
Impact of Repetitive DNA Elements on Snake Genome Biology and Evolution





Abstract
1. Background
2. Snake Genomes: A Model System for Studying Repetitive DNA in the Context of Chromosome Evolution Dynamics
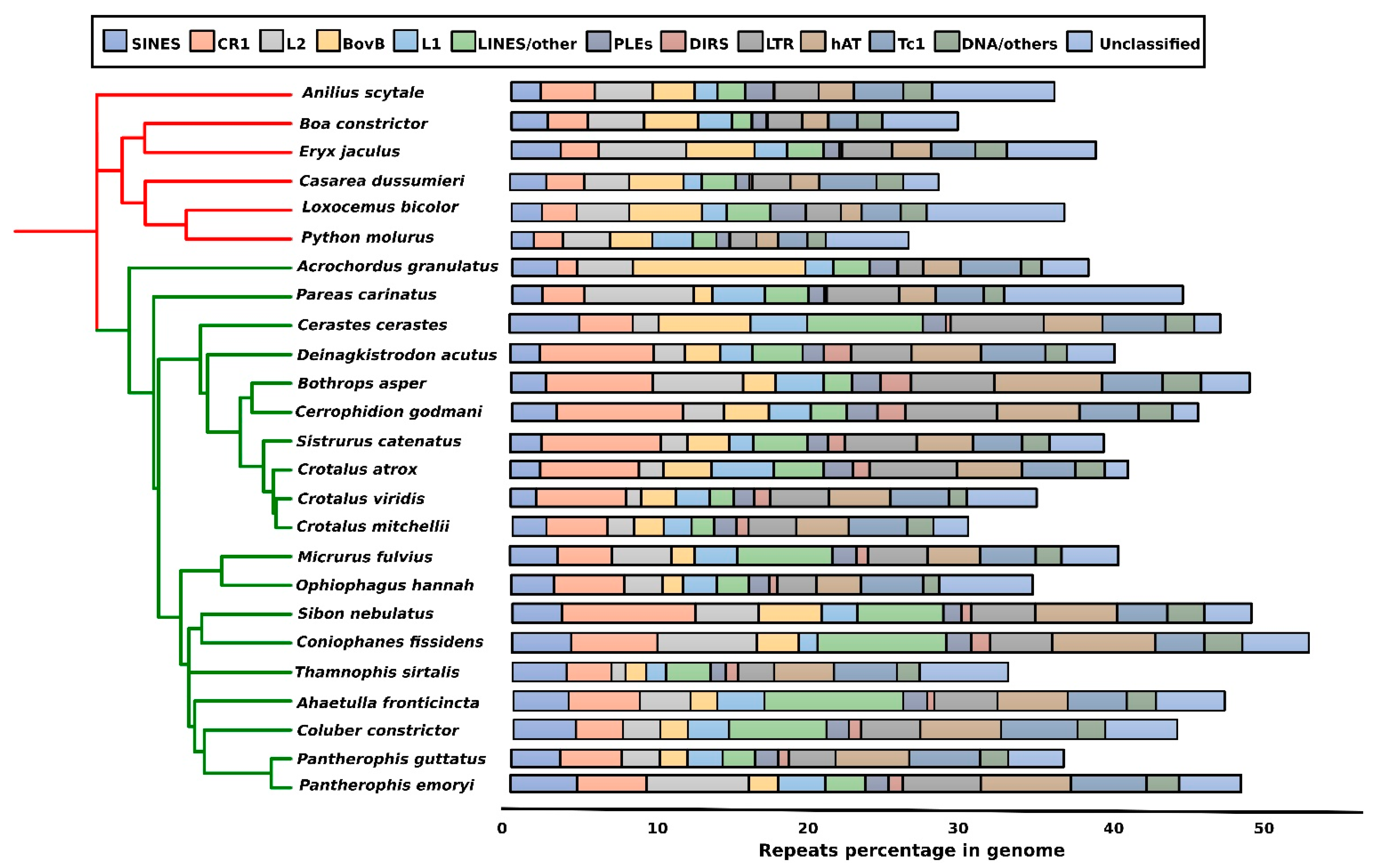
3. Repeat Abundance in Snake Genomes
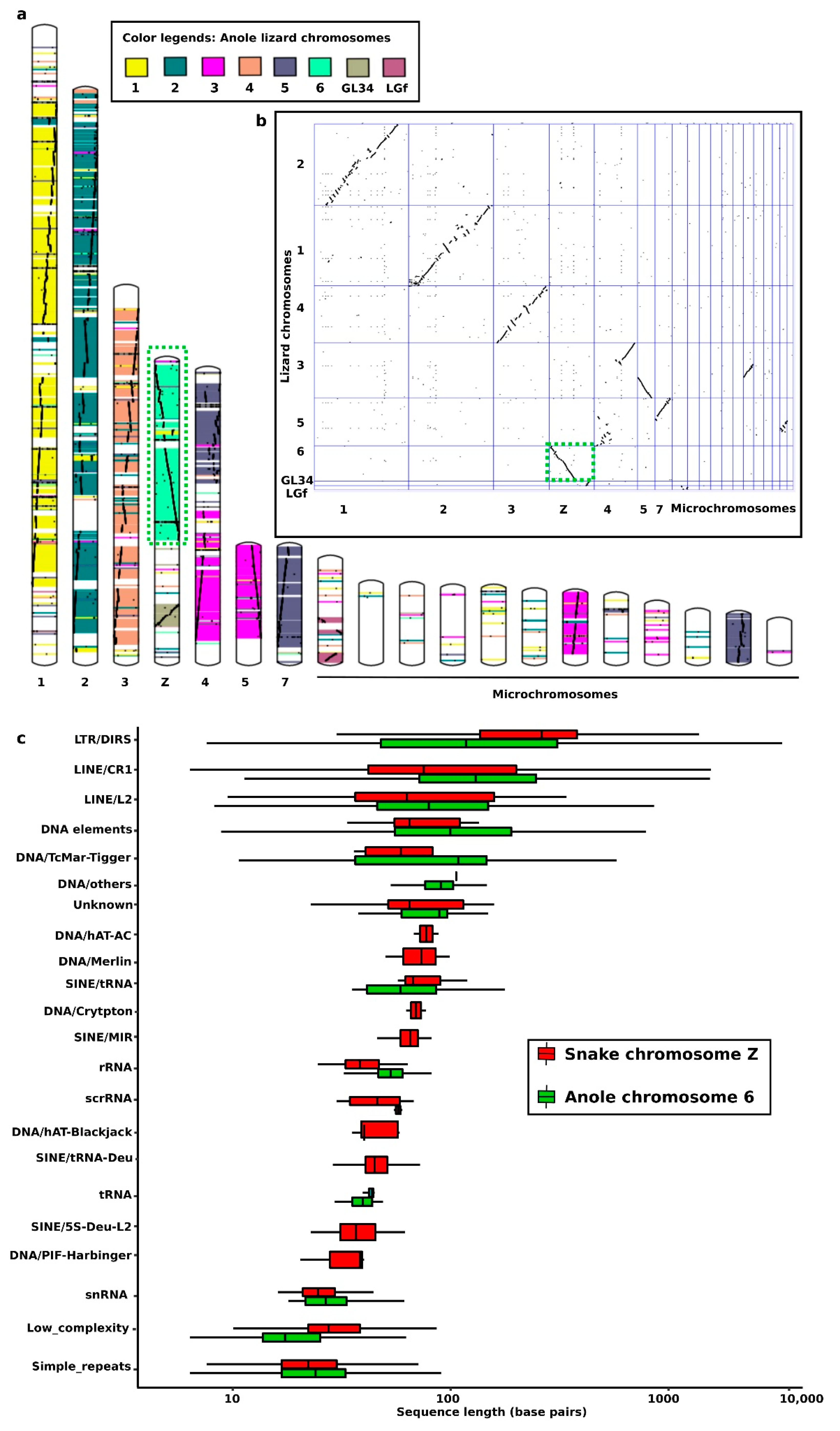
4. Snake Repeats in the Cytogenetics Era
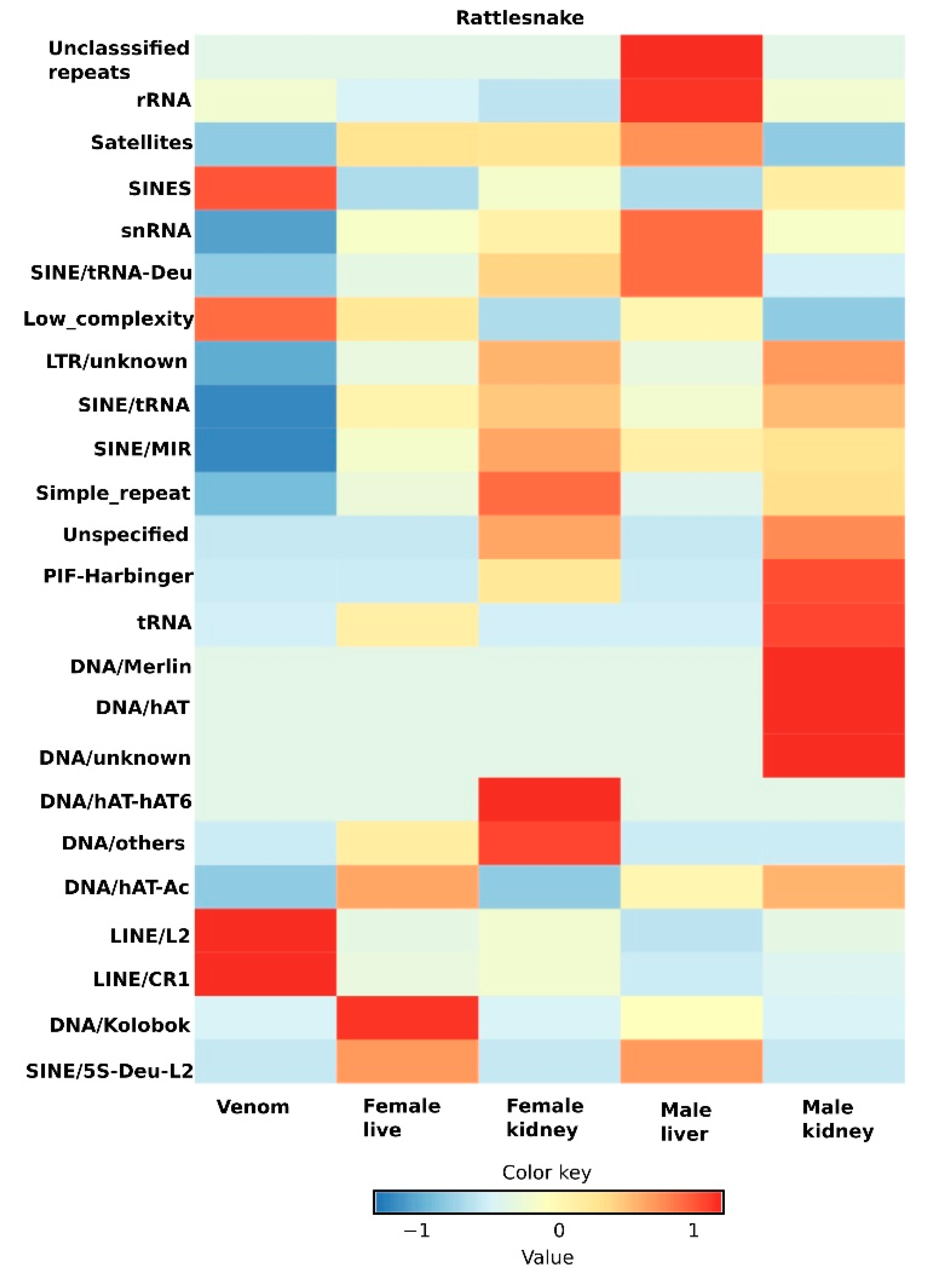
5. Transcriptomic Profiling and Expression Dynamics of Principal Repeats in Snakes
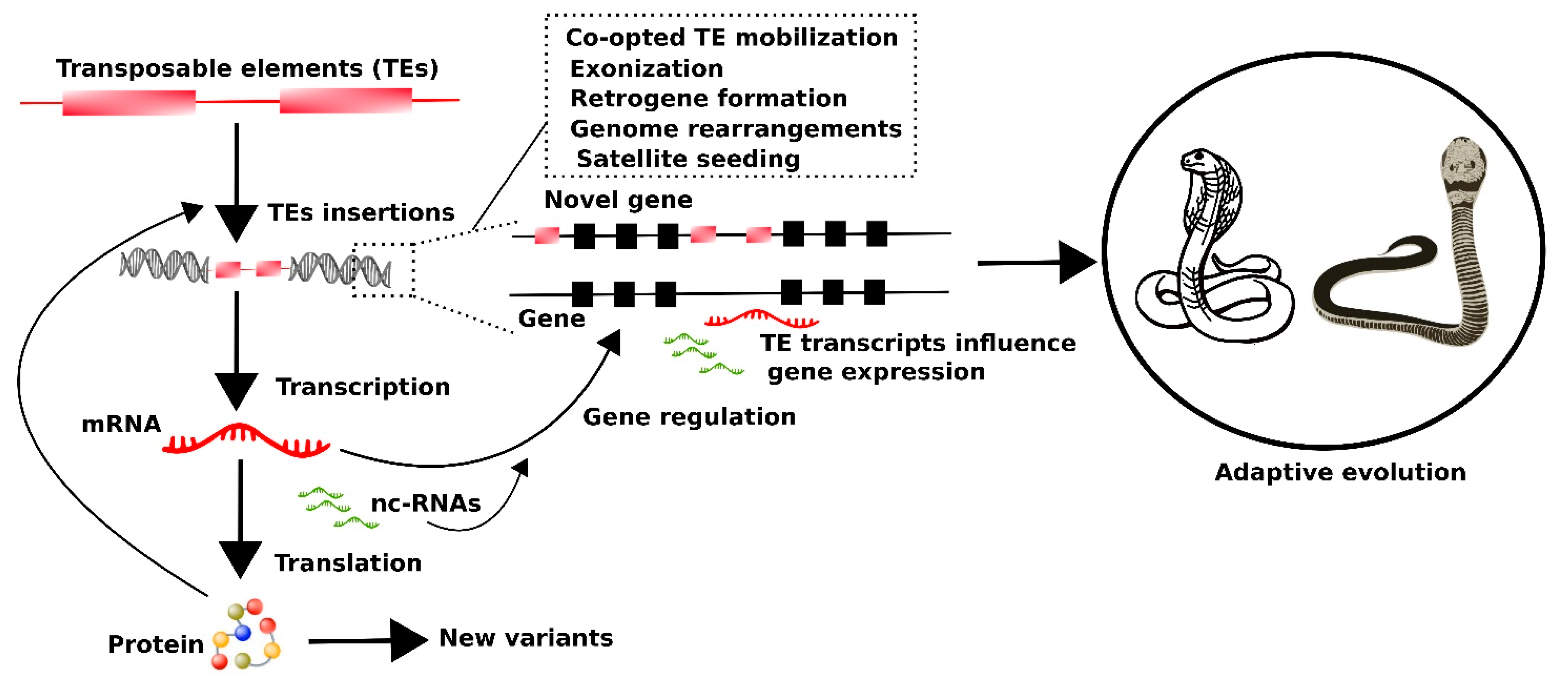
6. Putative Impact of Transposable Element Transcripts on the Evolutionary Dynamics of Snakes
7. Measuring the Expression of Repeat Elements: Modern Approaches and Challenges
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pyron, R.A.; Burbrink, F.T.; Wiens, J.J. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol. Biol. 2013, 13, 93. [Google Scholar] [CrossRef]
- Morinaga, G.; Bergmann, P.J. Evolution of fossorial locomotion in the transition from tetrapod to snake-like in lizards. Proc. Biol. Sci. 2020, 287, 20200192. [Google Scholar] [CrossRef]
- Greene, H.W. Snakes: The Evolution of Mystery in Nature; University of California Press: Berkeley, CA, USA, 1997; p. 351. [Google Scholar]
- Cundall, D.; Greene, H.W. Feeding in Snakes. In Feeding, Form, Function, and Evolution in Tetrapod Vertebrates; Schwenk, K., Ed.; Academic Press: San Diego, CA, USA, 2000; pp. 369–391. [Google Scholar]
- Simões, T.R.; Vernygora, O.; Caldwell, M.W.; Pierce, S.E. Megaevolutionary dynamics and the timing of evolutionary innovation in reptiles. Nat. Commun. 2020, 11, 3322. [Google Scholar] [CrossRef]
- Post, Y.; Puschhof, J.; Beumer, J.; Kerkkamp, H.M.; de Bakker, M.A.G.; Slagboom, J.; de Barbanson, B.; Wevers, N.R.; Spijkers, X.M.; Olivier, T.; et al. Snake venom gland organoids. Cell 2020, 180, 233–247. [Google Scholar] [CrossRef]
- Puschhof, J.; Post, Y.; Beumer, J.; Kerkkamp, H.M.; Bittenbinder, M.; Vonk, F.J.; Casewell, N.R.; Richardson, M.K.; Clevers, H. Derivation of snake venom gland organoids for in vitro venom production. Nat. Protoc. 2021, 16, 1494–1510. [Google Scholar] [CrossRef]
- Coates, M.; Ruta, M. Nice snake, shame about the legs. Trends Ecol. Evol. 2000, 15, 506–507. [Google Scholar] [CrossRef]
- Fry, B.G. From genome to “venome”: Molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 2005, 15, 403–420. [Google Scholar] [CrossRef]
- Schield, D.R.; Card, D.C.; Hales, N.R.; Perry, B.W.; Pasquesi, G.M.; Blackmon, H.; Adams, R.H.; Corbin, A.B.; Smith, C.F.; Ramesh, B.; et al. The origins and evolution of chromosomes, dosage compensation, and mechanisms underlying venom regulation in snakes. Genome Res. 2019, 29, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Isbister, G.K. Current research into snake antivenoms, their mechanisms of action and applications. Biochem. Soc. Trans. 2020, 48, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; De Koning, A.P.J.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef] [PubMed]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.R.; Kerkkamp, H.M.E.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef]
- Perry, B.W.; Card, D.C.; McGlothlin, J.W.; Pasquesi, G.I.M.; Adams, R.H.; Schield, D.R.; Hales, N.R.; Corbin, A.B.; Demuth, J.P.; Hoffmann, F.G.; et al. Molecular adaptations for sensing and securing prey and insight into amniote genome diversity from the garter Snake genome. Genome Biol. Evol. 2018, 10, 2110–2129. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Ren, J.L.; Deng, C.; Jiang, D.; Wang, J.; Qu, J.; Chang, J.; Yan, C.; Jiang, K.; Murphy, R.W.; et al. The genome of Shaw’s sea snake (Hydrophis curtus) reveals secondary adaptation to its marine environment. Mol. Biol. Evol. 2020, 37, 1744–1760. [Google Scholar] [CrossRef]
- Suryamohan, K.; Krishnankutty, S.P.; Guillory, J.; Jevit, M.; Schröder, M.S.; Wu, M.; Kuriakose, B.; Mathew, O.K.; Perumal, R.C.; Koludarov, I.; et al. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nat. Genet. 2020, 52, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; Rautsaw, R.M.; Strickland, J.L. The tiger rattlesnake genome reveals a complex genotype underlying a simple venom phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2014634118. [Google Scholar] [CrossRef]
- Cantor, T.E. Sketch of Undescribed Hooded Serpent with Fangs and Maxillar Teeth; Asiat. Res.; Bengal Military Orphan Press: Calcutta, India, 1836; pp. 87–94. [Google Scholar]
- Linnaeus, C. Systema Naturæ per Regna Tria Naturæ, Secundum Classes, Ordines, Genera, Species, cum Characteribus, Differentiis, Synonymis, Locis; Tomus, I., Ed.; Editio decima, reformata; Impensis Laurentii Salvii: Stockholm, Sweden, 1758; p. 824. [Google Scholar]
- Rafinesque, C.S. Further accounts of discoveries in natural history in the western states. Am. Mon. Mag. 1818, 4, 41. [Google Scholar]
- Kuhl, H. Beiträge zur Zoologie und Vergleichenden Anatomie; Hermannsche Buchhandlung: Frankfurt, Germany, 1820; p. 152. [Google Scholar]
- Yin, W.; Wang, Z.J.; Li, Q.Y.; Lian, J.M.; Zhou, Y.; Lu, B.Z.; Jin, L.J.; Qiu, P.X.; Zhang, P.; Zhu, W.B.; et al. Evolutionary trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; Hall, K.T.; Guibotsy Mboulas, M.L.; Gu, W.; Jason De Koning, A.P.; Fox, S.E.; Poole, A.W.; Vemulapalli, V.; Daza, J.M.; Mockler, T.; et al. Discovery of highly divergent repeat landscapes in snake genomes using high-throughput sequencing. Genome Biol. Evol. 2011, 3, 641–653. [Google Scholar] [CrossRef]
