Alvarez-Garcia, O.; Reixach, N.; Buxbaum, J.N.; Lotz, M.K. Transthyretin deposition promotes progression of osteoarthritis. Aging Cell 2017, 16, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-S.; Zhang, J.-R.; Zhao, Y.-L.; Li, Y.; Sun, Y.; Liu, T.; Wang, R.-T. Reduced prealbumin is associated with bone mineral density in women with osteoporosis. Nutrition 2017, 33, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Xiu, S.; Chhetri, J.K.; Sun, L.; Mu, Z.; Wang, L. Association of serum prealbumin with risk of osteoporosis in older adults with type 2 diabetes mellitus: A cross-sectional study. Ther. Adv. Chronic Dis. 2019, 10, 2040622319857361. [Google Scholar] [CrossRef] [PubMed]
- Ea, H.-K.; Nguyen, C.; Bazin, D.; Bianchi, A.; Guicheux, J.; Reboul, P.; Daudon, M.; Lioté, F. Articular cartilage calcification in osteoarthritis: Insights into crystal-induced stress. Arthritis Rheum. 2010, 63, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Stack, J.; McCarthy, G. Cartilage calcification and osteoarthritis: A pathological association? Osteoarthr. Cartil. 2020, 28, 1301–1302. [Google Scholar] [CrossRef]
- Chang, J.; Jackson, S.G.; Wardale, J.; Jones, S.W. Hypoxia Modulates the Phenotype of Osteoblasts Isolated from Knee Osteoarthritis Patients, Leading to Undermineralized Bone Nodule Formation. Arthritis Rheumatol. 2014, 66, 1789–1799. [Google Scholar] [CrossRef]
- Sharma, A.R.; Jagga, S.; Lee, S.-S.; Nam, J.-S. Interplay between Cartilage and Subchondral Bone Contributing to Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2013, 14, 19805–19830. [Google Scholar] [CrossRef]
- Nunes, A.F.; Saraiva, M.J.; Sousa, M. Transthyretin knockouts are a new mouse model for increased neuropeptide Y. FASEB J. 2005, 20, 166–168. [Google Scholar] [CrossRef]
- Nunes, A.F.; Liz, M.; Franquinho, F.; Teixeira, L.M.; Sousa, V.; Chenu, C.; Lamghari, M.; Sousa, M. Neuropeptide Y expression and function during osteoblast differentiation—Insights from transthyretin knockout mice. FEBS J. 2010, 277, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Franquinho, F.; Liz, M.; Nunes, A.F.; Neto, E.; Lamghari, M.; Sousa, M.M. Neuropeptide Y and osteoblast differentiation—The balance between the neuro-osteogenic network and local control. FEBS J. 2010, 277, 3664–3674. [Google Scholar] [CrossRef]
- Steinthorsdottir, V.; Thorleifsson, G.; Sulem, P.; Helgason, H.; Grarup, N.; Sigurdsson, A.; Helgadottir, H.; Johannsdottir, H.; Magnusson, O.T.; Gudjonsson, S.A.; et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat. Genet. 2014, 46, 294–298. [Google Scholar] [CrossRef]
- Bäck, N.; Luxmi, R.; Powers, K.G.; Mains, R.E.; Eipper, B.A. Peptidylglycine α-amidating monooxygenase is required for atrial secretory granule formation. Proc. Natl. Acad. Sci. USA 2020, 117, 17820–17831. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, P.; Perrone-Filardi, P. Dangerous relationships: Aortic stenosis and transthyretin cardiac amyloidosis. Eur. Heart J. 2017, 38, 2888–2889. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.; Castaño, A.; Burton, A.; Grodin, J.L. Transthyretin amyloid cardiomyopathy in women: Frequency, characteristics, and diagnostic challenges. Heart Fail. Rev. 2021, 26, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Mosterd, A.; Hoes, A.W. Clinical epidemiology of heart failure. Heart 2007, 93, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Sharma, A.; Chaddar, M.K.; Yadav, R.; Agnihotri, P.; Kar, A.; Biswas, S. Plasma Proteome Profiling of Coronary Artery Disease Patients: Downregulation of Transthyretin—An Important Event. Mediat. Inflamm. 2020, 2020, 3429541. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, P.; Xia, K.; Fang, H.; Jiang, M.; Xie, Q.; Yu, Z.; Yang, T. Association of Serum Prealbumin with Angiographic Severity in Patients with Acute Coronary Syndrome. Med. Sci. Monit. 2017, 23, 4041–4049. [Google Scholar] [CrossRef]
