
Prospects and Challenges for T Cell-Based Therapies of HCC

Abstract
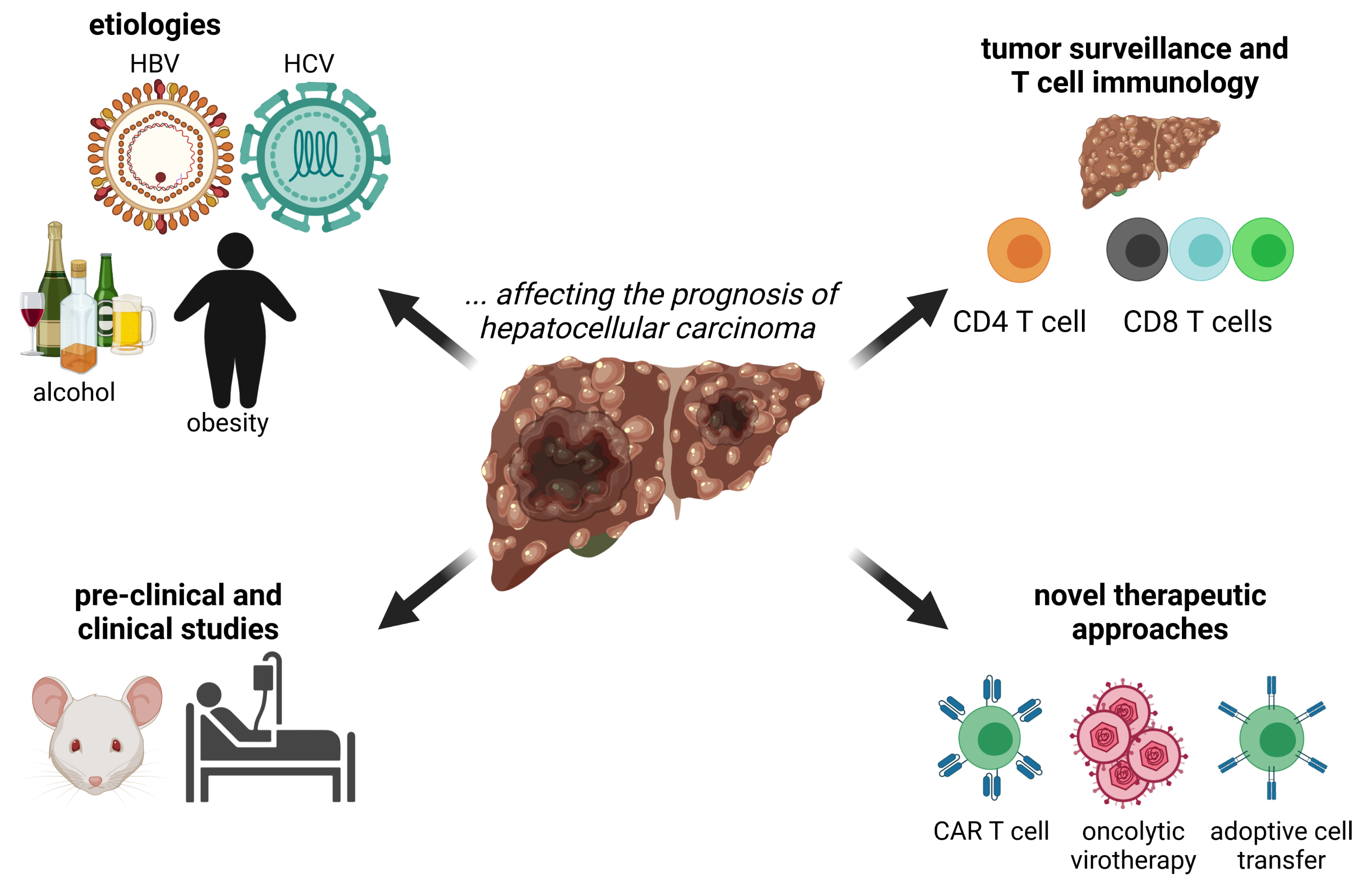
: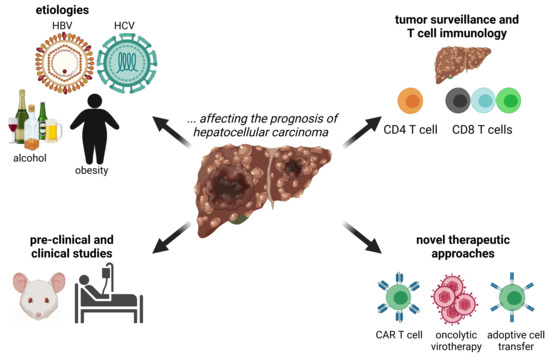
1. Introduction
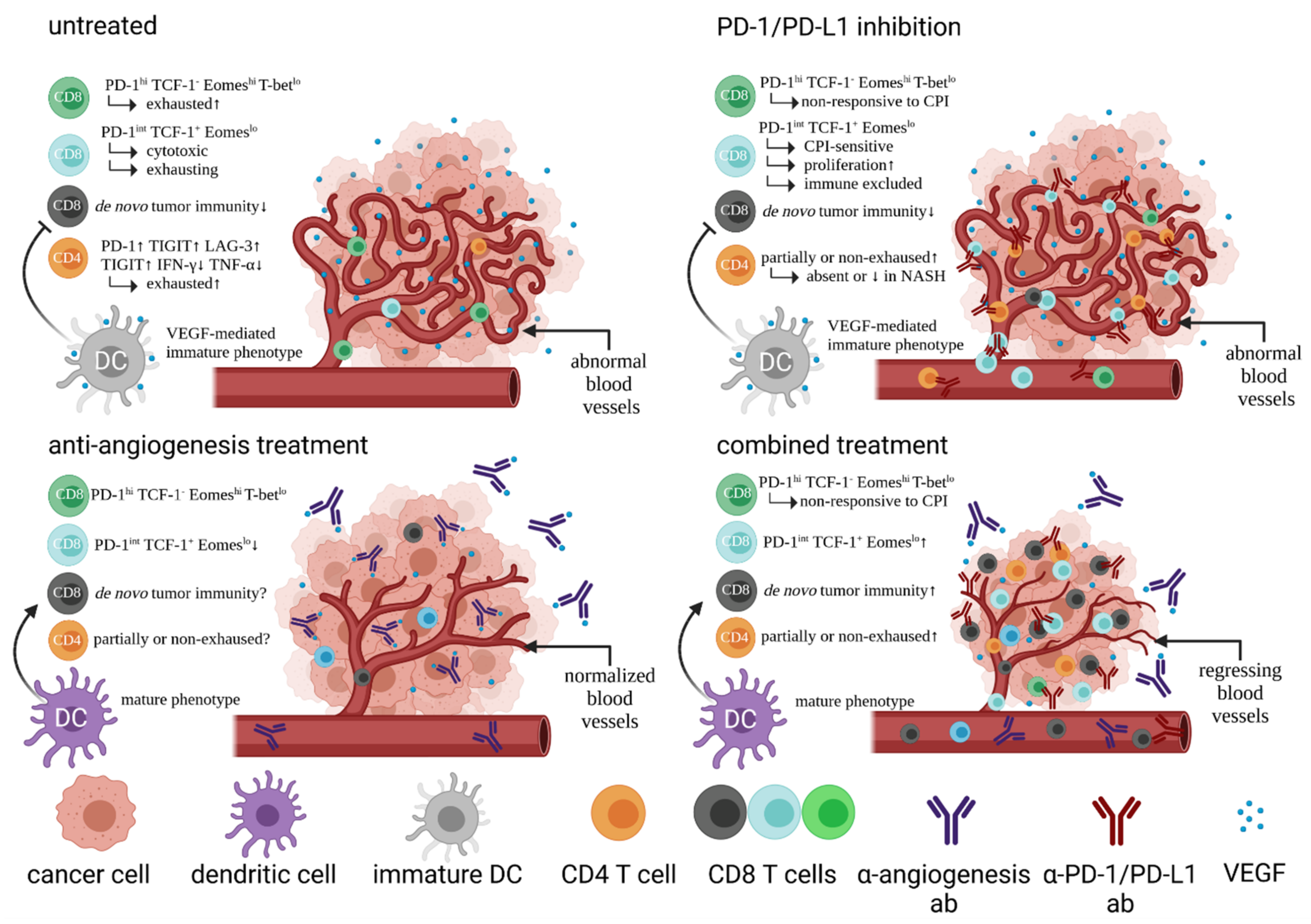
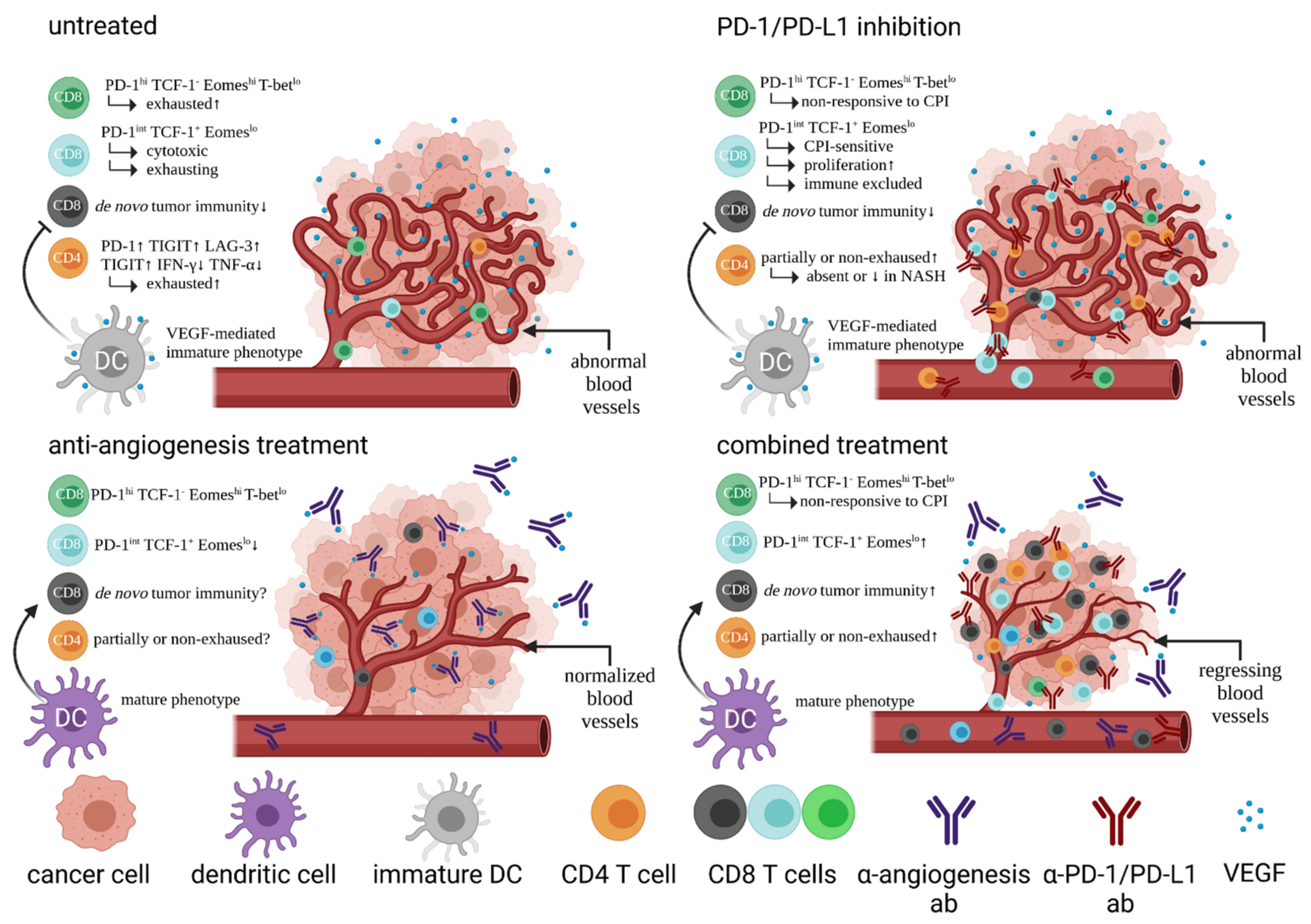
2. The Vade Mecum of HCC Treatment Is Now Based on Pillars of Cellular Immunity by Combining Checkpoint Inhibition with Anti-Angiogenesis Treatment
3. Biomarkers and Immunological Classification of HCC
4. The Immune Landscape of HCC
5. HCC Immune Surveillance by T Cells
6. Other Immunotherapeutic Approaches of HCC
7. Prospects and Challenges for T Cell-Based Therapies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | adoptive cell transfer |
| AFP | alpha-fetoprotein |
| APC | antigen-presenting cell |
| CAF | cancer-associated fibroblast |
| CAR | chimeric antigen receptor |
| CD | cluster of differentiation |
| CI | confidence interval |
| CPI | checkpoint inhibition |
| CTL | cytotoxic lymphocyte |
| CTLA-4 | cytotoxic T lymphocyte antigen 4 |
| DC | dendritic cell |
| FDA | United States Food and Drug Administration |
| FOXP3 | forkhead box protein P3 |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| GPC3 | glypican-3 |
| HCC | hepatocellular carcinoma |
| HIF-1 | hypoxia-inducible factor 1 |
| HLA | human leukocyte antigen |
| hTERT | human telomerase reverse transcriptase |
| ICAM | intercellular adhesion molecule |
| IFN-γ | interferon gamma |
| IHC | immunohistochemistry |
| IL | interleukin |
| iNKT | invariant NK T cell |
| LAG-3 | lymphocyte-activation gene 3 |
| LAMP | lysosomal associated membrane protein |
| MAIT cell | mucosal-associated invariant T cell |
| MDSC | myeloid-derived suppressor cell |
| MHC | major histocompatibility complex |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NY-ESO-1 | New York-esophageal squamous cell carcinoma-1 |
| ORR | objective response rate |
| OV | oncolytic virotherapy |
| PBMC | peripheral blood mononuclear cell |
| PD-1 | programmed cell death antigen 1 |
| PD-L1 | ligand for programmed cell death antigen 1 |
| Pexa-Vec | pexastimogene devacirepvec |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| RFA | radiofrequency ablation |
| ROS | reactive oxygen species |
| SART | squamous cell carcinoma antigen recognized by T cells |
| SIRT | selective internal radiation therapy |
| TAA | tumor-associated antigen |
| TACE | transarterial chemoembolization |
| TAM | tumor-associated macrophage |
| TCGA | The Cancer Genome Atlas |
| TCR | T cell receptor |
| Teff | effector T cell |
| Tex | exhausted CD8 T cells |
| TGF-β | tumor growth factor beta |
| TIGIT | T cell immunoreceptor with Ig and ITIM domains |
| TIM-3 | T cell immunoglobulin and mucin domain containing protein 3 |
| TKI | tyrosine kinase inhibitor |
| TMB | tumor mutational burden |
| TME | tumor microenvironment |
| Tmem | memory T cell |
| Tregs | regulatory T cells |
| T-vec | talimogene laherparepvec |
| VCAM | vascular cell adhesion molecule |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor. |
References
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Crocetti, L.; Bargellini, I.; Cioni, R. Loco-regional treatment of HCC: Current status. Clin. Radiol. 2017, 72, 626–635. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Chen, Z.H.; Hong, Y.F.; Chen, X.; Chen, J.; Lin, Q.; Lin, J.; Li, X.; Wen, J.Y.; Ruan, D.Y.; Dong, M.; et al. Comparison of five staging systems in predicting the survival rate of patients with hepatocellular carcinoma undergoing trans-arterial chemoembolization therapy. Oncol. Lett. 2018, 15, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Giannini, E.G.; Farinati, F.; Ciccarese, F.; Pecorelli, A.; Rapaccini, G.L.; Di Marco, M.; Benvegnù, L.; Caturelli, E.; Zoli, M.; Borzio, F.; et al. Prognosis of untreated hepatocellular carcinoma. Hepatology 2015, 61, 184–190. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Maker, A.V.; Phan, G.Q.; Attia, P.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Kammula, U.S.; Royal, R.E.; Haworth, L.R.; Levy, C.; et al. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: A phase I/II study. Ann. Surg. Oncol. 2005, 12, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, N.; Van Buuren, M.M.; Philips, D.; Velds, A.; Toebes, M.; Heemskerk, B.; Van Dijk, L.J.; Behjati, S.; Hilkmann, H.; El Atmioui, D.; et al. Tumor Exome Analysis Reveals Neoantigen-Specific T-Cell Reactivity in an Ipilimumab-Responsive Melanoma. J. Clin. Oncol. 2013, 31, e439–e442. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.C.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.H.; Harding, J.J.; Merle, P.; et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Long, G.V.; Atkinson, V.; Cebon, J.S.; Jameson, M.B.; Fitzharris, B.M.; McNeil, C.M.; Hill, A.G.; Ribas, A.; Atkins, M.B.; Thompson, J.A.; et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): An open-label, phase 1b trial. Lancet Oncol. 2017, 18, 1202–1210. [Google Scholar] [CrossRef] [Green Version]
- Pianko, M.J.; Moskowitz, A.J.; Lesokhin, A.M. Immunotherapy of Lymphoma and Myeloma: Facts and Hopes. Clin. Cancer Res. 2018, 24, 1002–1010. [Google Scholar] [CrossRef] [Green Version]
- Kuzume, A.; Chi, S.; Yamauchi, N.; Minami, Y. Immune-Checkpoint Blockade Therapy in Lymphoma. Int. J. Mol. Sci. 2020, 21, 5456. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Gordan, J.D.; Kennedy, E.B.; Abou-Alfa, G.K.; Beg, M.S.; Brower, S.T.; Gade, T.P.; Goff, L.; Gupta, S.; Guy, J.; Harris, W.P.; et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 4317–4345. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Shek, D.; Read, S.A.; Nagrial, A.; Carlino, M.S.; Gao, B.; George, J.; Ahlenstiel, G. Immune-Checkpoint Inhibitors for Advanced Hepatocellular Carcinoma: A Synopsis of Response Rates. Oncologist 2021. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Damen, C.A.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: The role of angiogenic factors. Cancer Res. 1996, 56, 1111–1117. [Google Scholar]
- Hellwig, S.M.; Damen, C.A.; van Adrichem, N.P.; Blijham, G.H.; Groenewegen, G.; Griffioen, A.W. Endothelial CD34 is suppressed in human malignancies: Role of angiogenic factors. Cancer Lett. 1997, 120, 203–211. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia Promotes Tumor Growth in Linking Angiogenesis to Immune Escape. Front. Immunol. 2012, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Voron, T.; Marcheteau, E.; Pernot, S.; Colussi, O.; Tartour, E.; Taieb, J.; Terme, M. Control of the Immune Response by Pro-Angiogenic Factors. Front. Oncol. 2014, 4, 70. [Google Scholar] [CrossRef] [Green Version]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: Implications for tumor immunotherapy. Cancer Immunol. Immunother. 2005, 54, 307–314. [Google Scholar] [CrossRef]
- Blank, C.; Mackensen, A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: An update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 2007, 56, 739–745. [Google Scholar] [CrossRef]