- Thongchum, R.; Singchat, W.; Laopichienpong, N.; Tawichasri, P.; Kraichak, E.; Prakhongcheep, O.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Suntrarachun, S.; et al. Diversity of PBI-DdeI satellite DNA in snakes correlates with rapid independent evolution and different functional roles. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Singchat, W.; Jehangir, M.; Panthum, T.; Srikulnath, K. Consequence of paradigm shift with repeat landscapes in reptiles: Powerful facilitators of chromosomal rearrangements for diversity and evolution. Genes 2020, 11, 827. [Google Scholar] [CrossRef]
- Canapa, A.; Barucca, M.; Biscotti, M.A.; Forconi, M.; Olmo, E. Transposons, genome size, and evolutionary insights in animals. Cytogenet. Genome Res. 2016, 147, 217–239. [Google Scholar] [CrossRef]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and diversity of transposable elements in vertebrate genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef]
- Fischer von Waldheim, G. Entomographia Imperii Rossici. IV. Orthoptera Imperii Rossici. Nouv. Mém. Soc. Imp. Nat. Moscou 1846, 8, 1–37. [Google Scholar]
- Palacios-Gimenez, O.M.; Koelman, J.; Palmada-Flores, M.; Bradford, T.M.; Jones, K.K.; Cooper, S.J.B.; Kawakami, T.; Suh, A. Comparative analysis of morabine grasshopper genomes reveals highly abundant transposable elements and rapidly proliferating satellite DNA repeats. BMC Biol. 2020, 18, 199. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Hoffman, J.I.; Schielzeth, H. Comparative analysis of genomic repeat content in gomphocerine grasshoppers reveals expansion of satellite DNA and helitrons in species with unusually large genomes. Genome Biol. Evol. 2020, 12, 1180–1193. [Google Scholar] [CrossRef]
- Tam, O.H.; Ostrow, L.W.; Gale Hammell, M. Diseases of the nERVous system: Retrotransposon activity in neurodegenerative disease. Mob. DNA 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- El-Hajjar, J.; Chatoo, W.; Hanna, R.; Nkanza, P.; Tétreault, N.; Tse, Y.C.; Wong, T.P.; Abdouh, M.; Bernier, G. Heterochromatic genome instability and neurodegeneration sharing similarities with Alzheimer’s disease in old Bmi1+/− mice. Sci. Rep. 2019, 9, 594. [Google Scholar] [CrossRef]
- Garrido-Ramos, M.A. Satellite DNA: An evolving topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Beridze, T. Satellite DNA; Springer Science and Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Razali, N.M.; Cheah, B.H.; Nadarajah, K. Transposable elements adaptive role in genome plasticity, pathogenicity and evolution in fungal phytopathogens. Int. J. Mol. Sci. 2019, 20, 3597. [Google Scholar] [CrossRef] [PubMed]
- Biscotti, M.A.; Canapa, A.; Forconi, M.; Olmo, E.; Barucca, M. Transcription of tandemly repetitive DNA: Functional roles. Chromosome Res. 2015, 23, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Lower, S.E.; Dion-Côté, A.M.; Clark, A.G.; Barbash, D.A. Special issue: Repetitive DNA sequences. Genes 2019, 10, 896. [Google Scholar] [CrossRef]
- Pasquesi, G.I.M.; Adams, R.H.; Card, D.C.; Schield, D.R.; Corbin, A.B.; Perry, B.W.; Reyes-Velasco, J.; Ruggiero, R.P.; Vandewege, M.W.; Shortt, J.A.; et al. Squamate reptiles challenge paradigms of genomic repeat element evolution set by birds and mammals. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Daron, J.; Glover, N.; Pingault, L.; Theil, S.; Jamilloux, V.; Paux, E.; Barbe, V.; Mangenot, S.; Alberti, A.; Wincker, P.; et al. Organization and evolution of transposable elements along the bread wheat chromosome 3B. Genome Biol. 2014, 15, 546. [Google Scholar] [CrossRef] [PubMed]
- Schrader, L.; Kim, J.W.; Ence, D.; Zimin, A.; Klein, A.; Wyschetzki, K.; Weichselgartner, T.; Kemena, C.; Stökl, J.; Schultner, E.; et al. Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Albertin, C.B.; Simakov, O.; Mitros, T.; Wang, Z.Y.; Pungor, J.R.; Edsinger-Gonzales, E.; Brenner, S.; Ragsdale, C.W.; Rokhsar, D.S. The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 2015, 524, 220–224. [Google Scholar] [CrossRef]
- Grabundzija, I.; Messing, S.A.; Thomas, J.; Cosby, R.L.; Bilic, I.; Miskey, C.; Gogol-Doring, A.; Kapitonov, V.; Diem, T.; Dalda, A.; et al. A Helitron transposon reconstructed from bats reveals a novel mechanism of genome shuffling in eukaryotes. Nat. Commun. 2016, 7, 10716. [Google Scholar] [CrossRef]
- Vicient, C.M.; Casacuberta, J.M. Impact of transposable elements on polyploid plant genomes. Ann. Bot. 2017, 120, 195–207. [Google Scholar] [CrossRef]
- Li, Z.W.; Hou, X.H.; Chen, J.F.; Xu, Y.C.; Wu, Q.; Gonzalez, J.; Guo, Y.L. Transposable elements contribute to the adaptation of Arabidopsis thaliana. Genome Biol. Evol. 2018, 10, 2140–2150. [Google Scholar] [CrossRef]
- Ganley, A.R.D. Concerted Evolution. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 126–130. [Google Scholar] [CrossRef]
- Galbraith, J.D.; Ludington, A.J.; Suh, A.; Sanders, K.L.; Adelson, D.L. New environment, new invaders—Repeated horizontal transfer of LINEs to sea snakes. Genome Biol. Evol. 2020, 12, 2370–2383. [Google Scholar] [CrossRef] [PubMed]
- Gamble, T.; Castoe, T.A.; Nielsen, S.V.; Banks, J.L.; Card, D.C.; Schield, D.R.; Schuett, G.W.; Booth, W. The discovery of XY sex chromosomes in a boa and python. Curr. Biol. 2017, 27, 2148–2153. [Google Scholar] [CrossRef]