- Wakabayashi, I.; Marumo, M.; Nonaka, D.; Lee, L.-J.; Mukai, J.; Ohki, M.; Tanaka, K.; Uchida, K. Cysteinylated transthyretin as a discriminator of cardiovascular risk in patients with diabetes mellitus. Clin. Chim. Acta 2017, 470, 46–50. [Google Scholar] [CrossRef]
- Hanson, J.L.; Arvanitis, M.; Koch, C.M.; Berk, J.L.; Ruberg, F.L.; Prokaeva, T.; Connors, L.H. Use of Serum Transthyretin as a Prognostic Indicator and Predictor of Outcome in Cardiac Amyloid Disease Associated with Wild-Type Transthyretin. Circ. Heart Fail. 2018, 11, e004000. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Henze, A.; Espe, K.M.; Wanner, C.; Krane, V.; Raila, J.; Hocher, B.; Schweigert, F.J.; Drechsler, C. Transthyretin Predicts Cardiovascular Outcome in Hemodialysis Patients with Type 2 Diabetes. Diabetes Care 2012, 35, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Heuschkel, M.A.; Skenteris, N.T.; Hutcheson, J.D.; Van Der Valk, D.D.; Bremer, J.; Goody, P.; Hjortnaes, J.; Jansen, F.; Bouten, C.V.; Bogaerdt, A.V.D.; et al. Integrative Multi-Omics Analysis in Calcific Aortic Valve Disease Reveals a Link to the Formation of Amyloid-Like Deposits. Cells 2020, 9, 2164. [Google Scholar] [CrossRef]
- Castano, A.; Bokhari, S.; Maurer, M.S. Unveiling wild-type transthyretin cardiac amyloidosis as a significant and potentially modifiable cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2595–2597. [Google Scholar] [CrossRef]
- Jensen, S.B.; Hindberg, K.; Solomon, T.; Smith, E.N.; Lapek, J.D.; Gonzalez, D.J.; Latysheva, N.; Frazer, K.A.; Braekkan, S.K.; Hansen, J.-B. Discovery of novel plasma biomarkers for future incident venous thromboembolism by untargeted synchronous precursor selection mass spectrometry proteomics. J. Thromb. Haemost. 2018, 16, 1763–1774. [Google Scholar] [CrossRef]
- Franz, T.; Rüggeberg, S.; Horn, P.; Li, X.; Vajkoczy, P. Detection of a γ-Carboxy-Glutamate as Novel Post-Translational Modification of Human Transthyretin. Protein Pept. Lett. 2008, 15, 43–46. [Google Scholar] [CrossRef]
- Parry, T.L.; Melehani, J.; Ranek, M.J.; Willis, M. Functional Amyloid Signaling via the Inflammasome, Necrosome, and Signalosome: New Therapeutic Targets in Heart Failure. Front. Cardiovasc. Med. 2015, 2, 25. [Google Scholar] [CrossRef]
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 2017, 135, 1357–1377. [Google Scholar] [CrossRef]
- Culotta, V.; Moon, J.C. Transthyretin Cardiac Amyloidosis: From Rare Monogenic Disease to Common Pathway in Heart Failure? Rev. Esp. Cardiol. 2016, 69, 888–889. [Google Scholar] [CrossRef]
- Mirzoyev, S.A.; Edwards, W.D.; Mohammed, S.F.; Donovan, J.L.; Roger, V.L.; Grogan, D.R.; Redfield, M.M. Cardiac Amyloid Deposition is Common in Elderly Patients with Heart Failure and Preserved Ejection Fraction. Circulation 2010, 122, A17926. [Google Scholar] [CrossRef]
- Da Silva, D.M.; Langer, H.; Graf, T. Inflammatory and Molecular Pathways in Heart Failure—Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis. Int. J. Mol. Sci. 2019, 20, 2322. [Google Scholar] [CrossRef]