- Bellone, M.; Calcinotto, A. Ways to Enhance Lymphocyte Trafficking into Tumors and Fitness of Tumor Infiltrating Lymphocytes. Front. Oncol. 2013, 3, 231. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yao, D.; Wang, L.; Wu, W.; Qiu, L.; Yao, M.; Yao, N.; Zhang, H.; Yu, D.; Ni, Q. Expression characteristics of HIF-1α and its clinical values in diagnosis and prognosis of hepatocellular carcinoma. Hepat. Mon. 2011, 11, 821–828. [Google Scholar] [CrossRef] [Green Version]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Boige, V.; Malka, D.; Bourredjem, A.; Dromain, C.; Baey, C.; Jacques, N.; Pignon, J.-P.; Vimond, N.; Bouvet-Forteau, N.; De Baere, T.; et al. Efficacy, Safety, and Biomarkers of Single-Agent Bevacizumab Therapy in Patients with Advanced Hepatocellular Carcinoma. Oncologist 2012, 17, 1063–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, A.B.; Cohen, E.I.; Ocean, A.; Lehrer, D.; Goldenberg, A.; Knox, J.J.; Chen, H.; Clark-Garvey, S.; Weinberg, A.; Mandeli, J.; et al. Phase II Trial Evaluating the Clinical and Biologic Effects of Bevacizumab in Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2008, 26, 2992–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Randomised efficacy and safety results for atezolizumab (Atezo) + bevacizumab (Bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC). Ann. Oncol. 2019, 30, v875. [Google Scholar] [CrossRef]
- Rahma, O.E.; Hodi, F.S. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin. Cancer Res. 2019, 25, 5449–5457. [Google Scholar] [CrossRef] [Green Version]
- Gnjatic, S.; Bronte, V.; Brunet, L.R.; Butler, M.O.; Disis, M.L.; Galon, J.; Hakansson, L.G.; Hanks, B.A.; Karanikas, V.; Khleif, S.N.; et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J. Immunother. Cancer 2017, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti–PD-1 Therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [Green Version]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376.e13. [Google Scholar] [CrossRef]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [Green Version]
- Löffler, M.W.; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; Muhlenbruch, L.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Hellström, I.; Hellström, K.E.; Pierce, G.E.; Yang, J.P.S. Cellular and Humoral immunity to Different Types of Human Neoplasms. Nature 1968, 220, 1352–1354. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Gao, Q.; Qiu, S.-J.; Fan, J.; Zhou, J.; Wang, X.-Y.; Xiao, Y.-S.; Xu, Y.; Li, Y.-W.; Tang, Z.-Y. Intratumoral Balance of Regulatory and Cytotoxic T Cells Is Associated With Prognosis of Hepatocellular Carcinoma After Resection. J. Clin. Oncol. 2007, 25, 2586–2593. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Scheiner, B.; Peck-Radosavljevic, M. Immunotherapy for advanced hepatocellular carcinoma: A focus on special subgroups. Gut 2021, 70, 204–214. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
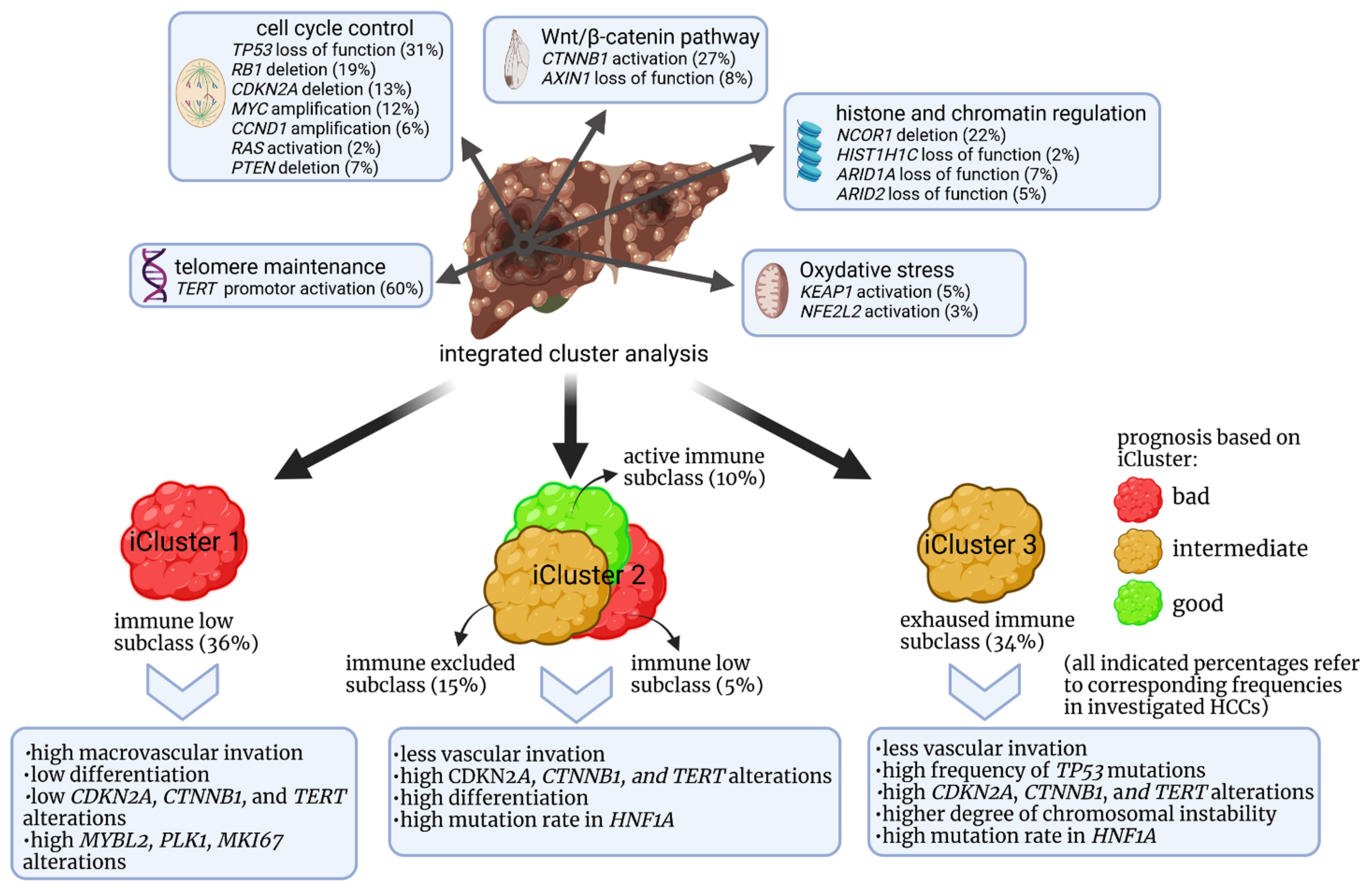
- Adeniji, N.; Dhanasekaran, R. Genomic Landscape of HCC. Curr. Hepatol. Rep. 2020, 19, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mao, M. Next generation sequencing reveals genetic landscape of hepatocellular carcinomas. Cancer Lett. 2013, 340, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; de Moura, M.C.; Putra, J.; Campreciós, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Lou, Y.; Yang, J.; Wang, J.; Feng, J.; Zhao, Y.; Wang, L.; Huang, X.; Fu, Q.; Ye, M.; et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut 2019, 68, 2019–2031. [Google Scholar] [CrossRef]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Cariani, E.; Missale, G. Immune landscape of hepatocellular carcinoma microenvironment: Implications for prognosis and therapeutic applications. Liver Int. 2019, 39, 1608–1621. [Google Scholar] [CrossRef] [Green Version]
- Foerster, F.; Hess, M.; Gerhold-Ay, A.; Marquardt, J.U.; Becker, D.; Galle, P.R.; Schuppan, D.; Binder, H.; Bockamp, E. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci. Rep. 2018, 8, 5351. [Google Scholar] [CrossRef]