- Li, S.F.; Su, T.; Cheng, G.Q.; Wang, B.X.; Li, X.; Deng, C.L.; Gao, W.J. Chromosome evolution in connection with repetitive sequences and epigenetics in plants. Genes 2017, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Singchat, W.; Jehangir, M.; Suntronpong, A.; Panthum, T.; Malaivijitnond, S.; Srikulnath, K. Dark matter of primate genomes: Satellite DNA repeats and their evolutionary dynamics. Cells 2020, 9, 2714. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Jehangir, M.; Cardoso, A.L.; Wolf, I.R.; Margarido, V.P.; Cabral-De-Mello, D.C.; O’Neill, R.; Valente, G.T.; Martins, C. B chromosomes of multiple species have intense evolutionary dynamics and accumulated genes related to important biological processes. BMC Genom. 2020, 21, 1–25. [Google Scholar] [CrossRef] [PubMed]
- King, D.G.; Soller, M.; Kashi, Y. Evolutionary tuning knobs. Endeavour 1997, 21, 36–40. [Google Scholar] [CrossRef]
- Trifonov, E.N. Elucidating sequence codes: Three codes for evolution. Ann. N. Y. Acad. Sci. 1999, 870, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N. Tuning function of tandemly repeating sequences: A molecular device for fast adaptation. In Evolutionary Theory and Processes: Modern Horizons; Springer: Dordrecht, Germany, 2004; pp. 115–138. [Google Scholar] [CrossRef]
- Gemmell, N.J. Repetitive DNA: Genomic dark matter matters. Nat. Rev. Genet. 2021. [Google Scholar] [CrossRef]
- Singer, M.F. Highly repeated sequences in mammalian genomes. Int. Rev. Cytol. 1982, 76, 67–112. [Google Scholar] [CrossRef]
- Barragán, M.J.L.; Martínez, S.; Marchal, J.A.; Bullejos, M.; Díaz de la Guardia, R.; Sánchez Bullejos, A.; Sánchez Baca, A. Highly repeated DNA sequences in three species of the genus Pteropus (Megachiroptera, Mammalia). Heredity 2002, 88, 366–370. [Google Scholar] [CrossRef][Green Version]
- Zhou, Y.; Shearwin-Whyatt, L.; Li, J.; Song, Z.; Hayakawa, T.; Stevens, D.; Fenelon, J.C.; Peel, E.; Cheng, Y.; Pajpach, F.; et al. Platypus and echidna genomes reveal mammalian biology and evolution. Nature 2021, 592, 756–762. [Google Scholar] [CrossRef]
- Capel, B. Vertebrate sex determination: Evolutionary plasticity of a fundamental switch. Nat. Rev. Genet. 2017, 18, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.E.; Ezaz, T. Understanding the evolution of reptile chromosomes through applications of combined cytogenetics and genomics approaches. Cytogenet. Genome Res. 2019, 157, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Pokorná, M.; Kratochvíl, L. Phylogeny of sex-determining mechanisms in squamate reptiles: Are sex chromosomes an evolutionary trap? Zool. J. Linn. Soc. 2009, 156, 168–183. [Google Scholar] [CrossRef]
- Sarre, S.D.; Ezaz, T.; Georges, A. Transitions between sex-determining systems in reptiles and amphibians. Annu. Rev. Genom. Hum. Genet. 2011, 12, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Holleley, C.E. Sex reversal triggers the rapid transition from genetic to temperature-dependent sex. Nature 2015, 523, 79–82. [Google Scholar] [CrossRef]
- Viana, P.F.; Ezaz, T.; de Bello Cioffi, M.; Liehr, T.; Al-Rikabi, A.; Goll, L.G.; Rocha, A.M.; Feldberg, E. Landscape of snake’ sex chromosomes evolution spanning 85 MYR reveals ancestry of sequences despite distinct evolutionary trajectories. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Emerson, J.J. Evolution: A Paradigm Shift in Snake Sex Chromosome Genetics. Curr. Biol. 2017, 27, R800–R803. [Google Scholar] [CrossRef]
- Ezaz, T.; Deakin, J.E. Repetitive Sequence and Sex Chromosome Evolution in Vertebrates. Adv. Evol. Biol. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Lepesant, J.M.J.; Cosseau, C.; Boissier, J.; Freitag, M.; Portela, J.; Climent, D.; Perrin, C.; Zerlotini, A.; Grunau, C. Chromatin structural changes around satellite repeats on the female sex chromosome in Schistosoma mansoni and their possible role in sex chromosome emergence. Genome Biol. 2012, 13, R14. [Google Scholar] [CrossRef]
- Lokody, I. Evolution: Transposons drive sex chromosome evolution. Nat. Rev. Genet. 2014, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Chalopin, D.; Volff, J.N.; Galiana, D.; Anderson, J.L.; Schartl, M. Transposable elements and early evolution of sex chromosomes in fish. Chromosome Res. 2015, 23, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Dechaud, C.; Volff, J.N.; Schartl, M.; Naville, M. Sex and the TEs: Transposable elements in sexual development and function in animals. Mob. DNA 2019, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E.S.; Charlesworth, B. The effects of recombination rate on the distribution and abundance of transposable elements. Genetics 2008, 178, 2169–2177. [Google Scholar] [CrossRef]
- Śliwińska, E.B.; Martyka, R.; Tryjanowski, P. Evolutionary interaction between W/Y chromosome and transposable elements. Genetica 2016, 144, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Vicoso, B.; Emerson, J.J.; Zektser, Y.; Mahajan, S.; Bachtrog, D. Comparative sex chromosome genomics in snakes: Differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biol. 2013, 11, e1001643. [Google Scholar] [CrossRef]
- Singchat, W.; O’Connor, R.E.; Tawichasri, P.; Suntronpong, A.; Sillapaprayoon, S.; Suntrarachun, S.; Muangmai, N.; Baicharoen, S.; Peyachoknagul, S.; Chanhome, L.; et al. Chromosome map of the Siamese cobra: Did partial synteny of sex chromosomes in the amniote represent “a hypothetical ancestral super-sex chromosome” or random distribution? BMC Genom. 2018, 19, 1–16. [Google Scholar] [CrossRef]