- Ami, D.A.; Mereghetti, P.; Leri, M.; Giorgetti, S.; Natalello, A.; Doglia, S.M.; Stefani, M.; Bucciantini, M. A FTIR microspectroscopy study of the structural and biochemical perturbations induced by natively folded and aggregated transthyretin in HL-1 cardiomyocytes. Sci. Rep. 2018, 8, 12508. [Google Scholar] [CrossRef]
- Andrikopoulou, E.; Bhambhvani, P. Nuclear imaging of cardiac amyloidosis. J. Nucl. Cardiol. 2017, 26, 505–508. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef]
- Stats, M.A.; Stone, J.R. Varying levels of small microcalcifications and macrophages in ATTR and AL cardiac amyloidosis: Implications for utilizing nuclear medicine studies to subtype amyloidosis. Cardiovasc. Pathol. 2016, 25, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular Calcification—New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Cannata-Andia, J.B.; Roman-Garcia, P.; Hruska, K. The connections between vascular calcification and bone health. Nephrol. Dial. Transplant. 2011, 26, 3429–3436. [Google Scholar] [CrossRef]
- Lerman, D.; Prasad, S.; Alotti, N. Calcific Aortic Valve Disease: Molecular Mechanisms and Therapeutic Approaches. Eur. Cardiol. Rev. 2015, 10, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, B.; Manian, U.; Sheyin, O.; Davey, R.; De, S. Stroke risk and atrial mechanical dysfunction in cardiac amyloidosis. ESC Heart Fail. 2020, 7, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.L.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus—Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef]
- Stabley, J.N.; Towler, D.A. Arterial Calcification in Diabetes Mellitus. Arter. Thromb. Vasc. Biol. 2017, 37, 205–217. [Google Scholar] [CrossRef]
- Dekki, N.; Refai, E.; Holmberg, R.; Köhler, M.; Jörnvall, H.; Berggren, P.-O.; Juntti-Berggren, L. Transthyretin binds to glucose-regulated proteins and is subjected to endocytosis by the pancreatic β-cell. Cell. Mol. Life Sci. 2011, 69, 1733–1743. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Shao, J.; Yin, Y.; Yin, X.; Ji, L.; Xin, Y.; Zou, J.; Yao, Y. Transthyretin Exerts Pro-Apoptotic Effects in Human Retinal Microvascular Endothelial Cells Through a GRP78-Dependent Pathway in Diabetic Retinopathy. Cell. Physiol. Biochem. 2017, 43, 788–800. [Google Scholar] [CrossRef]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef]
- Genereux, J.C.; Wiseman, R.L. Regulating extracellular proteostasis capacity through the unfolded protein response. Prion 2015, 9, 10–21. [Google Scholar] [CrossRef]
- Park, S.J. Protein–Nanoparticle Interaction: Corona Formation and Conformational Changes in Proteins on Nanoparticles. Int. J. Nanomed. 2020, 15, 5783–5802. [Google Scholar] [CrossRef]
- Ballet, T.; Boulangé, L.; Brechet, Y.; Bruckert, F.; Weidenhaupt, M. Protein conformational changes induced by adsorption onto material surfaces: An important issue for biomedical applications of material science. Bull. Pol. Acad. Sci. Tech. Sci. 2010, 58, 303–315. [Google Scholar] [CrossRef]
- Gebbink, M.F.; Bouma, B.; Maas, C.; Bouma, B.N. Physiological responses to protein aggregates: Fibrinolysis, coagulation and inflammation (new roles for old factors). FEBS Lett. 2009, 583, 2691–2699. [Google Scholar] [CrossRef] [PubMed]