- Rohr-Udilova, N.; Klinglmüller, F.; Schulte-Hermann, R.; Stift, J.; Herac, M.; Salzmann, M.; Finotello, F.; Timelthaler, G.; Oberhuber, G.; Pinter, M.; et al. Deviations of the immune cell landscape between healthy liver and hepatocellular carcinoma. Sci. Rep. 2018, 8, 6220. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From bench to bed: The tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 396. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.J.; Lee, Y.H.; Pan, L.; Lai, L.; Chua, C.; Wasser, M.; Lim, T.K.H.; Yeong, J.; Toh, H.C.; Lee, S.Y.; et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 2019, 68, 916–927. [Google Scholar] [CrossRef] [Green Version]
- Itoh, S.; Yoshizumi, T.; Yugawa, K.; Imai, D.; Yoshiya, S.; Takeishi, K.; Toshima, T.; Harada, N.; Ikegami, T.; Soejima, Y.; et al. Impact of Immune Response on Outcomes in Hepatocellular Carcinoma: Association With Vascular Formation. Hepatology 2020, 72, 1987–1999. [Google Scholar] [CrossRef]
- Zarour, H.M. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Sekyere, S.O.; Suneetha, P.V.; Kraft, A.R.M.; Zhang, S.; Dietz, J.; Sarrazin, C.; Manns, M.P.; Schlaphoff, V.; Cornberg, M.; Wedemeyer, H. A heterogeneous hierarchy of co-regulatory receptors regulates exhaustion of HCV-specific CD8 T cells in patients with chronic hepatitis C. J. Hepatol. 2015, 62, 31–40. [Google Scholar] [CrossRef]
- Ostroumov, D.; Duong, S.; Wingerath, J.; Woller, N.; Manns, M.P.; Timrott, K.; Kleine, M.; Ramackers, W.; Roessler, S.; Nahnsen, S.; et al. Transcriptome Profiling Identifies TIGIT as a Marker of T-Cell Exhaustion in Liver Cancer. Hepatology 2021, 73, 1399–1418. [Google Scholar] [CrossRef]
- Zheng, Q.; Xu, J.; Gu, X.; Wu, F.; Deng, J.; Cai, X.; Wang, G.; Li, G.; Chen, Z. Immune checkpoint targeting TIGIT in hepatocellular carcinoma. Am. J. Transl. Res. 2020, 12, 3212–3224. [Google Scholar] [PubMed]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.J.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Marraco, S.A.F.; Calderon-Copete, S.; Ferreira, D.P.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goding, S.R.; Wilson, K.A.; Xie, Y.; Harris, K.M.; Baxi, A.; Akpinarli, A.; Fulton, A.; Tamada, K.; Strome, S.E.; Antony, P.A. Restoring Immune Function of Tumor-Specific CD4+ T Cells during Recurrence of Melanoma. J. Immunol. 2013, 190, 4899–4909. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Liu, W.; Sanin, D.E.; Jia, G.; Tian, M.; Wang, H.; Zhu, B.; Lu, Y.; Qiao, T.; Wang, X.; et al. Heterogeneity of exhausted T cells in the tumor microenvironment is linked to patient survival following resection in hepatocellular carcinoma. OncoImmunology 2020, 9, 1746573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaoul, N.; Mancarella, S.; Lupo, L.; Giannelli, G.; Dituri, F. Impaired Anti-Tumor T cell Response in Hepatocellular Carcinoma. Cancers 2020, 12, 627. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [Green Version]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef]
- Kurioka, A.; Walker, L.J.; Klenerman, P.; Willberg, C.B. MAIT cells: New guardians of the liver. Clin. Transl. Immunol. 2016, 5, e98. [Google Scholar] [CrossRef] [PubMed]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Salomone, F.; Galvano, F.; Volti, G.L. Molecular Bases Underlying the Hepatoprotective Effects of Coffee. Nutrients 2017, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Dranoff, J.A. Coffee Consumption and Prevention of Cirrhosis: In Support of the Caffeine Hypothesis. Gene Expr. 2018, 18, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Feld, J.J.; Lavoie, É.G.; Fausther, M.; Dranoff, J.A. I drink for my liver, Doc: Emerging evidence that coffee prevents cirrhosis. F1000Research 2015, 4, 95. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Liang, J.; Ding, T.; Guo, Z.-W.; Yu, X.-J.; Hu, Y.-Z.; Zheng, L.; Xu, J. Expression pattern of tumour-associated antigens in hepatocellular carcinoma: Association with immune infiltration and disease progression. Br. J. Cancer 2013, 109, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.; Tauber, C.; Hensel, N.; Thimme, R. CD8+ T Cell Responses during HCV Infection and HCC. J. Clin. Med. 2021, 10, 991. [Google Scholar] [CrossRef] [PubMed]