- Singchat, W.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Indananda, C.; Duengkae, P.; Peyachoknagul, S.; O’Connor, R.E.; Griffin, D.K.; Srikulnath, K. Do sex chromosomes of snakes, monitor lizards, and iguanian lizards result from multiple fission of an “ancestral amniote super-sex chromosome”? Chromosome Res. 2020, 28, 209–228. [Google Scholar] [CrossRef]
- Singchat, W.; Ahmad, S.F.; Sillapaprayoon, S.; Muangmai, N.; Duengkae, P.; Peyachoknagul, S.; O’Connor, R.E.; Griffin, D.K.; Srikulnath, K. Partial amniote sex chromosomal linkage homologies shared on snake W sex chromosomes support the ancestral super-sex chromosome evolution in amniotes. Front. Genet. 2020, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Singchat, W.; Ahmad, S.F.; Laopichienpong, N.; Suntronpong, A.; Panthum, T.; Griffin, D.K.; Srikulnath, K. Snake W sex chromosome: The shadow of ancestral amniote super-sex chromosome. Cells 2020, 9, 2386. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. Sex Chromosomes and Sex-Linked Genes; Springer: Berlin, Germany, 1967. [Google Scholar]
- Mengden, G.A. Linear differentiation of the C-band pattern of the W chromosome in snakes and birds. Chromosoma 1981, 83, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Tarui, H.; Toriba, M.; Yamada, K.; Nishida-Umehara, C.; Agata, K.; Matsuda, Y. Evidence for different origin of sex chromosomes in snakes, birds, and mammals and step-wise differentiation of snake sex chromosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 18190–18195. [Google Scholar] [CrossRef] [PubMed]
- Viana, P.F.; Ezaz, T.; Cioffi, M.D.B.; Almeida, B.J.; Feldberg, E. Evolutionary insights of the ZW sex chromosomes in snakes: A new chapter added by the Amazonian puffing snakes of the genus Spilotes. Genes 2019, 10, 288. [Google Scholar] [CrossRef] [PubMed]
- Augstenová, B.; Mazzoleni, S.; Kratochvíl, L.; Rovatsos, M. Evolutionary dynamics of the W chromosome in caenophidian snakes. Genes 2017, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, A.B.S.M.; Milani, D.; Palacios-Gimenez, O.M.; Ruiz-Ruano, F.J.; Cabral-de-Mello, D.C. High dynamism for neo-sex chromosomes: Satellite DNAs reveal complex evolution in a grasshopper. Heredity 2020, 125, 124–137. [Google Scholar] [CrossRef]
- Nguyen, A.; Bachtrog, D. Toxic Y chromosome: Increased repeat expression and age-associated heterochromatin loss in male Drosophila with a young Y chromosome. PLoS Genet. 2021, 17, e1009438. [Google Scholar] [CrossRef]
- Kaiser, V.B.; Bachtrog, D. Evolution of sex chromosomes in insects. Annu. Rev. Genet. 2010, 44, 91–112. [Google Scholar] [CrossRef]
- Janes, D.E.; Valenzuela, N.; Ezaz, T.; Amemiya, C.; Edwards, S.V. Sex chromosome evolution in amniotes: Applications for bacterial artificial chromosome libraries. J. Biomed. Biotechnol. 2011, 6, 1–6. [Google Scholar] [CrossRef]
- Lesson, R.P. Catalogue des Reptiles qui font partie d’une Collection zoologique recueille dans l’Inde continentale ou en Afrique, et apportée en France par M. Lamare-Piquot. Bull. Sci. Nat. Géol. 1831, 25, 119–123. [Google Scholar]
- Shaw, G.; Nodder, F.P. The Naturalist’s Miscellany; Nodder & Co.: London, UK, 1797; Volume 8, pp. 255–300. [Google Scholar]
- Smith, A. Illustrations of the Zoology of South Africa, Reptilia; Smith, Elder, and Co.: London, UK, 1849. [Google Scholar]
- Tollis, M.; Boissinot, S. The evolutionary dynamics of transposable elements in eukaryote genomes. Genome Dyn. 2012, 7, 68–91. [Google Scholar] [CrossRef]
- Bracewell, R.; Chatla, K.; Nalley, M.J.; Bachtrog, D. Dynamic turnover of centromeres drives karyotype evolution in Drosophila. eLife 2019, 8, e49002. [Google Scholar] [CrossRef]
- Hartley, G.; O’Neill, R.J. Centromere repeats: Hidden gems of the genome. Genes 2019, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Srikulnath, K.; Matsubara, K.; Uno, Y.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S. Karyological characterization of the butterfly lizard (Leiolepis reevesii rubritaeniata, Agamidae, Squamata) by molecular cytogenetic approach. Cytogenet. Genome Res. 2009, 125, 213–223. [Google Scholar] [CrossRef]
- Srikulnath, K.; Nishida, C.; Matsubara, K.; Uno, Y.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S.; Matsuda, Y. Karyotypic evolution in squamate reptiles: Comparative gene mapping revealed highly conserved linkage homology between the butterfly lizard (Leiolepis reevesii rubritaeniata, Agamidae, Lacertilia) and the Japanese four-striped rat snake (Elaphe quadrivirgata, Colubridae, Serpentes). Chromosome Res. 2009, 17, 975–986. [Google Scholar] [CrossRef]
- Srikulnath, K.; Uno, Y.; Nishida, C.; Matsuda, Y. Karyotype evolution in monitor lizards: Cross-species chromosome mapping of cDNA reveals highly conserved synteny and gene order in the Toxicofera clade. Chromosome Res. 2013, 21, 805–819. [Google Scholar] [CrossRef]
- Srikulnath, K.; Matsubara, K.; Uno, Y.; Nishida, C.; Olsson, M.; Matsuda, Y. Identification of the linkage group of the Z sex chromosomes of the sand lizard (Lacerta agilis, Lacertidae) and elucidation of karyotype evolution in lacertid lizards. Chromosoma 2014, 123, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Srikulnath, K.; Uno, Y.; Nishida, C.; Ota, H.; Matsuda, Y. Karyotype reorganization in the Hokou gecko (Gekko hokouensis, Gekkonidae): The process of microchromosome disappearance in Gekkota. PLoS ONE 2015, 10, e0134829. [Google Scholar] [CrossRef]
- Cohen, M.M.; Clark, H.F. The somatic chromosomes of five crocodilian species. Cytogenet. Genome Res. 1967, 6, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Belterman, R.H.R.; De Boer, L.E.M. A karyological study of 55 species of birds, including karyotypes of 39 species new to cytology. Genetica 1984, 65, 39–82. [Google Scholar] [CrossRef]