- Cesarman-Maus, G.C.; Hajjar, K.A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 2005, 129, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, M.; Suriyaphol, P.; Paprotka, K.; Spath, L.; Ochsenhirt, V.; Schmitt, A.; Han, S.-R.; Husmann, M.; Gerl, V.B.; Bhakdi, S.; et al. Enzymatic Modification of Low-Density Lipoprotein in the Arterial Wall. Arter. Thromb. Vasc. Biol. 2004, 24, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Miszta, A.; Huskens, D.; Donkervoort, D.; Roberts, M.; Wolberg, A.; de Laat, B. Assessing Plasmin Generation in Health and Disease. Int. J. Mol. Sci. 2021, 22, 2758. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, P.; Luttun, A.; Martin-Ventura, J.L.; Lupu, F.; Carmeliet, P.; Collen, D.; Anglès-Cano, E.; Lijnen, H.R. Plasminogen activation: A mediator of vascular smooth muscle cell apoptosis in atherosclerotic plaques. J. Thromb. Haemost. 2005, 4, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.K.; Strickland, S. A critical role for plasminogen in inflammation. J. Exp. Med. 2020, 217, e20191865. [Google Scholar] [CrossRef]
- Mangione, P.P.; Verona, G.; Corazza, A.; Marcoux, J.; Canetti, D.; Giorgetti, S.; Raimondi, S.; Stoppini, M.; Esposito, M.; Relini, A.; et al. Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J. Biol. Chem. 2018, 293, 14192–14199. [Google Scholar] [CrossRef] [PubMed]
- Maia, L. Emerging CNS involvement in FAP-TTR long survival patients. Orphanet J. Rare Dis. 2015, 10, I14. [Google Scholar] [CrossRef]
- Schrutka, L.; Avanzini, N.; Seirer, B.; Rettl, R.; Dachs, T.; Duca, F.; Binder, C.; Dalos, D.; Eslam, R.B.; Bonderman, D. Bleeding events in patients with cardiac amyloidosis. Eur. Heart J. 2020, 41, 2122. [Google Scholar] [CrossRef]
- Bouma, B.; Maas, C.; Hazenberg, B.P.C.; Lokhorst, H.M.; Gebbink, M.F.B.G. Increased plasmin-α2-antiplasmin levels indicate activation of the fibrinolytic system in systemic amyloidoses. J. Thromb. Haemost. 2007, 5, 1139–1142. [Google Scholar] [CrossRef]
- Constantinescu, P.; Brown, R.A.; Wyatt, A.; Ranson, M.; Wilson, M.R. Amorphous protein aggregates stimulate plasminogen activation, leading to release of cytotoxic fragments that are clients for extracellular chaperones. J. Biol. Chem. 2017, 292, 14425–14437. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.D.C.; Ribeiro-Silva, C.; Ribeiro, R.; Gilberto, S.; Gomes, R.; Ferreira, A.; Mateus, E.P.; Barroso, E.; Coelho, A.; Ponces-Freire, A.; et al. Transthyretin Amyloidosis: Chaperone Concentration Changes and Increased Proteolysis in the Pathway to Disease. PLoS ONE 2015, 10, e0125392. [Google Scholar] [CrossRef] [PubMed]
- Zamolodchikov, D.; Berk-Rauch, H.E.; Oren, D.A.; Stor, D.S.; Singh, P.K.; Kawasaki, M.; Aso, K.; Strickland, S.; Ahn, H.J. Biochemical and structural analysis of the interaction between β-amyloid and fibrinogen. Blood 2016, 128, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Yaprak, E.; Kasap, M.; Akpinar, G.; Islek, E.E.; Sinanoglu, A. Abundant proteins in platelet-rich fibrin and their potential contribution to wound healing: An explorative proteomics study and review of the literature. J. Dent. Sci. 2018, 13, 386–395. [Google Scholar] [CrossRef]
- Dickinson, G.; Vega, I.E.; Wahl, K.J.; Orihuela, B.; Beyley, V.; Rodriguez, E.N.; Everett, R.; Bonaventura, J.; Rittschof, D. Barnacle cement: A polymerization model based on evolutionary concepts. J. Exp. Biol. 2009, 212, 3499–3510. [Google Scholar] [CrossRef]
- Chan, N.C.; Weitz, J.I. Recent advances in understanding, diagnosing and treating venous thrombosis. F1000Research 2020, 9, 1206. [Google Scholar] [CrossRef]
- Kwaan, H.; Lindholm, P. The Central Role of Fibrinolytic Response in COVID-19—A Hematologist’s Perspective. Int. J. Mol. Sci. 2021, 22, 1283. [Google Scholar] [CrossRef]
