
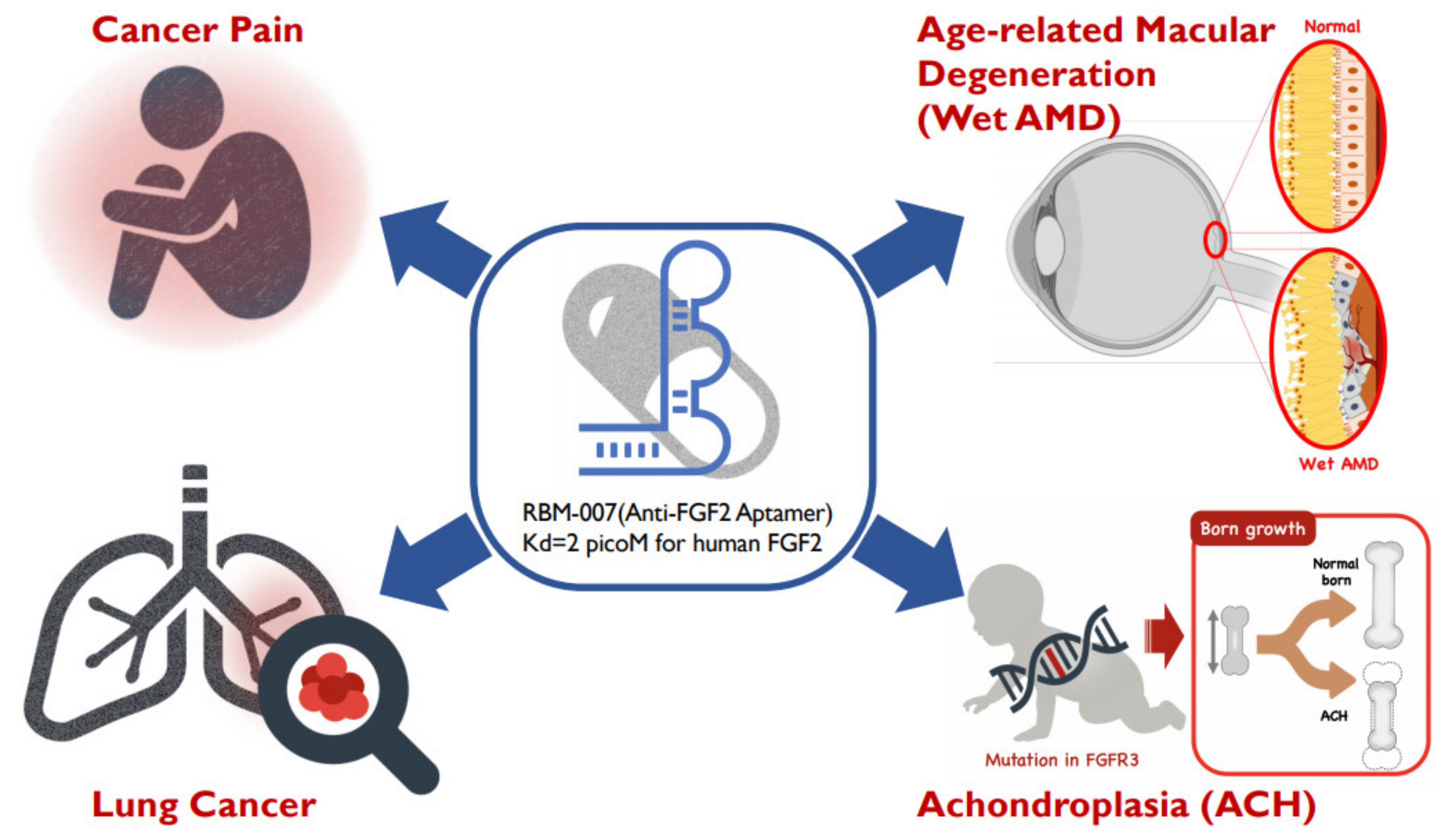
Multiple Therapeutic Applications of RBM-007, an Anti-FGF2 Aptamer

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Anti-FGF2 Aptamer
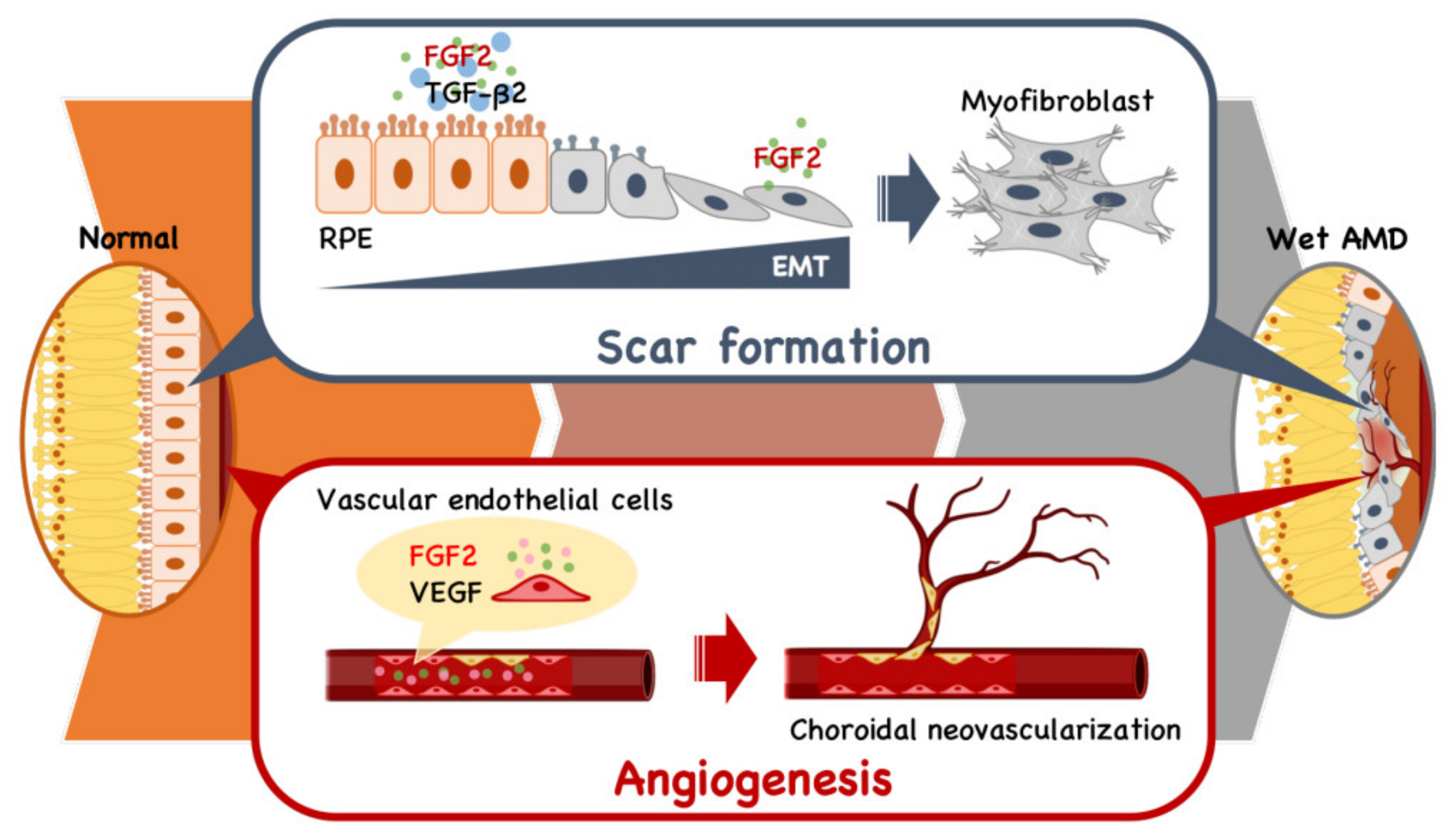
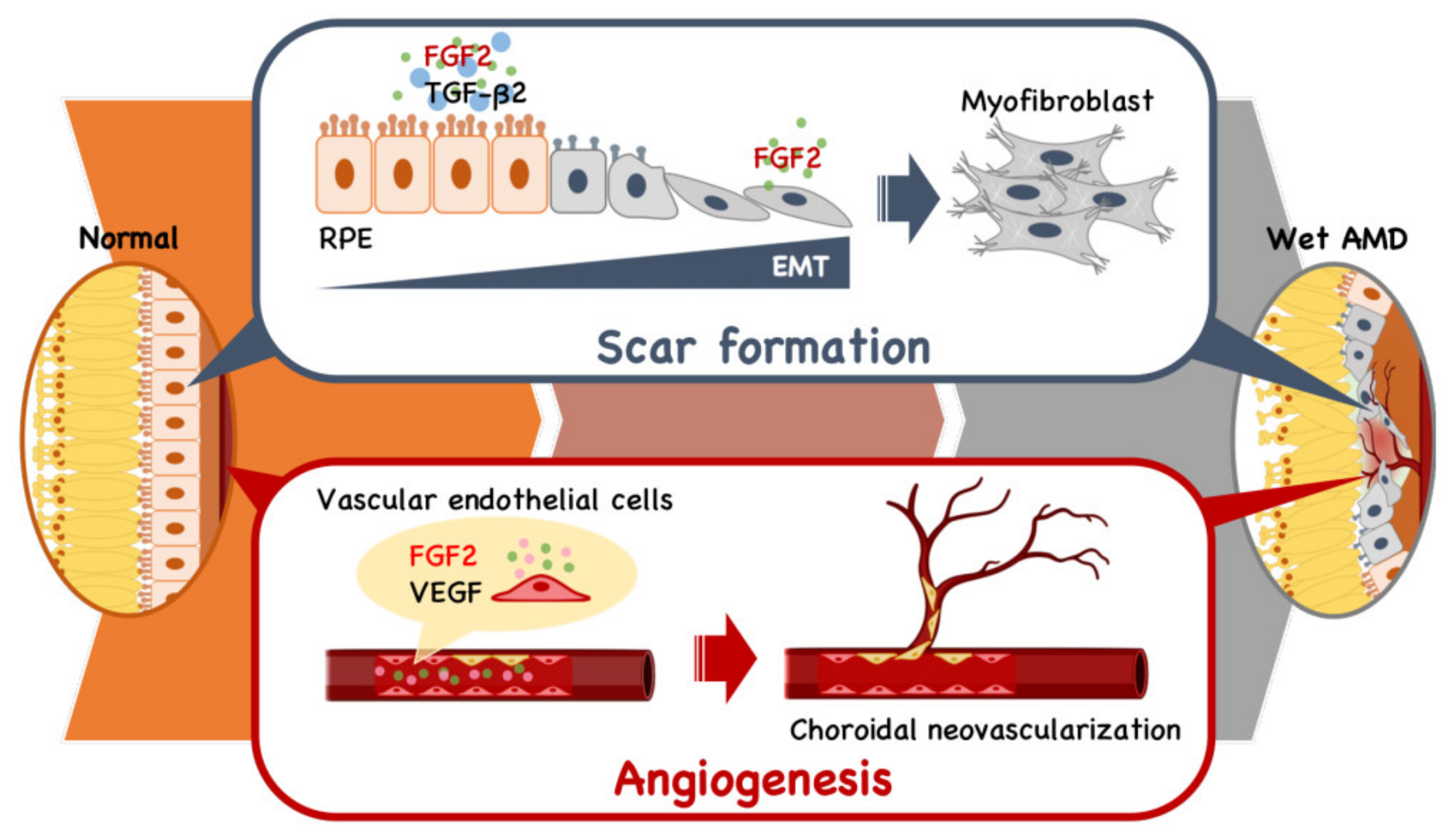
3. Therapeutic Application in Age-Related Macular Degeneration
4. Therapeutic Application in Achondroplasia
5. Therapeutic Application in Cancer Pain
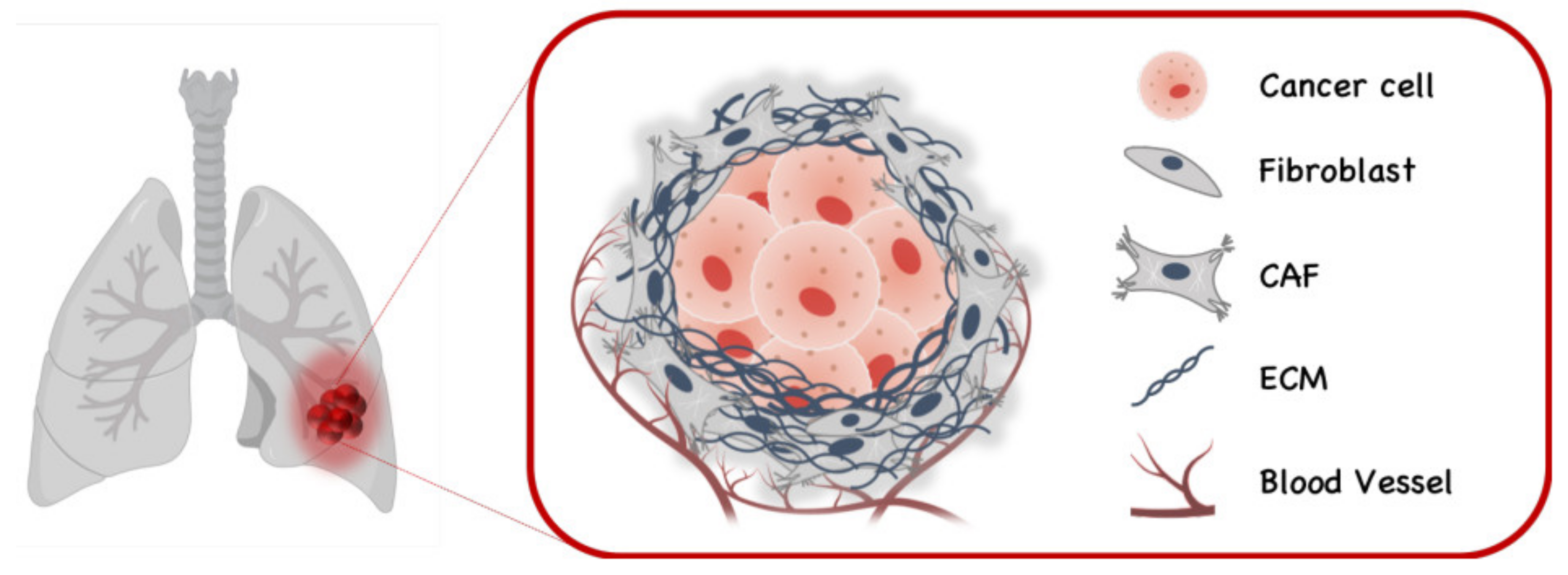
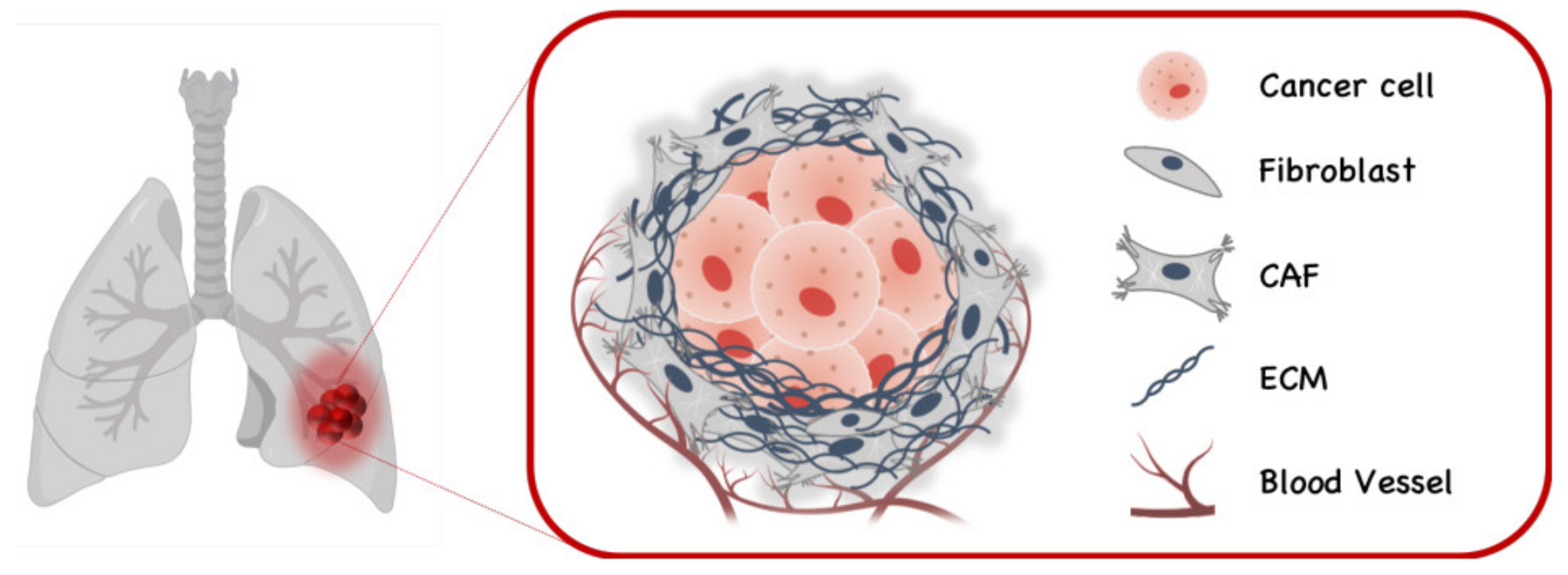
6. Therapeutic Application in Lung Cancer
7. Conclusion and Perspectives
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krejci, P.; Prochazkova, J.; Bryja, V.; Kozubik, A.; Wilcox, W.R. Molecular pathology of the fibroblast growth factor family. Hum. Mutat. 2011, 30, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Marie, P.J.; Miraoui, H.; Severe, N. FGF/FGFR signaling in bone formation: Progress and perspectives. Growth Factors 2012, 30, 117–123. [Google Scholar] [CrossRef]
- Okada-Ban, M.; Thiery, J.P.; Jouanneau, J. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 2000, 32, 263–267. [Google Scholar] [CrossRef]
- Eda, H.; Aoki, K.; Marumo, K.; Fujii, K.; Ohkawa, K. FGF-2 signaling induces downregulation of TAZ protein in osteoblastic MC3T3-E1 cells. Biochem. Biophys. Res. Commun. 2008, 366, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Nakamura, K.; Tabata, Y.; Ikada, Y.; Aoyama, I.; Anzai, J.; Nakamura, T.; Hiyama, Y.; Tamura, M. Acceleration of fracture healing in nonhuman primates by fibroblast growth factor-2. J. Clin. Endocrinol. Metab. 2001, 86, 875–880. [Google Scholar] [CrossRef]