- Valleley, E.M.A.; Harrison, C.J.; Cook, Y.; Ferguson, M.W.J.; Sharpe, P.T. The karyotype of Alligator mississippiensis, and chromosomal mapping of the ZFY/X homologue, Zfc. Chromosoma 1994, 103, 502–507. [Google Scholar] [CrossRef]
- Matsuda, Y.; Nishida-Umehara, C.; Tarui, H.; Kuroiwa, A.; Yamada, K.; Isobe, T.; Ando, J.; Fujiwara, A.; Hirao, Y.; Nishimura, O.; et al. Highly conserved linkage homology between birds and turtles: Bird and turtle chromosomes are precise counterparts of each other. Chromosome Res. 2005, 13, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Olmo, E. Rate of chromosome changes and speciation in reptiles. Genetica 2005, 125, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Nishida-Umehara, C.; Ishijima, J.; Tsuda, Y.; Ota, H.; Matsuda, Y. Different origins of bird and reptile sex chromosomes inferred from comparative mapping of chicken Z-linked genes. Cytogenet. Genome Res. 2007, 117, 92–102. [Google Scholar] [CrossRef]
- Kawagoshi, T.; Nishida, C.; Ota, H.; Kumazawa, Y.; Endo, H.; Matsuda, Y. Molecular structures of centromeric heterochromatin and karyotypic evolution in the Siamese crocodile (Crocodylus siamensis) (Crocodylidae, Crocodylia). Chromosome Res. 2008, 16, 1119–1132. [Google Scholar] [CrossRef]
- Kawagoshi, T.; Uno, Y.; Matsubara, K.; Matsuda, Y.; Nishida, C. The ZW micro-sex chromosomes of the chinese soft-shelled turtle (Pelodiscus sinensis, Trionychidae, Testudines) have the same origin as chicken chromosome 15. Cytogenet. Genome Res. 2009, 125, 125–131. [Google Scholar] [CrossRef]
- Kawagoshi, T.; Nishida, C.; Matsuda, Y. The origin and differentiation process of X and Y chromosomes of the black marsh turtle (Siebenrockiella crassicollis, Geoemydidae, Testudines). Chromosome Res. 2012, 20, 95–110. [Google Scholar] [CrossRef][Green Version]
- Kasai, F.; O’Brien, P.C.M.; Ferguson-Smith, M.A. Reassessment of genome size in turtle and crocodile based on chromosome measurement by flow karyotyping: Close similarity to chicken. Biol. Lett. 2012, 8, 631–635. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.E.; Kiazim, L.; Skinner, B.; Fonseka, G.; Joseph, S.; Jennings, R.; Larkin, D.M.; Griffin, D.K. Patterns of microchromosome organization remain highly conserved throughout avian evolution. Chromosoma 2019, 128, 21–29. [Google Scholar] [CrossRef]
- Burt, D.W. Origin and evolution of avian microchromosomes. Cytogenet. Genome Res. 2002, 96, 97–112. [Google Scholar] [CrossRef]
- Warren, W.C.; Hillier, L.D.W.; Tomlinson, C.; Minx, P.; Kremitzki, M.; Graves, T.; Markovic, C.; Bouk, N.; Pruitt, K.D.; Thibaud-Nissen, F.; et al. A new chicken genome assembly provides insight into avian genome structure. G3 Genes Genomes Genet. 2017, 7, 109–117. [Google Scholar] [CrossRef]
- Nanda, I.; Schrama, D.; Feichtinger, W.; Haaf, T.; Schartl, M.; Schmid, M. Distribution of telomeric (TTAGGG)n sequences in avian chromosomes. Chromosoma 2002, 111, 215–227. [Google Scholar] [CrossRef]
- Srikulnath, K.; Uno, Y.; Matsubara, K.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S.; Nishida, C.; Matsuda, Y. Chromosomal localization of the 18S-28S and 5S rRNA genes and (TTAGGG)n sequences of butterfly lizards (Leiolepis belliana belliana and Leiolepis boehmei. Agamidae, Squamata). Genet. Mol. Biol. 2011, 34, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Srikulnath, K.; Azad, B.; Singchat, W.; Ezaz, T. Distribution and amplification of interstitial telomeric sequences (ITSs) in Australian dragon lizards support frequent chromosome fusions in Iguania. PLoS ONE 2019, 14, e0212683. [Google Scholar] [CrossRef] [PubMed]
- Rovatsos, M.; Kratochvíl, L.; Altmanová, M.; Pokorná, M.J. Interstitial telomeric motifs in squamate reptiles: When the exceptions outnumber the rule. PLoS ONE 2015, 10, e0134985. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, A.V. Micro vs. macro: A review of structure and functions of avian micro- and macrochromosomes. Genetika 1996, 32, 597–608. [Google Scholar]
- Fillon, V. The chicken as a model to study microchromosomes in birds: A review. Genet. Sel. Evol. 1998, 30, 209. [Google Scholar] [CrossRef]
- Smith, J.; Paton, I.R.; Bruley, C.K.; Windsor, D.; Burke, D.; Ponce De Leon, F.A.; Burt, D.W. Integration of the genetic and physical maps of the chicken macrochromosomes. Anim. Genet. 2000, 31, 20–27. [Google Scholar] [CrossRef]
- Yamada, K.; Nishida-Umehara, C.; Matsuda, Y. Molecular and cytogenetic characterization of site-specific repetitive DNA sequences in the Chinese soft-shelled turtle (Pelodiscus sinensis, Trionychidae). Chromosome Res. 2005, 13, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; O’Meally, D.; Azad, B.; Georges, A.; Sarre, S.D.; Graves, J.A.M.; Matsuda, Y.; Ezaz, T. Amplification of microsatellite repeat motifs is associated with the evolutionary differentiation and heterochromatinization of sex chromosomes in Sauropsida. Chromosoma 2016, 125, 111–123. [Google Scholar] [CrossRef]
- Gregory, T.R. Animal Genome Size Database. 2017. Available online: http://www.genomesize.com/ (accessed on 10 May 2021).
- Elliott, T.A.; Gregory, T.R. What’s in a genome? The C-value enigma and the evolution of eukaryotic genome content. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140331. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker. 2020. Available online: https://www.repeatmasker.org/ (accessed on 10 May 2021).