- Zinellu, A.; Mangoni, A.A. Serum Prealbumin Concentrations, COVID-19 Severity, and Mortality: A Systematic Review and Meta-Analysis. Front. Med. 2021, 8, 638529. [Google Scholar] [CrossRef]
- Schulz, C.; Engelmann, B.; Massberg, S. Crossroads of coagulation and innate immunity: The case of deep vein thrombosis. J. Thromb. Haemost. 2013, 11, 233–241. [Google Scholar] [CrossRef]
- Schuliga, M. The Inflammatory Actions of Coagulant and Fibrinolytic Proteases in Disease. Mediat. Inflamm. 2015, 2015, 437695. [Google Scholar] [CrossRef]
- Trimaille, A.; Thachil, J.; Marchandot, B.; Curtiaud, A.; Leonard-Lorant, I.; Carmona, A.; Matsushita, K.; Sato, C.; Sattler, L.; Grunebaum, L.; et al. D-Dimers Level as a Possible Marker of Extravascular Fibrinolysis in COVID-19 Patients. J. Clin. Med. 2020, 10, 39. [Google Scholar] [CrossRef]
- Colombo, G.; Clerici, M.; Altomare, A.; Rusconi, F.; Giustarini, D.; Portinaro, N.; Garavaglia, M.L.; Rossi, R.; Dalle-Donne, I.; Milzani, A.D.G. Thiol oxidation and di-tyrosine formation in human plasma proteins induced by inflammatory concentrations of hypochlorous acid. J. Proteom. 2017, 152, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J. The Dual Role of Myeloperoxidase in Immune Response. Int. J. Mol. Sci. 2020, 21, 8057. [Google Scholar] [CrossRef] [PubMed]
- Macedo, B.; Batista, A.R.; Ferreira, N.; Almeida, M.; Saraiva, M.J. Anti-apoptotic treatment reduces transthyretin deposition in a transgenic mouse model of Familial Amyloidotic Polyneuropathy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Bandeira-Melo, C.; Chimelli, L.; Waddington-Cruz, M.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. Inflammatory profiling of patients with familial amyloid polyneuropathy. BMC Neurol. 2019, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.K.; Chen, Z.-L.; Norris, E.; Revenko, A.S.; MacLeod, A.R.; Strickland, S. Blood-derived plasminogen drives brain inflammation and plaque deposition in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E9687–E9696. [Google Scholar] [CrossRef]
- Azevedo Estefania, P.; Foguel, D. The Role of Inflammation in Amyloid Diseasesitle. In Amyloid Diseases; IntechOpen: London, UK, 2018. [Google Scholar]
- Klebanoff, S.J.; Kettle, T.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef]
- Tang, W.W.; Wu, Y.; Nicholls, S.; Hazen, S.L. Plasma Myeloperoxidase Predicts Incident Cardiovascular Risks in Stable Patients Undergoing Medical Management for Coronary Artery Disease. Clin. Chem. 2011, 57, 33–39. [Google Scholar] [CrossRef]
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Münzel, T.; Simoons, M.L.; Hamm, C.W. Myeloperoxidase Serum Levels Predict Risk in Patients With Acute Coronary Syndromes. Circulation 2003, 108, 1440–1445. [Google Scholar] [CrossRef]
- Sukhorukov, V.; Khotina, V.; Ekta, M.B.; Ivanova, E.; Sobenin, I.; Orekhov, A. Endoplasmic Reticulum Stress in Macrophages: The Vicious Circle of Lipid Accumulation and Pro-Inflammatory Response. Biomedicines 2020, 8, 210. [Google Scholar] [CrossRef]
- Boukais, K.; Bayles, R.; Borges, L.D.F.; Louedec, L.; Boulaftali, Y.; Ho-Tin-Noé, B.; Arocas, V.; Bouton, M.-C.; Michel, J.-B. Uptake of Plasmin-PN-1 Complexes in Early Human Atheroma. Front. Physiol. 2016, 7, 273. [Google Scholar] [CrossRef]
- Lepedda, A.J.; Formato, M. Oxidative Modifications in Advanced Atherosclerotic Plaques: A Focus on In Situ Protein Sulfhydryl Group Oxidation. Oxidative Med. Cell. Longev. 2020, 2020, 6169825. [Google Scholar] [CrossRef] [PubMed]