- Nakagawa, N.; Yasuda, H.; Yano, K.; Mochizuki, S.; Kobayashi, N.; Fujimoto, H.; Shima, N.; Morinaga, T.; Chikazu, D.; Kawaguchi, H.; et al. Basic fibroblast growth factor induces osteoclast formation by reciprocally regulating the production of osteoclast differentiation factor and osteoclastogenesis inhibitory factor in mouse osteoblastic cells. Biochem. Biophys. Res. Commun. 1999, 265, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Chikazu, D.; Nakamura, K.; Kumegawa, M.; Hakeda, Y. Direct and indirect actions of fibroblast growth factor 2 on osteoclastic bone resorption in cultures. J. Bone Miner. Res. 2000, 15, 466–473. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Yoshitake, Y.; Matuo, Y.; Sasaki, H.; Nishikawa, K. Monoclonal antibodies against heparin-binding growth factor II/basic fibroblast growth factor that block its biological activity: Invalidity of the antibodies for tumor angiogenesis. Proc. Natl. Acad. Sci. USA 1989, 86, 9911–9915. [Google Scholar] [CrossRef] [Green Version]
- Rege, A.A.; Bjercke, R.J.; Erichsen, D.; Owens, R.; Stephan, C.C.; Brock, T.A. Development of novel monoclonal antibodies for the analysis of functional sites in FGF-2. Growth Factors 1999, 16, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Kopff, C.; Konrad, J.; Riedel, A.; Gessner, C.; Wirtz, H. Influence of basic fibroblast growth factor on the proliferation of non-small cell lung cancer cell lines. Lung Cancer 2004, 44, 167–174. [Google Scholar] [CrossRef]
- Hori, A.; Sasada, R.; Matsutani, E.; Naito, K.; Sakura, Y.; Fujita, T.; Kozai, Y. Suppression of solid tumor growth by immunoneutralizing monoclonal antibody against human basic fibroblast growth factor. Cancer Res. 1991, 51, 6180–6184. [Google Scholar]
- Wang, L.; Park, H.; Chhim, S.; Ding, Y.; Jiang, W.; Queen, C.; Kim, K.J. A novel monoclonal antibody to fibroblast growth factor 2 effectively inhibits growth of hepatocellular carcinoma xenografts. Mol. Cancer Ther. 2012, 11, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Nonaka, Y.; Miyakawa, S.; Fujiwara, M.; Nakamura, Y. Dual therapeutic action of a neutralizing anti-FGF2 aptamer in bone diseases and bone cancer pain. Mol. Ther. 2016, 24, 1974–1986. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Ellington, A.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Rudnicka, A.R.; Kapetanakis, V.V.; Jarrar, Z.; Wathem, A.K.; Wormald, R.; Flecher, A.E.; Cook, D.G.; Owen, C.G. Incidence of late-stage age-related macular degeneration in american whites: Systematic review and meta-analysis. Am. J. Ophthalmol. 2015, 160, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Blaauwgeers, H.G.; Holtkamp, G.M.; Rutten, H.; Witmer, A.N.; Koolwijk, P.; Partanen, T.A.; Alitalo, K.; Kroon, M.E.; Kijlstra, A.; van Hinsbergh, V.W.M.; et al. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Am. J. Pathol. 1990, 155, 421–428. [Google Scholar] [CrossRef]
- Drolet, D.W.; Green, L.S.; Gold, L.; Janjic, N. Fit for the eye: Aptamers in ocular disorders. Nucleic Acid Ther. 2016, 26, 127–146. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.M.; Kaiser, P.K.; Michels, M.; Soubrane, G.; Heier, J.S.; Kim, R.Y.; Sy, J.P.; Schneider, S.; ANCHOR Study Group. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1432–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashani, A.H.; Lebkowski, J.S.; Rahhal, F.M.; Avery, R.L.; Salehi-Had, H.; Dang, W.; Lin, C.-M.; Mitra, D.; Zhu, D.; Thomas, B.B.; et al. A bioengineered retinal pigment epithelial monolayer for advanced, dry age-related macular degeneration. Sci. Trans. Med. 2018, 10, eaao4097. [Google Scholar] [CrossRef] [Green Version]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON a multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. VIEW 1 and VIEW 2 study groups: Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Fine, S.L.; Ying, G.S.; Jaffe, G.J.; Grunwald, J.E.; Toth, C.; Redford, M.; Ferris, F.L., 3rd. Comparison of age-related macular degeneration treatments trials (CATT) research group: Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: Two-year results. Ophthalmology 2012, 119, 1388–1398. [Google Scholar] [CrossRef] [Green Version]