- Alfoldi, J.; Di-Palma, F.; Grabherr, M.; Williams, C.; Kong, L.; Mauceli, E.; Russell, P.; Lowe, C.B.; Glor, R.E.; Jaffe, J.D. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011, 477, 587–591. [Google Scholar] [CrossRef]
- Puinongpo, W.; Singchat, W.; Petpradub, S.; Kraichak, E.; Nunome, M.; Laopichienpong, N.; Thongchum, R.; Intarasorn, T.; Sillapaprayoon, S.; Indananda, C.; et al. Existence of Bov-B line retrotransposons in snake lineages reveals recent multiple horizontal gene transfers with copy number variation. Genes 2020, 11, 1241. [Google Scholar] [CrossRef]
- Boissinot, S.; Bourgeois, Y.; Manthey, J.D.; Ruggiero, R.P. The mobilome of reptiles: Evolution, structure, and function. Cytogenet. Genome Res. 2019, 157, 21–33. [Google Scholar] [CrossRef]
- Suh, A.; Churakov, G.; Ramakodi, M.P.; Platt, R.N.; Jurka, J.; Kojima, K.K.; Caballero, J.; Smit, A.F.; Vliet, K.A.; Hoffmann, F.G.; et al. Multiple lineages of ancient CR1 retroposons shaped the early genome evolution of amniotes. Genome Biol. Evol. 2014, 7, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, A.M. Phylogenomic investigation of CR1 LINE diversity in reptiles. Syst. Biol. 2006, 55, 902–911. [Google Scholar] [CrossRef]
- Badenhorst, D.; Hillier, L.D.W.; Literman, R.; Montiel, E.E.; Radhakrishnan, S.; Shen, Y.; Minx, P.; Janes, D.E.; Warren, W.C.; Edwards, S.V.; et al. Physical mapping and refinement of the painted turtle genome (Chrysemys picta) inform amniote genome evolution and challenge turtle-bird chromosomal conservation. Genome Biol. Evol. 2015, 7, 2038–2050. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Olmo, E. Evolution of genome size and DNA base composition in reptiles. Genetica 1981, 57, 39–50. [Google Scholar] [CrossRef]
- Olmo, E. Genomic composition of reptiles: Evolutionary perspectives. J. Herpetol. 1984, 18, 20. [Google Scholar] [CrossRef]
- Kerkkamp, H.M.I.; Kini, R.M.; Pospelov, A.S.; Vonk, F.J.; Henkel, C.V.; Richardson, M.K. Snake genome sequencing: Results and future prospects. Toxins 2016, 8, 360. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef]
- Kordiš, D.; Gubenšek, F. Unusual horizontal transfer of a long interspersed nuclear element between distant vertebrate classes. Proc. Natl. Acad. Sci. USA 1998, 95, 10704–10709. [Google Scholar] [CrossRef] [PubMed]
- Ivancevic, A.M.; Kortschak, R.D.; Bertozzi, T.; Adelson, D.L. Horizontal transfer of BovB and L1 retrotransposons in eukaryotes. Genome Biol. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dunemann, S.M.; Wasmuth, J.D. Horizontal transfer of a retrotransposon between parasitic nematodes and the common shrew. Mob. DNA 2019, 10, 24. [Google Scholar] [CrossRef]
- Sobecky, P.A.; Hazen, T.H. Horizontal gene transfer and mobile genetic elements in marine systems. Methods Mol. Biol. 2009, 532, 435–453. [Google Scholar] [CrossRef]
- Zhang, H.H.; Peccoud, J.; Xu, M.R.X.; Zhang, X.G.; Gilbert, C. Horizontal transfer and evolution of transposable elements in vertebrates. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Lacépède, B.G.E. Mémoire sur plusieurs animaux de la Nouvelle-Hollande dont la description n’a pas encore été publiée. Ann. Mus. Natl. Hist. Nat. 1804, 4, 184–211. [Google Scholar]
- Gaunt, S.J. Hox cluster genes and collinearities throughout the tree of animal life. Int. J. Dev. Biol. 2018, 62, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Linnaeus, C. Systema Naturæ per Regna Tria Naturæ, Secundum Classes, Ordines, Genera, Species, cum Characteribus, Differentiis, Synonymis, Locis; Tomus, I., Ed.; Editio duodecima, reformata; Laurentii Salvii: Stockholm, Sweden, 1766; pp. 1–532. [Google Scholar]
- Ullate-Agote, A.; Milinkovitch, M.C.; Tzika, A.C. The genome sequence of the corn snake (Pantherophis guttatus), a valuable resource for EvoDevo studies in squamates. Int. J. Dev. Biol. 2014, 58, 881–888. [Google Scholar] [CrossRef]
- Günther, A. Catalogue of Colubrine Snakes of the British Museum; British Museum (Natural History): London, UK, 1858; Volume I–XVI, pp. 1–281. [Google Scholar]
- Neff, B.D.; Gross, M.R. Microsatellite evolution in vertebrates: Inference from AC dinucleotide repeats. Evolution 2001, 55, 1717–1733. [Google Scholar] [CrossRef]
- Le Chevalier, H.; Marí-Mena, N.; Carro, B.; Prunier, J.G.; Bossu, C.; Darnet, E.; Souchet, J.; Guillaume, O.; Calvez, O.; Bertrand, R. Isolation and characterization of fourteen polymorphic microsatellite markers in the viperine snake Natrix maura. Ecol. Evol. 2019, 9, 11227–11231. [Google Scholar] [CrossRef]
- Shibata, H.; Chijiwa, T.; Oda-Ueda, N. The habu genome reveals accelerated evolution of venom protein genes. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Mengden, G.A.; Stock, A.D. Chromosomal evolution in Serpentes; a comparison of G and C chromosome banding patterns of some colubrid and boid genera. Chromosoma 1980, 79, 53–64. [Google Scholar] [CrossRef]
- Pinkel, D.; Straume, T.; Gray, J.W. Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc. Natl. Acad. Sci. USA 1986, 83, 2934–2938. [Google Scholar] [CrossRef]
- Kannan, T.P.; Zilfalil, B.A. Cytogenetics: Past, present and future. Malays. J. Med. Sci. 2009, 16, 4–9. [Google Scholar] [PubMed]
- Porter, C.; Hamilton, M.; Sites, J.; Baker, R. Location of ribosomal DNA in chromosomes of squamate reptiles: Systematic and evolutionary implications. Herpetologica 1991, 47, 271–280. [Google Scholar]
- Viana, P.F.; Ribeiro, L.B.; Souza, G.M.; Chalkidis, H.D.M.; Gross, M.C.; Feldberg, E. Is the karyotype of neotropical boid snakes really conserved? Cytotaxonomy, chromosomal rearrangements and karyotype organization in the Boidae family. PLoS ONE 2016, 11, 160274. [Google Scholar] [CrossRef] [PubMed]
- Trajtengertz, I.; Beçak, M.L.; Ruiz, I.R.G. Ribosomal cistrons in Bothrops neuwiedi (Serpentes) subspecies from Brazil. Genome 1995, 38, 601–606. [Google Scholar] [CrossRef]
- Lacerda, J.B. Leçons sur le Venin des Serpents du Brésil et sur la Méthode de Traitement des Morsures Venimeuses par le Permanganate de Potasse; Lombaerts: Rio de Janeiro, Brazil, 1884; p. 194. [Google Scholar]
- Falcione, C.; Hernando, A.; Bressa, M.J. Comparative cytogenetic analysis in erythrolamprus snakes (Serpentes: Dipsadidae) from Argentina. An. Acad. Bras. Ciênc. 2018, 90, 1417–1429. [Google Scholar] [CrossRef]
- Shaw, G. General Zoology, or Systematic Natural History; Kearsley, G., Ed.; Thomas Davison: London, UK, 1802; Volume 3, Part 2; pp. 313–615. [Google Scholar]
- Oguiura, N.; Ferrarezzi, H.; Batistic, R.F. Cytogenetics and molecular data in snakes: A phylogenetic approach. Cytogenet. Genome Res. 2009, 127, 128–142. [Google Scholar] [CrossRef]
- Wagler, J. Serpentum Brasiliensium species novae, ou histoire naturelle des espèces nouvelles de serpens. In Animalia Nova Sive Species Novae; NAtrix bahiensis: 27, Monaco; de Spix, J., Ed.; Typis Franc. Seraph. Hübschmanni: Munich, Germany, 1824; p. 75. [Google Scholar]
- Hallowell, E. Report upon the Reptilia of the North Pacific Exploring Expedition, under command of Capt. John Rogers. Proc. Acad. Nat. Sci. Phila. 1860, 12, 480–510. [Google Scholar]
- Matsubara, K.; Uno, Y.; Srikulnath, K.; Seki, R.; Nishida, C.; Matsuda, Y. Molecular cloning and characterization of satellite DNA sequences from constitutive heterochromatin of the habu snake (Protobothrops flavoviridis, Viperidae) and the Burmese python (Python bivittatus, Pythonidae). Chromosoma 2015, 124, 529–539. [Google Scholar] [CrossRef]
- Schneider, J.G. Historiae Amphibiorum Naturalis et Literariae; Friedrich Frommann: Jena, Germany, 1801; p. 374. [Google Scholar]
- Panicker, S.G.; Singh, L. Banded krait minor satellite (Bkm) contains sex and species-specific repetitive DNA. Chromosoma 1994, 103, 40–45. [Google Scholar] [CrossRef]
- Singh, L.; Purdom, I.F.; Jones, K.W. Satellite DNA and evolution of sex chromosomes. Chromosoma 1976, 59, 43–62. [Google Scholar] [CrossRef]
- Singh, L.; Purdom, I.F.; Jones, K.W. Sex chromosome associated satellite DNA: Evolution and conservation. Chromosoma 1980, 79, 137–157. [Google Scholar] [CrossRef] [PubMed]
- Mank, J.E.; Ellegren, H. Parallel divergence and degradation of the avian W sex chromosome. Trends Ecol. Evol. 2007, 22, 389–391. [Google Scholar] [CrossRef]
- Peters, W. Über Eine Neue Schildkrötenart, Cinosternon Effeldtii: Und Einige Andere Neue oder Weniger Bekannte Amphibien; Hansebooks GmbH: Norderstedt, Germany, 1873; pp. 603–618. [Google Scholar]
- Duméril, A.M.C.; Bibron, G.; Duméril, A.H.A. Erpétologie Générale ou Histoire Naturelle Complète des Reptiles; Tome septième: Paris, France, 1834. [Google Scholar]
- O’Meally, D.; Patel, H.R.; Stiglec, R.; Sarre, S.D.; Georges, A.; Marshall Graves, J.A.; Ezaz, T. Non-homologous sex chromosomes of birds and snakes share repetitive sequences. Chromosome Res. 2010, 18, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.E.; Potter, S.; O’Neill, R.; Ruiz-Herrera, A.; Cioffi, M.B.; Eldridge, M.D.B.; Fukui, K.; Marshall Graves, J.A.; Griffin, D.; Grutzner, F.; et al. Chromosomics: Bridging the gap between genomes and chromosomes. Genes 2019, 10, 627. [Google Scholar] [CrossRef] [PubMed]
- Meštrović, N.; Mravinac, B.; Pavlek, M.; Vojvoda-Zeljko, T.; Šatović, E.; Plohl, M. Structural and functional liaisons between transposable elements and satellite DNAs. Chromosome Res. 2015, 23, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Günther, A. On a collection of reptiles from China. Ann. Mag. Nat. Hist. 1888, 1, 165–172. [Google Scholar] [CrossRef][Green Version]