- Harpel, P.C.; Chang, V.T.; Borth, W. Homocysteine and other sulfhydryl compounds enhance the binding of lipoprotein(a) to fibrin: A potential biochemical link between thrombosis, atherogenesis, and sulfhydryl compound metabolism. Proc. Natl. Acad. Sci. USA 1992, 89, 10193–10197. [Google Scholar] [CrossRef] [PubMed]
- Likozar, A.R.; Zavrtanik, M.; Šebeštjen, M. Lipoprotein(a) in atherosclerosis: From pathophysiology to clinical relevance and treatment options. Ann. Med. 2020, 52, 162–177. [Google Scholar] [CrossRef]
- Dato, V.A.; Chiabrando, G.A. The Role of Low-Density Lipoprotein Receptor-Related Protein 1 in Lipid Metabolism, Glucose Homeostasis and Inflammation. Int. J. Mol. Sci. 2018, 19, 1780. [Google Scholar] [CrossRef]
- Alemi, M.; Gaiteiro, C.; Ribeiro, C.A.; Santos, L.M.; Gomes, J.; Oliveira, S.M.; Couraud, P.-O.; Weksler, B.; Romero, I.; Saraiva, M.J.; et al. Transthyretin participates in beta-amyloid transport from the brain to the liver- involvement of the low-density lipoprotein receptor-related protein 1? Sci. Rep. 2016, 6, 20164. [Google Scholar] [CrossRef]
- Kudinov, V.A.; Alekseeva, O.Y.; Torkhovskaya, T.I.; Baskaev, K.K.; Artyushev, R.I.; Saburina, I.N.; Markin, S.S. High-Density Lipoproteins as Homeostatic Nanoparticles of Blood Plasma. Int. J. Mol. Sci. 2020, 21, 8737. [Google Scholar] [CrossRef]
- Kuai, R.; Li, D.; Chen, Y.E.; Moon, J.J.; Schwendeman, A. High-Density Lipoproteins (HDL)—Nature’s Multi-Functional Nanoparticles. ACS Nano 2016, 10, 3015–3041. [Google Scholar] [CrossRef]
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and antithrombotic actions of HDL. Circ. Res. 2006, 98, 1352–1364. [Google Scholar] [CrossRef]
- Tabet, F.; Vickers, K.C.; Torres, L.F.C.; Wiese, C.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef]
- Shao, J.; Fan, G.; Yin, X.; Gu, Y.; Wang, X.; Xin, Y.; Yao, Y. A novel transthyretin/STAT4/miR-223-3p/FBXW7 signaling pathway affects neovascularization in diabetic retinopathy. Mol. Cell. Endocrinol. 2019, 498, 110541. [Google Scholar] [CrossRef]
- Shao, J.; Zhang, Y.; Fan, G.; Xin, Y.; Yao, Y. Transcriptome analysis identified a novel 3-LncRNA regulatory network of transthyretin attenuating glucose induced hRECs dysfunction in diabetic retinopathy. BMC Med Genom. 2019, 12, 134. [Google Scholar] [CrossRef]
- Fogelman, A.M. Further evidence that high-density lipoprotein is a chameleon-like lipoprotein: Figure. Eur. Heart J. 2015, 36, 3017–3019. [Google Scholar] [CrossRef][Green Version]
- Liz, M.; Gomes, C.M.; Saraiva, M.J.; Sousa, M. ApoA-I cleaved by transthyretin has reduced ability to promote cholesterol efflux and increased amyloidogenicity. J. Lipid Res. 2007, 48, 2385–2395. [Google Scholar] [CrossRef]
- Westermark, P.; Mucchiano, G.; Marthin, T.; Johnson, K.H.; Sletten, K. Apolipoprotein A1-derived amyloid in human aortic atherosclerotic plaques. Am. J. Pathol. 1995, 147, 1186–1192. [Google Scholar]
- Begue, F.; Tanaka, S.; Mouktadi, Z.; Rondeau, P.; Veeren, B.; Diotel, N.; Tran-Dinh, A.; Robert, T.; Vélia, E.; Mavingui, P.; et al. Altered high-density lipoprotein composition and functions during severe COVID-19. Sci. Rep. 2021, 11, 2291. [Google Scholar] [CrossRef]
- Glushchenko, A.V.; Jacobsen, D.W. Molecular Targeting of Proteins by L-Homocysteine: Mechanistic Implications for Vascular Disease. Antioxid. Redox Signal. 2007, 9, 1883–1898. [Google Scholar] [CrossRef]