- Daniel, E.; Toth, C.A.; Grunwald, J.E.; Jaffe, G.J.; Martin, D.F.; Fine, S.L.; Huang, J.; Ying, G.; Hagstrom, S.A.; Winter, K.; et al. Risk of scar in the comparison of age-related macular degeneration treatments trials. Ophthalmology 2014, 121, 656–666. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.Y.; Oubraham, H.; Uzzan, J.; Dobois, L.; Tadayoni, R. Causes of unsuccessful ranibizumab treatment in exudative age-related macular degeneration in clinical settings. Retina 2012, 32, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Schultz, G.S.; Grant, A.B. Neovascular growth factors. Eye 1991, 1, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Vinding, T. Occurrence of drusen, pigmentary changes and exudative changes in the macula with reference to age-related macular degeneration an epidemiological study of 1000 aged individuals. Acta Ophthalmol. 1990, 68, 410–414. [Google Scholar] [CrossRef]
- Tomanek, R.J.; Sandra, A.; Zheng, W.; Brock, T.; Bjercke, R.J.; Holifield, J.S. Vascular endothelial growth factor and basic fibroblast growth factor differentially modulate early postnatal coronary angiogenesis. Circ. Res. 2001, 88, 1135–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgore, F.; Lip, G.Y.H.; Blann, A.D. Basic fibrobrast growth factor induces the secretion of vascular endothelial growth factor by human aortic smooth muscle cells but not by endothelial cells. Eur. J. Clin. Investig. 2003, 33, 833–839. [Google Scholar] [CrossRef]