- Fujimi, T.J.; Tsuchiya, T.; Tamiya, T. A comparative analysis of invaded sequences from group IA phospholipase A2 genes provides evidence about the divergence period of genes groups and snake families. Toxicon 2002, 40, 873–884. [Google Scholar] [CrossRef]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S. Repetitive DNA in eukaryotic genomes. Chromosome Res. 2015, 23, 415–420. [Google Scholar] [CrossRef]
- Sundaram, V.; Wysocka, J. Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190347. [Google Scholar] [CrossRef]
- Zeng, L.; Pederson, S.M.; Kortschak, R.D.; Adelson, D.L. Transposable elements and gene expression during the evolution of amniotes. Mob. DNA 2018, 9, 17. [Google Scholar] [CrossRef]
- Boeva, V. Analysis of genomic sequence motifs for deciphering transcription factor binding and transcriptional regulation in eukaryotic cells. Front. Genet. 2016, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Sun, X.; Liu, H.; Xie, J. MicroRNA genes derived from repetitive elements and expanded by segmental duplication events in mammalian genomes. PLoS ONE 2011, 6, e17666. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, Z.; Krause, H.M. Long noncoding RNAs and repetitive elements: Junk or intimate evolutionary partners? Trends Genet. 2019, 35, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinf. 2009, 7, 147–154. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef]
- Durban, J.; Sasa, M.; Calvete, J.J. Venom gland transcriptomics and microRNA profiling of juvenile and adult yellow-bellied sea snake, Hydrophis platurus, from Playa del Coco (Guanacaste, Costa Rica). Toxicon 2018, 153, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Torvik, V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005, 21, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Mariño-Ramírez, L.; Jordan, I.K. Origin and evolution of human microRNAs from transposable elements. Genetics 2007, 176, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Jordan, I.K. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA 2008, 14, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Holton, N.W.; Williams, J.D.; Hernan, W.L.; Bishop, I.P.; Dembosky, J.A.; Elste, J.E.; Gregoire, N.S.; Kim, J.-A.; Koehler, W.W.; et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob. Genet. Elem. 2011, 1, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Gim, J.-A.; Ha, H.-S.; Ahn, K.; Kim, D.-S.; Kim, H.-S. Genome-wide identification and classification of microRNAs derived from repetitive elements. Genom. Inform. 2014, 12, 261. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Platt, R.N.; Vandewege, M.W.; Tabassum, R.; Hsu, C.Y.; Isberg, S.R.; Peterson, D.G.; Finger, J.W.; Kieran, T.J.; Glenn, T.C.; et al. Identification and characterization of microRNAs (miRNAs) and their transposable element origins in the saltwater crocodile, Crocodylus porosus. Anal. Biochem. 2020, 602, 113781. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Johnson, R.; Guigó, R. The RIDL hypothesis: Transposable elements as functional domains of long noncoding RNAs. RNA 2014, 20, 959–976. [Google Scholar] [CrossRef]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef]
- Schrader, L.; Schmitz, J. The impact of transposable elements in adaptive evolution. Mol. Ecol. 2019, 28, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Volff, J.N. Turning junk into gold: Domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays 2006, 28, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.R.; Greene, W.K. Transposable elements: Powerful facilitators of evolution. Bioessays 2009, 31, 703–714. [Google Scholar] [CrossRef]
- Fedoroff, N.V. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Prentis, P.J.; Wilson, J.R.U.; Dormontt, E.E.; Richardson, D.M.; Lowe, A.J. Adaptive evolution in invasive species. Trends Plant Sci. 2008, 13, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Barrón, M.G.; Fiston-Lavier, A.S.; Petrov, D.A.; González, J. Population genomics of transposable elements in drosophila. Annu. Rev. Genet. 2014, 48, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Stapley, J.; Santure, A.W.; Dennis, S.R. Transposable elements as agents of rapid adaptation may explain the genetic paradox of invasive species. Mol. Ecol. 2015, 24, 2241–2252. [Google Scholar] [CrossRef]
- Faino, L.; Seidl, M.F.; Shi-Kunne, X.; Pauper, M.; Van Den Berg, G.C.M.; Wittenberg, A.H.J.; Thomma, B.P.H.J. Transposons passively and actively contribute to evolution of the two-speed genome of a fungal pathogen. Genome Res. 2016, 26, 1091–1100. [Google Scholar] [CrossRef]
- Kim, S.; Mun, S.; Kim, T.; Lee, K.H.; Kang, K.; Cho, J.Y.; Han, K. Transposable element-mediated structural variation analysis in dog breeds using whole-genome sequencing. Mamm. Genome 2019, 30, 289–300. [Google Scholar] [CrossRef]
- Mun, S.; Kim, S.; Lee, W.; Kang, K.; Meyer, T.J.; Han, B.G.; Han, K.; Kim, H.S. A study of transposable element-associated structural variations (TASVs) using a de novo-assembled Korean genome. Exp. Mol. Med. 2021, 53, 615–630. [Google Scholar] [CrossRef]
- Dong, S.; Raffaele, S.; Kamoun, S. The two-speed genomes of filamentous pathogens: Waltz with plants. Curr. Opin. Genet. Dev. 2015, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, I.; Duboule, D. Snakes: Hatching of a model system for Evo-Devo? Int. J. Dev. Biol. 2014, 58, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Cao, X. The effect of transposable elements on phenotypic variation: Insights from plants to humans. Sci. China Life Sci. 2016, 59, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Scheib, H.; van der Weerd, L.; Young, B.; McNaughtan, J.; Ryan Ramjan, S.F.; Vidal, N.; Poelmann, R.E.; Norman, J.A. Evolution of an arsenal: Structural and functional diversification of the venom system in the advanced snakes (Caenophidia). Mol. Cell. Proteom. 2008, 7, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.; Peona, V.; Guichard, E.; Taccioli, C.; Boattini, A. Transposable Elements Activity is Positively Related to Rate of Speciation in Mammals. J. Mol. Evol. 2018, 86, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.K.; Feschotte, C. The evolutionary history of human DNA transposons: Evidence for intense activity in the primate lineage. Genome Res. 2007, 17, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.A.; Feschotte, C.; Pagan, H.J.T.; Smith, J.D.; Pritham, E.J.; Arensburger, P.; Atkinson, P.W.; Craig, N.L. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res. 2008, 18, 717–728. [Google Scholar] [CrossRef]
- Platt, R.N.; Vandewege, M.W.; Kern, C.; Schmidt, C.J.; Hoffmann, F.G.; Ray, D.A. Large numbers of novel miRNAs originate from DNA transposons and are coincident with a large species radiation in bats. Mol. Biol. Evol. 2014, 31, 1536–1545. [Google Scholar] [CrossRef]
- Feiner, N. Accumulation of transposable elements in hox gene clusters during adaptive radiation of anolis lizards. Proc. R. Soc. B Biol. Sci. 2016, 283. [Google Scholar] [CrossRef]
- Miller, W.J.; McDonald, J.F.; Pinsker, W. Molecular domestication of mobile elements. Genetica 1997, 100, 261–270. [Google Scholar] [CrossRef]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017, 33, 817–831. [Google Scholar] [CrossRef]
- Lev-Maor, G.; Sorek, R.; Shomron, N.; Ast, G. The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science 2003, 300, 1288–1291. [Google Scholar] [CrossRef]
- Amit, M.; Sela, N.; Keren, H.; Melamed, Z.; Muler, I.; Shomron, N.; Izraeli, S.; Ast, G. Biased exonization of transposed elements in duplicated genes: A lesson from the TIF-IA gene. BMC Mol. Biol. 2007, 8, 1–15. [Google Scholar] [CrossRef]
- Krull, M.; Petrusma, M.; Makalowski, W.; Brosius, J.; Schmitz, J. Functional persistence of exonized mammalian-wide interspersed repeat elements (MIRs). Genome Res. 2007, 17, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Kaessmann, H.; Vinckenbosch, N.; Long, M. RNA-based gene duplication: Mechanistic and evolutionary insights. Nat. Rev. Genet. 2009, 10, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Casola, C.; Betrán, E. The genomic impact of gene retrocopies: What have we learned from comparative genomics, population genomics, and transcriptomic analyses? Genome Biol. Evol. 2017, 9, 1351–1373. [Google Scholar] [CrossRef] [PubMed]
- Gray, Y.H.M. It takes two transposons to tango: Transposable-element-mediated chromosomal rearrangements. Trends Genet. 2000, 16, 461–468. [Google Scholar] [CrossRef]
- Weil, C.F. Too many ends: Aberrant transposition. Genes Dev. 2009, 23, 1032–1036. [Google Scholar] [CrossRef]
- Robberecht, C.; Voet, T.; Esteki, M.Z.; Nowakowska, B.A.; Vermeesch, J.R. Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res. 2013, 23, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Startek, M.; Szafranski, P.; Gambin, T.; Campbell, I.M.; Hixson, P.; Shaw, C.A.; Stankiewicz, P.; Gambin, A. Genome-wide analyses of LINE-LINE-mediated nonallelic homologous recombination. Nucleic Acids Res. 2015, 43, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef]
- Kent, T.V.; Uzunović, J.; Wright, S.I. Coevolution between transposable elements and recombination. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160458. [Google Scholar] [CrossRef] [PubMed]
- Di-Poï, N.; Montoya-Burgos, J.I.; Miller, H.; Pourquié, O.; Milinkovitch, M.C.; Duboule, D. Changes in Hox genes structure and function during the evolution of the squamate body plan. Nature 2010, 464, 99–103. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- St. Laurent, G.; Wahlestedt, C.; Kapranov, P. The landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef]