- Hiroishi, K.; Eguchi, J.; Baba, T.; Shimazaki, T.; Ishii, S.; Hiraide, A.; Sakaki, M.; Doi, H.; Uozumi, S.; Omori, R.; et al. Strong CD8+ T-cell responses against tumor-associated antigens prolong the recurrence-free interval after tumor treatment in patients with hepatocellular carcinoma. J. Gastroenterol. 2010, 45, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Boon, T.; Van Der Bruggen, P. Human tumor antigens recognized by T lymphocytes. J. Exp. Med. 1996, 183, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. A New Era for Cancer Immunotherapy Based on the Genes that Encode Cancer Antigens. Immunity 1999, 10, 281–287. [Google Scholar] [CrossRef] [Green Version]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.-T. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Nakamoto, Y.; Marukawa, Y.; Arai, K.; Yamashita, T.; Tsuji, H.; Kuzushima, K.; Takiguchi, M.; Kaneko, S. Cytotoxic T cell responses to human telomerase reverse transcriptase in patients with hepatocellular carcinoma. Hepatology 2006, 43, 1284–1294. [Google Scholar] [CrossRef]
- Kaji, K.; Mizukoshi, E.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Fushimi, K.; Nakagawa, H.; Yamada, K.; Terashima, T.; Kitahara, M.; et al. Cellular Immune Responses for Squamous Cell Carcinoma Antigen Recognized by T Cells 3 in Patients with Hepatocellular Carcinoma. PLoS ONE 2017, 12, e0170291. [Google Scholar] [CrossRef]
- Thimme, R.; Neagu, M.; Boettler, T.; Neumann-Haefelin, C.; Kersting, N.; Geissler, M.; Makowiec, F.; Obermaier, R.; Hopt, U.T.; Blum, H.E.; et al. Comprehensive analysis of the α-fetoprotein-specific CD8+ T cell responses in patients with hepatocellular carcinoma. Hepatology 2008, 48, 1821–1833. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Li, L.; Rao, X.; Wen, Z.; Ding, X.; Wang, X.; Xu, W.; Meng, C.; Yi, Y.; Guan, Y.; Chen, Y.; et al. Implications of driver genes associated with a high tumor mutation burden identified using next-generation sequencing on immunotherapy in hepatocellular carcinoma. Oncol. Lett. 2020, 19, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/β-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Galarreta, M.R.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti–PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Sekyere, S.O.; Schlevogt, B.; Mettke, F.; Kabbani, M.; Deterding, K.; Wirth, T.C.; Vogel, A.; Manns, M.P.; Falk, C.S.; Cornberg, M.; et al. HCC Immune Surveillance and Antiviral Therapy of Hepatitis C Virus Infection. Liver Cancer 2019, 8, 41–65. [Google Scholar] [CrossRef]
- Knocke, S.; Fleischmann-Mundt, B.; Saborowski, M.; Manns, M.P.; Kühnel, F.; Wirth, T.C.; Woller, N. Tailored Tumor Immunogenicity Reveals Regulation of CD4 and CD8 T Cell Responses against Cancer. Cell Rep. 2016, 17, 2234–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Heinrich, B.; Brown, Z.J.; Diggs, L.P.; Vormehr, M.; Ma, C.; Subramanyam, V.; Rosato, U.; Ruf, B.; Walz, J.S.; McVey, J.C.; et al. Steatohepatitis Impairs T-cell–Directed Immunotherapies Against Liver Tumors in Mice. Gastroenterology 2021, 160, 331–345.e6. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef] [Green Version]