- Malabanan, K.P.; Kanellakis, P.; Bobik, A.; Khachigian, L.M. Activation transcription factor-4 induced by fibroblast growth factor-2 regulates vascular endothelial growth factor-A transcription in vascular smooth muscle cells and mediates intimal thickening in rat arteries following balloon injury. Circ. Res. 2008, 103, 378–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Bråkenhielm, E.; Pawliuk, R.; Wariaro, D.; Post, M.J.; Wahlberg, E.; Leboulch, P.; Cao, Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat. Med. 2003, 9, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Birsner, A.E.; Benny, O.; D’Amato, R.J. The corneal micropocket assay: A model of angiogenesis in the mouse eye. J. Vis. Exp. 2014, 90, e51375. [Google Scholar] [CrossRef] [Green Version]
- Strutz, F.; Zeisberg, M.; Ziyadeh, F.N.; Yang, C.Q.; Kalluri, R.; Müller, G.A.; Neilson, E.G. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002, 61, 1714–1728. [Google Scholar] [CrossRef] [Green Version]
- Cordeiro, M.F.; Reichel, M.B.; Gay, J.A.; D’Esposita, F.; Alexander, R.A.; Khaw, P.T. Transforming growth factor-beta1, -beta2, and -beta3 in vivo: Effects on normal and mitomycin C-modulated conjunctival scarring. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1975–1982. [Google Scholar]
- Nassar, K.; Lüke, J.; Kamal, M.; Abd El-Nabi, E.; Soliman, M.; Rohrbach, M.; Grisanti, S. The novel use of decorin in prevention of the development of proliferative vitreoretinopathy (PVR). Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, M.F.; Mead, A.; Ali, R.R.; Alexander, R.A.; Murray, S.; Chen, C.; York-Defalco, C.; Dean, N.M.; Schultz, G.S.; Khaw, P.T. Novel antisense oligonucleotides targeting TGF-beta inhibit in vivo scarring and improve surgical outcome. Gene Ther. 2003, 10, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.; Yoshida, S.; Nakao, S.; Nakama, T.; Kita, T.; Asato, R.; Sassa, Y.; Arita, R.; Miyazaki, M.; Enaida, H.; et al. Periostin promotes the generation of fibrous membranes in proliferative vitreoretinopathy. FASEB J. 2014, 28, 131–142. [Google Scholar] [CrossRef]