- Lim, A.; Sengupta, S.; McComb, M.E.; Théberge, R.; Wilson, W.G.; Costello, C.; Jacobsen, D.W. In Vitro and in Vivo Interactions of Homocysteine with Human Plasma Transthyretin. J. Biol. Chem. 2003, 278, 49707–49713. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, A.; Ramaswamy, V.; Kuruvilla, S.; Sehgal, P.K.; Balakrishnan, K. Calcified atherosclerotic plaque—Where exactly is the calcium and what does it contain? Indian J. Thorac. Cardiovasc. Surg. 2012, 28, 6–14. [Google Scholar] [CrossRef]
- Korosoglou, G.; Giusca, S.; Katus, H.A. The coronary calcium paradox: Yet another step towards the differentiation between stable and rupture-prone coronary plaques? Atherosclerosis 2018, 274, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Herrington David, M.; Chunhong, M.; Sarah, P.; ZongminG, F.; Guoqiang, Y.; Lulu, C.; Vidya, V.; Yi, F.; Yizhi, W.; Tim, H.; et al. Proteomic Architecture of Human Coronary and Aortic Atherosclerosis. bioRxiv 2017. [Google Scholar] [CrossRef]
- Liang, W.; Ward, L.J.; Karlsson, H.; Ljunggren, S.A.; Li, W.; Lindahl, M.; Yuan, X.-M. Distinctive proteomic profiles among different regions of human carotid plaques in men and women. Sci. Rep. 2016, 6, 26231. [Google Scholar] [CrossRef]
- Mautner, S.L.; Lin, F.; Mautner, G.C.; Roberts, W.C. Comparison in women versus men of composition of atherosclerotic plaques in native coronary arteries and in saphenous veins used as aortocoronary conduits. J. Am. Coll. Cardiol. 1993, 21, 1312–1318. [Google Scholar] [CrossRef]
- \Wendorff, C.; Wendorff, H.; Pelisek, J.; Tsantilas, P.; Zimmermann, A.; Zernecke, A.; Kuehnl, A.; Eckstein, H.-H. Carotid Plaque Morphology Is Significantly Associated with Sex, Age, and History of Neurological Symptoms. Stroke 2015, 46, 3213–3219. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Shao, J.; Yao, Y. Transthyretin represses neovascularization in diabetic retinopathy. Mol. Vis. 2016, 22, 1188–1197. [Google Scholar]
- Fan, G.; Gu, Y.; Zhang, J.; Xin, Y.; Shao, J.; Giampieri, F.; Battino, M. Transthyretin Upregulates Long Non-Coding RNA MEG3 by Affecting PABPC1 in Diabetic Retinopathy. Int. J. Mol. Sci. 2019, 20, 6313. [Google Scholar] [CrossRef]
- Cehofski, L.J.; Kruse, A.; Alsing, A.N.; Nielsen, J.E.; Pedersen, S.; Kirkeby, S.; Honoré, B.; Vorum, H. Intravitreal bevacizumab upregulates transthyretin in experimental branch retinal vein occlusion. Mol. Vis. 2018, 24, 759–766. [Google Scholar]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Navarro, E.; Mallén, A.; Cruzado, J.M.; Torras, J.; Hueso, M. Unveiling ncRNA regulatory axes in atherosclerosis progression. Clin. Transl. Med. 2020, 9, e5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, B.; Yang, Y.-X.; Jia, Q.-J.; Zhang, A.; Qi, Z.-W.; Zhang, J.-P. Long Noncoding RNAs in Pathological Cardiac Remodeling: A Review of the Update Literature. BioMed Res. Int. 2019, 2019, 7159592. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; He, Y.; Li, N.; Fang, X.; Shang, T.; Zhang, H.; Zheng, X. Long noncoding RNA MEG3 suppressed endothelial cell proliferation and migration through regulating miR-21. Am. J. Transl. Res. 2017, 9, 3326–3335. [Google Scholar] [PubMed]
- Zhang, Y.; Liu, X.; Bai, X.; Lin, Y.; Li, Z.; Fu, J.; Li, M.; Zhao, T.; Yang, H.; Xu, R.; et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J. Pineal Res. 2018, 64, e12449. [Google Scholar] [CrossRef]
- Fusaro, M.; Cianciolo, G.; Brandi, M.L.; Ferrari, S.; Nickolas, T.L.; Tripepi, G.; Plebani, M.; Zaninotto, M.; Iervasi, G.; La Manna, G.; et al. Vitamin K and Osteoporosis. Nutrients 2020, 12, 3625. [Google Scholar] [CrossRef]
- Elshaikh, A.O.; Shah, L.; Mathew, C.J.; Lee, R.; Jose, M.T.; Cancarevic, I. Influence of Vitamin K on Bone Mineral Density and Osteoporosis. Cureus 2020, 12, e10816. [Google Scholar] [CrossRef]