- Tiegs, G.; Lohse, A.W. Immune tolerance: What is unique about the liver. J. Autoimmun. 2010, 34, 1–6. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Kitahara, M.; Mizukoshi, E.; Terashima, T.; Nakagawa, H.; Horii, R.; Iida, N.; Arai, K.; Yamashita, T.; Sakai, Y.; Yamashita, T.; et al. Safety and Long-Term Outcome of Intratumoral Injection of OK432-Stimulated Dendritic Cells for Hepatocellular Carcinomas After Radiofrequency Ablation. Transl. Oncol. 2020, 13, 100777. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Mayr, C.; Kiesslich, T.; Stintzing, S.; Neureiter, D. Immunmodulatory Treatment Strategies of Hepatocellular Carcinoma: From Checkpoint Inhibitors Now to an Integrated Approach in the Future. Cancers 2021, 13, 1558. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Zender, L.; Schulte, B.; Mundt, B.; Plentz, R.; Rudolph, K.L.; Manns, M.; Kubicka, S.; Kühnel, F. A telomerase-dependent conditionally replicating adenovirus for selective treatment of cancer. Cancer Res. 2003, 63, 3181–3188. [Google Scholar] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woller, N.; Gürlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Moehler, M.; Heo, J.; Lee, H.C.; Tak, W.Y.; Chao, Y.; Paik, S.W.; Yim, H.J.; Byun, K.S.; Baron, A.; Ungerechts, G.; et al. Vaccinia-based oncolytic immunotherapy Pexastimogene Devacirepvec in patients with advanced hepatocellular carcinoma after sorafenib failure: A randomized multicenter Phase IIb trial (TRAVERSE). OncoImmunology 2019, 8, 1615817. [Google Scholar] [CrossRef] [Green Version]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation with Antitumor Lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Alexander, M.; Culos, K.; Roddy, J.; Shaw, R.; Bachmeier, C.; Shigle, T.L.; Mahmoudjafari, Z. Chimeric Antigen Receptor T Cell Therapy: A Comprehensive Review of Clinical Efficacy, Toxicity, and Best Practices for Outpatient Administration. Transplant. Cell. Ther. 2021. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2016, 7, 690. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, N.; Zhang, Y.-F.; Fu, H.; Feng, M.; Schneider, D.; Su, L.; Wu, X.; Zhou, J.; Mackay, S.; et al. Persistent Polyfunctional Chimeric Antigen Receptor T Cells That Target Glypican 3 Eliminate Orthotopic Hepatocellular Carcinomas in Mice. Gastroenterology 2020, 158, 2250–2265.e20. [Google Scholar] [CrossRef]
- Shirakawa, H.; Suzuki, H.; Shimomura, M.; Kojima, M.; Gotohda, N.; Takahashi, S.; Nakagohri, T.; Konishi, M.; Kobayashi, N.; Kinoshita, T.; et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009, 100, 1403–1407. [Google Scholar] [CrossRef]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef]
- Rochigneux, P.; Chanez, B.; De Rauglaudre, B.; Mitry, E.; Chabannon, C.; Gilabert, M. Adoptive Cell Therapy in Hepatocellular Carcinoma: Biological Rationale and First Results in Early Phase Clinical Trials. Cancers 2021, 13, 271. [Google Scholar] [CrossRef] [PubMed]
- Verdegaal, E.M.E.; de Miranda, N.F.C.C.; Visser, M.; Harryvan, T.; Van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; Van Der Minne, C.E.; Schotte, R.; Spits, R.H.; et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Giraud, J.; Chalopin, D.; Blanc, J.-F.; Saleh, M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 2021, 12, 655697. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H.; Ribas, A.; Dissette, V.B.; Lee, Y.; Yang, J.Q.; De La Rocha, P.; Duran, S.D.; Hernandez, J.; Seja, E.; Potter, D.M.; et al. A Phase I/II Trial Testing Immunization of Hepatocellular Carcinoma Patients with Dendritic Cells Pulsed with Four α-Fetoprotein Peptides. Clin. Cancer Res. 2006, 12, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Mizukoshi, E.; Kobayashi, E.; Tamai, T.; Hamana, H.; Ozawa, T.; Kishi, H.; Kitahara, M.; Yamashita, T.; Arai, K.; et al. Association Between High-Avidity T-Cell Receptors, Induced by α-Fetoprotein—Derived Peptides, and Anti-Tumor Effects in Patients With Hepatocellular Carcinoma. Gastroenterology 2017, 152, 1395–1406.e10. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Forner, A.; Korangy, F.; N’Kontchou, G.; Barget, N.; Ayuso, C.; Ormandy, L.A.; Manns, M.P.; Beaugrand, M.; Bruix, J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010, 10, 209. [Google Scholar] [CrossRef] [Green Version]
- Mizukoshi, E.; Nakagawa, H.; Kitahara, M.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Fushimi, K.; Kobayashi, E.; Kishi, H.; Muraguchi, A.; et al. Immunological features of T cells induced by human telomerase reverse transcriptase-derived peptides in patients with hepatocellular carcinoma. Cancer Lett. 2015, 364, 98–105. [Google Scholar] [CrossRef]
- Sawada, Y.; Yoshikawa, T.; Nobuoka, D.; Shirakawa, H.; Kuronuma, T.; Motomura, Y.; Mizuno, S.; Ishii, H.; Nakachi, K.; Konishi, M.; et al. Phase I Trial of a Glypican-3–Derived Peptide Vaccine for Advanced Hepatocellular Carcinoma: Immunologic Evidence and Potential for Improving Overall Survival. Clin. Cancer Res. 2012, 18, 3686–3696. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Nakatsugawa, M.; Suzuki, S.; Shirakawa, H.; Nobuoka, D.; Sakemura, N.; Motomura, Y.; Tanaka, Y.; Hayashi, S.-I.; Nakatsura, T. HLA-A2-restricted glypican-3 peptide-specific CTL clones induced by peptide vaccine show high avidity and antigen-specific killing activity against tumor cells. Cancer Sci. 2011, 102, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Leboeuf, C.; Mailly, L.; Wu, T.; Bour, G.; Durand, S.; Brignon, N.; Ferrand, C.; Borg, C.; Tiberghien, P.; Thimme, R.; et al. In Vivo Proof of Concept of Adoptive Immunotherapy for Hepatocellular Carcinoma Using Allogeneic Suicide Gene-modified Killer Cells. Mol. Ther. 2014, 22, 634–644. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Wang, X.; Zheng, L.; Lyu, T.; Figini, M.; Wang, B.; Procissi, D.; Shangguan, J.; Sun, C.; Pan, L.; et al. MRI-guided interventional natural killer cell delivery for liver tumor treatment. Cancer Med. 2018, 7, 1860–1869. [Google Scholar] [CrossRef] [Green Version]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Teufel, A.; Zhan, T.; Härtel, N.; Bornschein, J.; Ebert, M.P.; Schulte, N. Management of immune related adverse events induced by immune checkpoint inhibition. Cancer Lett. 2019, 456, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Adkins, S. CAR T-Cell Therapy: Adverse Events and Management. J. Adv. Pract. Oncol. 2019, 10, 21–28. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
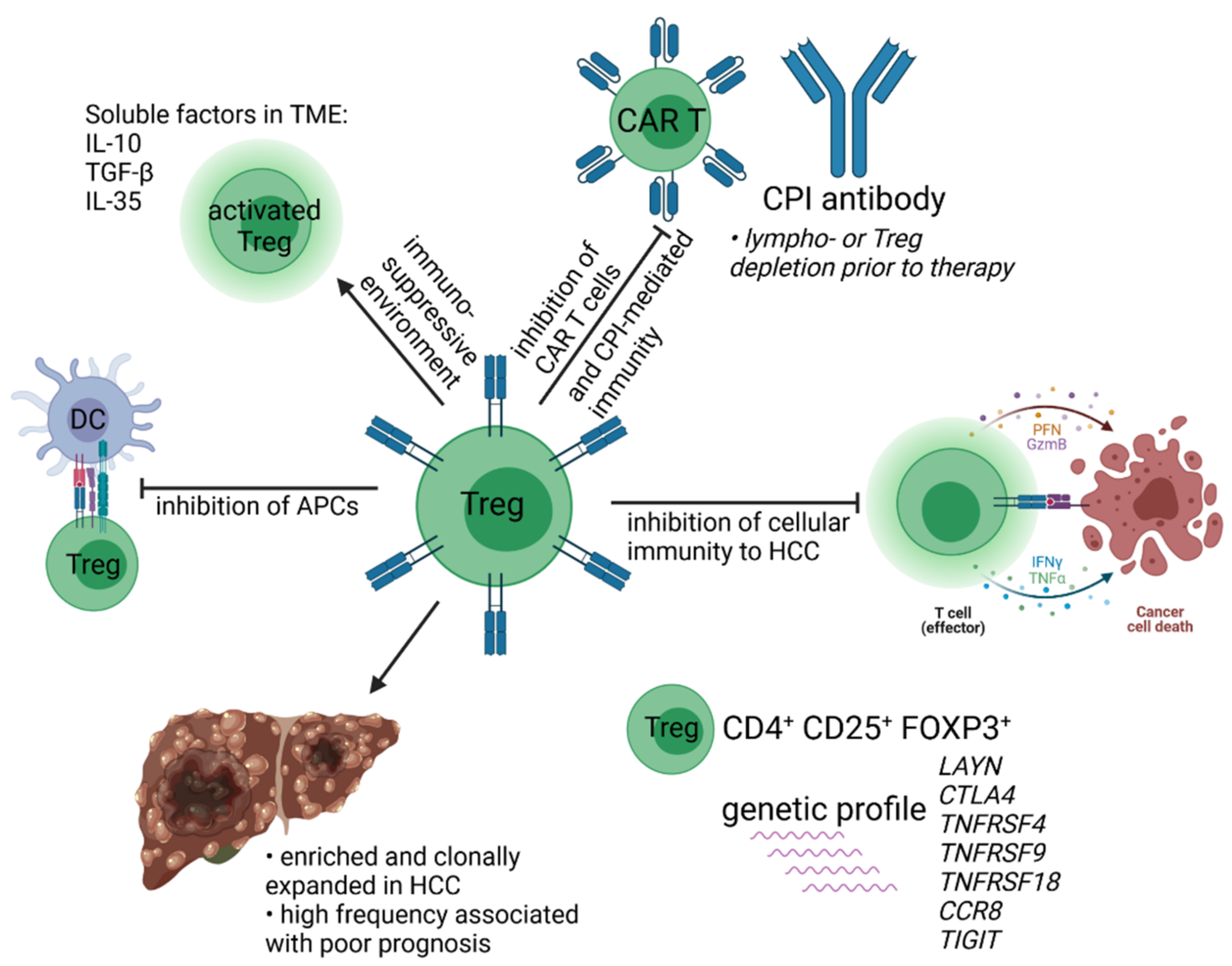
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Woller, N.; Knocke, S.; Mundt, B.; Gürlevik, E.; Strüver, N.; Kloos, A.; Boozari, B.; Schache, P.; Manns, M.P.; Malek, N.P.; et al. Virus-induced tumor inflammation facilitates effective DC cancer immunotherapy in a Treg-dependent manner in mice. J. Clin. Investig. 2011, 121, 2570–2582. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III–IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T Cell Phenotype | T Cell Function | Prognosis in HCC Patients | Study |
|---|---|---|---|
| FOXP3+ CD45+ including other lineages | Suppress CD8-mediated immunity; expression of TGF-β, VEGF, and IL-10 | poor | [55] |
| PD-1hi CD4 Treg and PD-1hi CD8 Trm | More suppressive and exhausted in HBV-related HCC, reversible by CPI | poor for Treg better for Trm | [63] |
| tumor-infiltrating CD8 T cell | immune defence against tumor progression | good | [54,64] |