- Matsuda, Y.; Nonaka, Y.; Futakawa, S.; Imai, H.; Akita, K.; Nishihata, T.; Fujiwara, M.; Ali, Y.; Bhisitkul, R.B.; Nakamura, Y. Anti-angiogenic and anti-scarring dual action of an anti-fibroblast growth factor 2 aptamer in animal models of retinal disease. Mol. Ther. Nucl. Acids 2019, 17, 819–828. [Google Scholar] [CrossRef] [Green Version]
- U.S. National Library Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT03633084 (accessed on 16 August 2018).
- U.S. National Library Medicine. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04200248 (accessed on 16 December 2019).
- Orioli, I.M.; Castilla, E.E.; Barbosa-Neto, J.G. The birth prevalence rates for the skeletal dysplasias. J. Med. Genet. 1986, 23, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Sun, A.; Mekikian, P.B.; Martin, J.; Rimoin, D.L.; Lachman, R.S.; Wilcox, W.R. FGFR3 mutation frequency in 324 cases from the International Skeletal Dysplasia Registry. Mol. Genet. Genom. Med. 2014, 2, 497–503. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Horton, W.; Hristova, K. Physical basis behind achondroplasia, the most common form of human dwarfism. J. Biol. Chem. 2010, 285, 30103–30114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M.; Legeai-Mallet, L. Achondroplasia: Development, pathogenesis, and therapy. Dev. Dyn. 2017, 246, 291–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komla-Ebri, D.; Dambroise, E.; Kramer, I.; Benoist-Lasselin, C.; Kaci, N.; Gall, C.L.; Martin, L.; Busca, P.; Barbault, F.; Graus-Porta, D.; et al. Tyrosine kinase inhibitor NVP-BGJ398 functionally improves FGFR3-related dwarfism in mouse model. J. Clin. Investig. 2016, 126, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Yorifuji, T.; Higuchi, S.; Kawakita, R. Growth Hormone Treatment for Achondroplasia. Pediatr. Endocrinol. Rev. 2018, 16, 123–128. [Google Scholar] [PubMed]