- Shioi, A.; Morioka, T.; Shoji, T.; Emoto, M. The Inhibitory Roles of Vitamin K in Progression of Vascular Calcification. Nutrients 2020, 12, 583. [Google Scholar] [CrossRef]
- Bang, O.Y.; Fujimura, M.; Kim, S.-K. The Pathophysiology of Moyamoya Disease: An Update. J. Stroke 2016, 18, 12–20. [Google Scholar] [CrossRef]


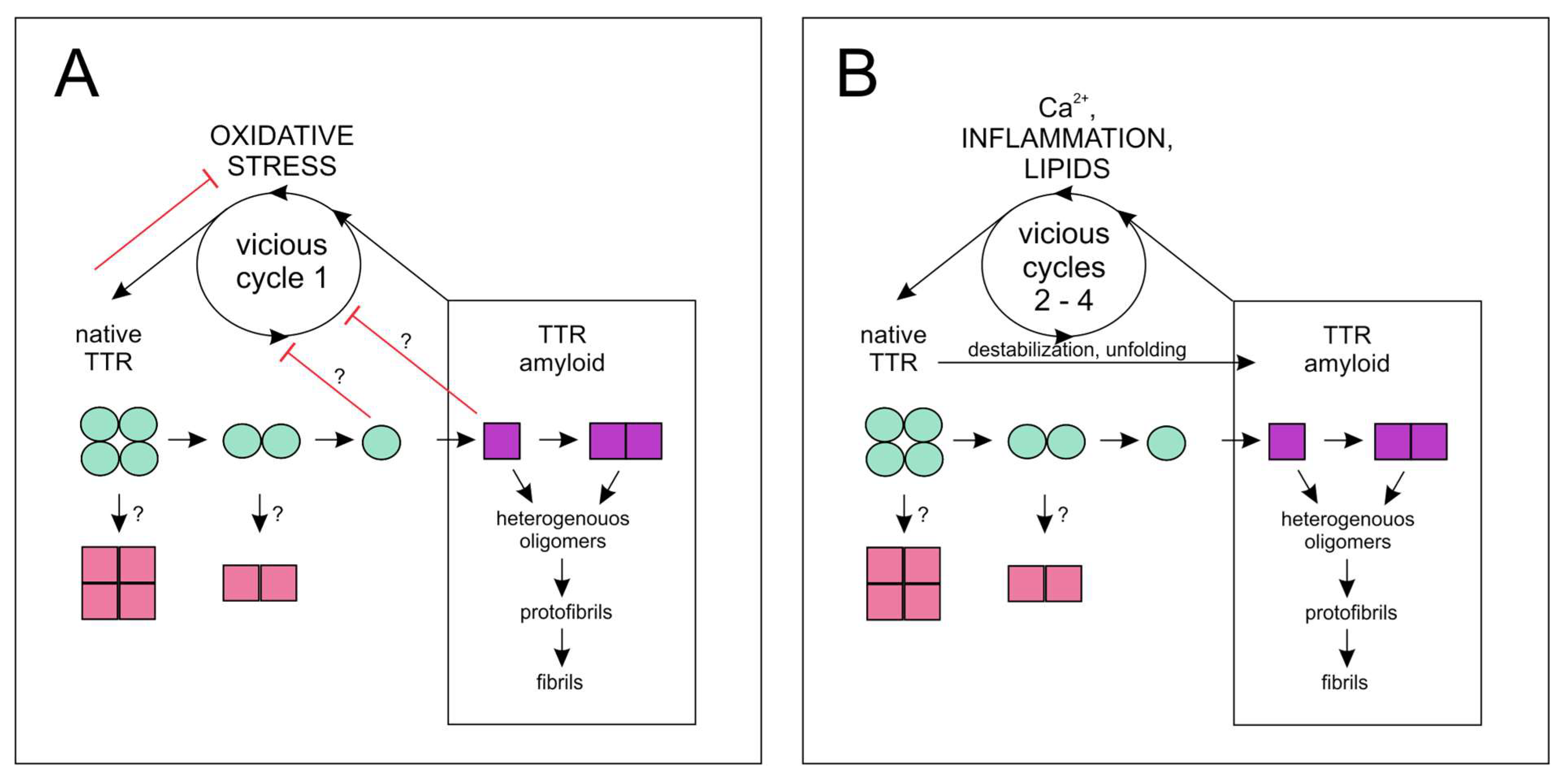{kind=link}
| Disease | TTR | Selected Effects | Other Factors/Conditions Involved | References |
|---|---|---|---|---|
| Rheumatoid arthritis | deregulation in plasma, different post-translational modification of TTR cysteine residue 10 | inflammation of the synovial membrane | gelsolin, angiotensinogen, lipopolysaccharide-binding protein, protein S100-A9 | [129,130] |
| Juvenile idiopathic arthritis | upregulation in plasma and synovial fluid, oxidized and aggregated TTR forms | chronic autoimmune disorder | innate immunity | [132] |
| Osteoarthritis | TTR proteolysis, reduced levels of the truncated form, deposition of TTR amyloid in articular cartilage | changed expression of catabolic and inflammatory genes, hypoxia | [126,133,134] | |
| Osteoporosis | low plasma levels | reduced bone mineral density | type 2 diabetes mellitus | [135,136] |
| Disease | TTR | Selected Symptoms/Effects | Other Factors/Conditions Involved | References |
|---|---|---|---|---|
| Cardiovascular disease | negative correlation, low plasma levels, positive correlation with the cysteinylated form of TTR | increased mortality, angiographic severity (coronary artery stenosis), vascular stiffness, arterial blockage by atherosclerotic plaque, acute coronary syndrome, mortality | type 2 diabetes mellitus acute coronary syndrome, oxidative stress and changes in the levels of proteins involved in blood coagulation, iron homeostasis, anti-oxidant and immune response, cell-matrix adhesion, response to Ca2+, plasmin and thrombin inhibition, HDL remodeling | [149,150,151,154] |
| Calcific aortic valve disease | amyloid deposits | calcified aortic valve | differentially expressed molecules of transcriptome, proteome, and miRNA, protein–protein interaction network, amyloid β precursor protein | [155] |
| ATTR cardiac amyloidosis | amyloid deposits, cardiac amyloid infiltration, lowered plasma levels | heart failure with preserved ejection fraction, aortic stenosis, left ventricular wall thickness | increased cardiomyocyte stiffness related to abnormal Ca2+ homeostasis preceded by osteoarticular disorders | [146,147,156] |
| Thromboembolism | upregulated plasma level | elevated thrombin generation and hypofibrinolytic state | reduced DJ-1 activity | [157] |
| Moya-moya disease | γ-carboxylation of glutamic acid residue 42 | extensive vascularization, stenosis | [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieczorek, E.; Ożyhar, A. Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases. Cells 2021, 10, 1768. https://doi.org/10.3390/cells10071768
Wieczorek E, Ożyhar A. Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases. Cells. 2021; 10(7):1768. https://doi.org/10.3390/cells10071768
Chicago/Turabian StyleWieczorek, Elżbieta, and Andrzej Ożyhar. 2021. "Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases" Cells 10, no. 7: 1768. https://doi.org/10.3390/cells10071768
APA StyleWieczorek, E., & Ożyhar, A. (2021). Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases. Cells, 10(7), 1768. https://doi.org/10.3390/cells10071768