| CD8 T cell with high expression of inhibitory receptors: PD-1, TIGIT, TIM-3, LAG-3 | exhaustion phenotype (Tex) | poor (for fully exhausted T cells) | [66,67] |
| PD-1int TCF-1+ Eomeslo CD8 T cell | early Tex, proliferative capacity, responsive to CPI | good under CPI treatment | [71,72] |
| PD-1hi TCF-1− Tbetlo Eomeshi CD8 T cell | terminally exhausted Tex, non-responsive to CPI | poor | [71,72] |
| CD8 T cell cluster SLC4A10 | MAIT cells | poor, if frequency is low in HCC tissue | [79] |
| CD8 T cell cluster CX3CR1, FCGR3A, FGFBP2 | effector T cells | n/d | [79] |
| CD8 T cell cluster CTLA4, PDCD1, HAVCR2 | terminally exhausted Tex | n/d | [79] |
| CD8 T cell cluster CTLA4, PDCD1, HAVCR2, GZMK | Early Tex with putative cytotoxicity | n/d | [79] |
| CD4 Treg cluster FOXP3, CTLA4, TNFRSF18, TNFRSF4, and CCR8 | T reg | no correlation found in this study | [79] |
| Tex and Treg cluster LAYN | suppressive function | poor when LAYN expression is high | [79] |
| Prospects | Challenges | |
|---|---|---|
| Identification and isolation of tu-specific T cells: TAAs and neoantigens | TAAs well defined for most HLA types neoantigens drive tumor responses, high frequency of tu-specific T cells | low frequency of TAA-specific T cells, monoclonal patient specific, time consuming id process, not clinically applicable yet, monoclonal |
| T cell expansion: in vivo and ex vivo | in vivo by CPI yields polyclonal responses, no id process necessary, rapid induction of immunity, agents off-the-shelf ex vivo from blood, well accessible source, putatively less influenced by the TME ex vivo from tumor-tissue, tumor-spec. T cells enriched | frequent non-response, temporal response, adverse events ex vivo from blood, requires id or modification by CARs, low initial frequency, monoclonal, adverse events ex vivo from tumor-tissue, time consuming culture/selection, exhausted T cells, limited availability, still experimental, adverse events |
| T cell homing | local administration: effective in targeted tumors systemic administration: simple route of application | less effective in non-targeted tumor nodules systemic transfer requires proper homing, putatively less effective than direct targeting in CAR-T cell therapies |
| safety | monitoring of adverse events well established majority of adverse events manageable | adverse events display high diversity of autoimmune disorders adverse events can pose life threatening complications in some patients |
| TME management: targeted intervention oncolytic virotherapy | complementing tumor immune cycle, expansion of response time, facilitates immunotherapies broadening spectrum of T cell responses, virus inflammation dampens tolerance induction of tumors, facilitates and promotes immunotherapies | precise intervention or characterization of tumor attributes required accessibility of tumors for OV injection required |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woller, N.; Engelskircher, S.A.; Wirth, T.; Wedemeyer, H. Prospects and Challenges for T Cell-Based Therapies of HCC. Cells 2021, 10, 1651. https://doi.org/10.3390/cells10071651
Woller N, Engelskircher SA, Wirth T, Wedemeyer H. Prospects and Challenges for T Cell-Based Therapies of HCC. Cells. 2021; 10(7):1651. https://doi.org/10.3390/cells10071651
Chicago/Turabian StyleWoller, Norman, Sophie Anna Engelskircher, Thomas Wirth, and Heiner Wedemeyer. 2021. "Prospects and Challenges for T Cell-Based Therapies of HCC" Cells 10, no. 7: 1651. https://doi.org/10.3390/cells10071651
APA StyleWoller, N., Engelskircher, S. A., Wirth, T., & Wedemeyer, H. (2021). Prospects and Challenges for T Cell-Based Therapies of HCC. Cells, 10(7), 1651. https://doi.org/10.3390/cells10071651