- Gudernova, I.; Vesela, I.; Balek, L.; Buchtova, M.; Dosedelova, H.; Kunova, M.; Pivnicka, J.; Jelinkova, I.; Roubalova, L.; Kozubik, A.; et al. Multikinase activity of fibroblast growth factor receptor (FGFR) inhibitors SU5402, PD173074, AZD1480, AZD4547 and BGJ398 compromises the use of small chemicals targeting FGFR catalytic activity for therapy of short-stature syndromes. Hum. Mol. Genet. 2016, 25, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Bosakova, M.; Nonaka, Y.; Hruba, E.; Yasuda, K.; Futakawa, S.; Kubota, T.; Fafilek, B.; Gregor, T.; Abraham, S.P.; et al. RNA aptamer restores defective bone growth in FGFR3-related skeletal dysplasia. Sci. Trans. Med. 2021, 13, eaba4226. [Google Scholar] [CrossRef]
- Savarirayan, R.; Irving, M.; Bacino, C.A.; Bostwick, B.; Charrow, J.; Cormier-Daire, V.; Le Quan Sang, K.-H.; Dickson, P.; Harmatz, P.; Phillips, J.; et al. C-Type Natriuretic Peptide Analogue Therapy in Children with Achondroplasia. N. Engl. J. Med. 2019, 381, 25–35. [Google Scholar] [CrossRef]
- Wendt, D.J.; Dvorak-Ewell, M.; Bullens, S.; Lorget, F.; Bell, S.M.; Peng, J.; Castillo, S.; Aoyagi-Scharber, M.; O’Neill, C.A.; Krejci, P.; et al. Neutral endopeptidase-resistant C-type natriuretic peptide variant represents a new therapeutic approach for treatment of fibroblast growth factor receptor 3-related dwarfism. J. Pharmacol. Exp. Ther. 2015, 353, 132–149. [Google Scholar] [CrossRef] [Green Version]
- Pejchalova, K.; Krejci, P.; Wilcox, W.R. C-natriuretic peptide: An important regulator of cartilage. Mol. Genet. Metab. 2007, 92, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Japic Clinical Trials Information. Available online: https://www.clinicaltrials.jp/cti-user/trial/ShowDirect.jsp?directLink=0p52SZqEULFxxBQ0QuD.Vg (accessed on 12 May 2021).