- Lech, M.; Anders, H.J. Expression profiling by real-time quantitative polymerase chain reaction (RT-qPCR). Methods Mol. Biol. 2014, 1169, 133–142. [Google Scholar] [CrossRef]
- Urbanek, M.O.; Nawrocka, A.U.; Krzyzosiak, W.J. Small RNA detection by in situ hybridization methods. Int. J. Mol. Sci. 2015, 16, 13259–13286. [Google Scholar] [CrossRef]
- Huang, Q.; Mao, Z.; Li, S.; Hu, J.; Zhu, Y. A non-radioactive method for small RNA detection by Northern blotting. Rice 2014, 7, 26. [Google Scholar] [CrossRef]
- Conti, A.; Carnevali, D.; Bollati, V.; Fustinoni, S.; Pellegrini, M.; Dieci, G. Identification of RNA polymerase III-transcribed Alu loci by computational screening of RNA-Seq data. Nucleic Acids Res. 2015, 43, 817–835. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Babarinde, I.A.; Li, Y.; Hutchins, A.P. Computational methods for mapping, assembly and quantification for coding and non-coding transcripts. Comput. Struct. Biotechnol. J. 2019, 17, 628–637. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.D. Transposable element detection from whole genome sequence data. Mob. DNA 2015, 6, 1–9. [Google Scholar] [CrossRef]
- O’Neill, K.; Brocks, D.; Hammell, M.G. Mobile genomics: Tools and techniques for tackling transposons. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190345. [Google Scholar] [CrossRef]
- Goerner-Potvin, P.; Bourque, G. Computational tools to unmask transposable elements. Nat. Rev. Genet. 2018, 19, 688–704. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 15, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, M.; Mohammed Ismail, W.; Rho, M.; Fox, G.C.; Oh, S.; Tang, H. MGEScan: A Galaxy-based system for identifying retrotransposons in genomes. Bioinformatics 2016, 15, 2502–2504. [Google Scholar] [CrossRef]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 1, 351–358. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef] [PubMed]
- Goubert, C.; Modolo, L.; Vieira, C.; Moro, C.V.; Mavingui, P.; Boulesteix, M. De novo assembly and annotation of the Asian tiger mosquito (Aedes albopictus) repeatome with dnaPipeTE from raw genomic reads and comparative analysis with the yellow fever mosquito (Aedes aegypti). Genome Biol. Evol. 2015, 7, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Nielsen, R.; Wu, Y. REPdenovo: Inferring de novo repeat motifs from short sequence reads. PLoS ONE 2016, 11, e0150719. [Google Scholar] [CrossRef]
- Guo, R.; Li, Y.R.; He, S.; Ou-Yang, L.; Sun, Y.; Zhu, Z. RepLong: De novo repeat identification using long read sequencing data. Bioinformatics 2018, 34, 1099–1107. [Google Scholar] [CrossRef]
- Koch, P.; Platzer, M.; Downie, B.R. RepARK—De novo creation of repeat libraries from whole-genome NGS reads. Nucleic Acids Res. 2014, 42, e80. [Google Scholar] [CrossRef]
- Criscione, S.W.; Zhang, Y.; Thompson, W.; Sedivy, J.M.; Neretti, N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genom. 2014, 15, 583. [Google Scholar] [CrossRef]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.H.; Yalamanchili, H.K.; Guo, C.; Shulman, J.M.; Liu, Z. An ultra-fast and scalable quantification pipeline for transposable elements from next generation sequencing data. Pac. Symp. Biocomput. 2018, 23, 168–179. [Google Scholar]
- Valdebenito-Maturana, B.; Riadi, G. TEcandidates: Prediction of genomic origin of expressed transposable elements using RNA-seq data. Bioinformatics 2018, 15, 3915–3916. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Common Name | Assembly ID | Assembly Level | Genome Size (Gbp) | Year of Release |
|---|---|---|---|---|---|
| Crotalus horridus | Timber rattlesnake | ASM162548v1 | Contig | 1.520 | 2016 |
| Crotalus pyrrhus | CrotMitch1.0 | Contig | 1.127 | 2014 | |
| Crotalus tigris | Tiger rattlesnake | ASM1654583v1 | Contig | 1.612 | 2021 |
| Crotalus viridis viridis | Prairie rattlesnake | UTA_CroVir_3.0 | Chromosome | 1.340 | 2019 |
| Emydocephalus ijimae | EmyIji_1.0 | Scaffold | 1.625 | 2019 | |
| Hydrophis cyanocinctus | Asian annulated sea snake | ASM402372v1 | Scaffold | 1.390 | 2019 |
| Hydrophis hardwickii | Hardwick’s sea snake | ASM402376v1 | Scaffold | 1.296 | 2019 |
| Hydrophis melanocephalus | hydMel_1.0 | Scaffold | 1.403 | 2019 | |
| Laticauda colubrina | Yellow-lipped sea krait | latCor_2.0 | Scaffold | 2.039 | 2020 |
| Laticauda laticaudata | Blue-ringed sea krait | latLat_1.0 | Scaffold | 1.559 | 2019 |
| Naja naja | Indian cobra | Nana_v5 | Chromosome | 1.769 | 2019 |
| Notechis scutatus | Mainland tiger snake | TS10Xv2-PRI | Scaffold | 1.666 | 2018 |
| Ophiophagus hannah | King cobra | OphHan1.0 | Scaffold | 1.594 | 2013 |
| Pantherophis guttatus | UNIGE_PanGut_3.0 | Scaffold | 1.707 | 2020 | |
| Pantherophis obsoletus | Western rat snake | UNIGE_PanObs_1.0 | Scaffold | 1.692 | 2020 |
| Protobothrops flavoviridis | Habu | HabAm_1.0 | Scaffold | 1.413 | 2018 |
| Protobothrops mucrosquamatus | Chinese habu | P.Mucros_1.0 | Scaffold | 1.674 | 2016 |
| Pseudonaja textilis | Eastern brown snake | EBS10Xv2-PRI | Scaffold | 1.590 | 2018 |
| Ptyas mucosa | Dhaman | UNIGE_Pmuc_v1.0 | Scaffold | 1.721 | 2020 |
| Python bivittatus | Burmese python | Python_molurus_bivittatus-5.0. | Scaffold | 1.435 | 2013 |
| Thamnophis elegans | Western terrestrial garter snake | rThaEle1.pri | Chromosome | 1.672 | 2019 |
| Thamnophis sirtalis | Thamnophis_sirtalis-6.0 | Scaffold | 1.425 | 2015 | |
| Thermophis baileyi | DSBC_Tbai_1.0 | Scaffold | 1.748 | 2018 | |
| Vipera berus berus | Common viper | Vber.be_1.0 | Scaffold | 1.532 | 2014 |
| Application | Tool | Repeat Annotation Approach | Features | Input Format | Year | Reference |
|---|---|---|---|---|---|---|
| Repeats annotations and determination of their genomic abundance | RepeatMasker | Homology-based detection nhmmer, cross_match, ABBlast/WUBlast,RMBlast | Identifies all types of interspersed repeats and low-complexity DNA sequences | FASTA assembled contigs or scaffolds | 2013 | Smit (2020) [123] |
| TRF | Stochastic model of tandem repeats by percent identity and frequency of insertions and deletions | Detects tandem repeats in range from 1 to 2000 bp | FASTA | 1999 | Benson (1999) [242] | |
| MGEScan | Hmmer | Identifies LTR and non-LTR retroelements | FASTA | 2016 | Lee et al. (2016) [243] | |
| LTRdigest | Local alignment and hidden Markov model-based method | Determines the position of potential LTR retrotransposons or ERV insertions | LTR annotation in GFF3 format | 2009 | Steinbiss et al. (2009) [244] | |
| RECON | Pairwise-based similarity and BLAST-based mapping | Identifies and classifies de novo repeat sequence families | FASTA | 2002 | Bao and Eddy (2002) [245] | |
| RepeatScout | lmer frequency similarity-based mapping with consensus | Identifies novel repeat families | FASTA | 2005 | Price et al. (2005) [246] | |
| RepeatModeler2 | A pipeline of multiple algorithm runs (RECON, RepeatScout, and LtrHarvest/Ltr_retriever) | Produces high-quality libraries of TE families and detects LTR structure | FASTA | 2020 | Flynn et al. (2020) [247] | |
| RepeatExplorer2 | Graph-based reads clustering and characterization of repeats | Assembles repeats and detects novel satellites and TEs | Unassembled short reads in FASTQ | 2020 | Novák et al. (2020) [248] | |
| dnaPipeTE | RepeatMasker-based mapping | Quantifies the proportion of TEs in unassembled small datasets | FASTQ reads | 2015 | Goubert et al. (2015) [249] | |
| REPdenovo | k-mer counting and de novo assembly of repeats | Generates longer repeats | Paired-end FASTQ reads | 2016 | Chu et al. (2016) [250] | |
| RepLong | Constructs a network of read overlaps based on pairwise alignment | Identifies novel repetitive elements in the genome using PacBio long reads | FASTA | 2018 | Guo et al. (2018) [251] | |
| RepARK | Abundant k-mers-based analysis of NGS reads | Detects repetitive motifs and annotates TE classes | FASTQ reads | 2014 | Koch et al. (2014) [252] | |
| Repeats expression | RepEnrich | Bowtie2-based sensitive mapping of RNA sequences | Assigns repetitive elements into families and subfamilies and identify transcripts | ChIP-seq and RNA-seq FASTQ reads | 2014 | Criscione et al. (2014) [253] |
| TEtranscripts | Uniq and multimode mapping | Accurate differential expression analysis of repeats | GTF files, RNA-seq alignments BAM files | 2015 | Jin et al. (2015) [254] | |
| SalmonTE | k-mer-based quasi-mapping | Fast and scalable quantification of TE transcripts | FASTQ files and phenotype data | 2018 | Jeong et al. (2018) [255] | |
| TEcandidates | Bowtie2-based mapping | Differential expression analysis of TEs | FASTQ or FASTA | 2018 | Valdebenito-Maturana et al. (2020) [256] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.F.; Singchat, W.; Panthum, T.; Srikulnath, K. Impact of Repetitive DNA Elements on Snake Genome Biology and Evolution. Cells 2021, 10, 1707. https://doi.org/10.3390/cells10071707
Ahmad SF, Singchat W, Panthum T, Srikulnath K. Impact of Repetitive DNA Elements on Snake Genome Biology and Evolution. Cells. 2021; 10(7):1707. https://doi.org/10.3390/cells10071707
Chicago/Turabian StyleAhmad, Syed Farhan, Worapong Singchat, Thitipong Panthum, and Kornsorn Srikulnath. 2021. "Impact of Repetitive DNA Elements on Snake Genome Biology and Evolution" Cells 10, no. 7: 1707. https://doi.org/10.3390/cells10071707
APA StyleAhmad, S. F., Singchat, W., Panthum, T., & Srikulnath, K. (2021). Impact of Repetitive DNA Elements on Snake Genome Biology and Evolution. Cells, 10(7), 1707. https://doi.org/10.3390/cells10071707