- Wang, L.X.; Wang, Z.J. Animal and cellular models of chronic pain. Adv. Drug Deliv. Rev. 2003, 55, 949–965. [Google Scholar] [CrossRef]
- Seghezzi, G.; Patel, S.; Ren, C.J.; Gualandris, A.; Pintucci, G.; Robbins, E.S.; Shapiro, R.L.; Galloway, A.C.; Rifkin, D.B.; Mignatti, P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: An Autocrine mechanism contributing to angiogenesis. J. Cell Biol. 1998, 141, 1659–1673. [Google Scholar] [CrossRef]
- Ferhat, L.; Represa, A.; Zouaoui-Aggoun, D.; Ferhat, W.; Ben-Ari, Y.; Khrestchatisky, M. FGF-2 induces nerve growth factor expression in cultured rat hippocampal neurons. Eur. J. Neurosci. 1997, 9, 1282–1289. [Google Scholar] [CrossRef]
- Spranger, M.; Lindholm, D.; Bandtlow, C.; Heumann, R.; Gnahn, H.; Naher-Noe, M.; Thoenen, H. Regulation of nerve growth factor (NGF) synthesis in the rat central nervous system: Comparison between the effects of interleukin-I and various growth factors in astrocyte cultures and in vivo. Eur. J. Neurosci. 1990, 2, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Iwata, T.; Leung, H.Y. Mechanisms of FGFR-mediated carcinogenesis. Biochim. Biophys. Acta. 2012, 1823, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Desai, A.; Adjei, A.A. FGFR Signaling as a Target for Lung Cancer Therapy. J. Thorac. Oncol. 2016, 11, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, J.; Yasuda, H.; Nonaka, Y.; Fujiwara, M.; Nakamura, Y.; Soejima, K.; Betsuyaku, T. FGF2 aptamer inhibits the growth of FGF2-FGFR pathway driven lung cancer cells. Biochem. Biophys. Res. Commun. 2018, 503, 1330–13134. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Sugiyama, S.; Sakamoto, T.; Miyakawa, S.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; Mori, Y.; Nakamura, Y.; et al. Conformational plasticity of RNA for target recognition as revealed by the 2.15 Å crystal structure of a human IgG-aptamer complex. Nucl. Acids Res. 2010, 38, 7822–7829. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y. Aptamers as therapeutic middle molecules. Biochimie 2018, 145, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Oladipupo, S.S.; Smith, C.; Santeford, A.; Park, C.; Sene, A.; Wiley, L.A.; Osei-Owusu, P.; Hsu, J.; Zapata, N.; Liu, F.; et al. Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 13379–13384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Santeford, A.; Ban, N.; Lee, T.J.; Smith, C.; Smith, C.; Ornitz, D.M.; Apte, R.S. FGF2-induced STAT3 activation regulates pathologic neovascularization. Exp. Eye Res. 2019, 187, 107775. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, Y. Multiple Therapeutic Applications of RBM-007, an Anti-FGF2 Aptamer. Cells 2021, 10, 1617. https://doi.org/10.3390/cells10071617
Nakamura Y. Multiple Therapeutic Applications of RBM-007, an Anti-FGF2 Aptamer. Cells. 2021; 10(7):1617. https://doi.org/10.3390/cells10071617
Chicago/Turabian StyleNakamura, Yoshikazu. 2021. "Multiple Therapeutic Applications of RBM-007, an Anti-FGF2 Aptamer" Cells 10, no. 7: 1617. https://doi.org/10.3390/cells10071617
APA StyleNakamura, Y. (2021). Multiple Therapeutic Applications of RBM-007, an Anti-FGF2 Aptamer. Cells, 10(7), 1617. https://doi.org/10.3390/cells10071617