
Immunoproteasome Function in Normal and Malignant Hematopoiesis


Abstract
1. Introduction
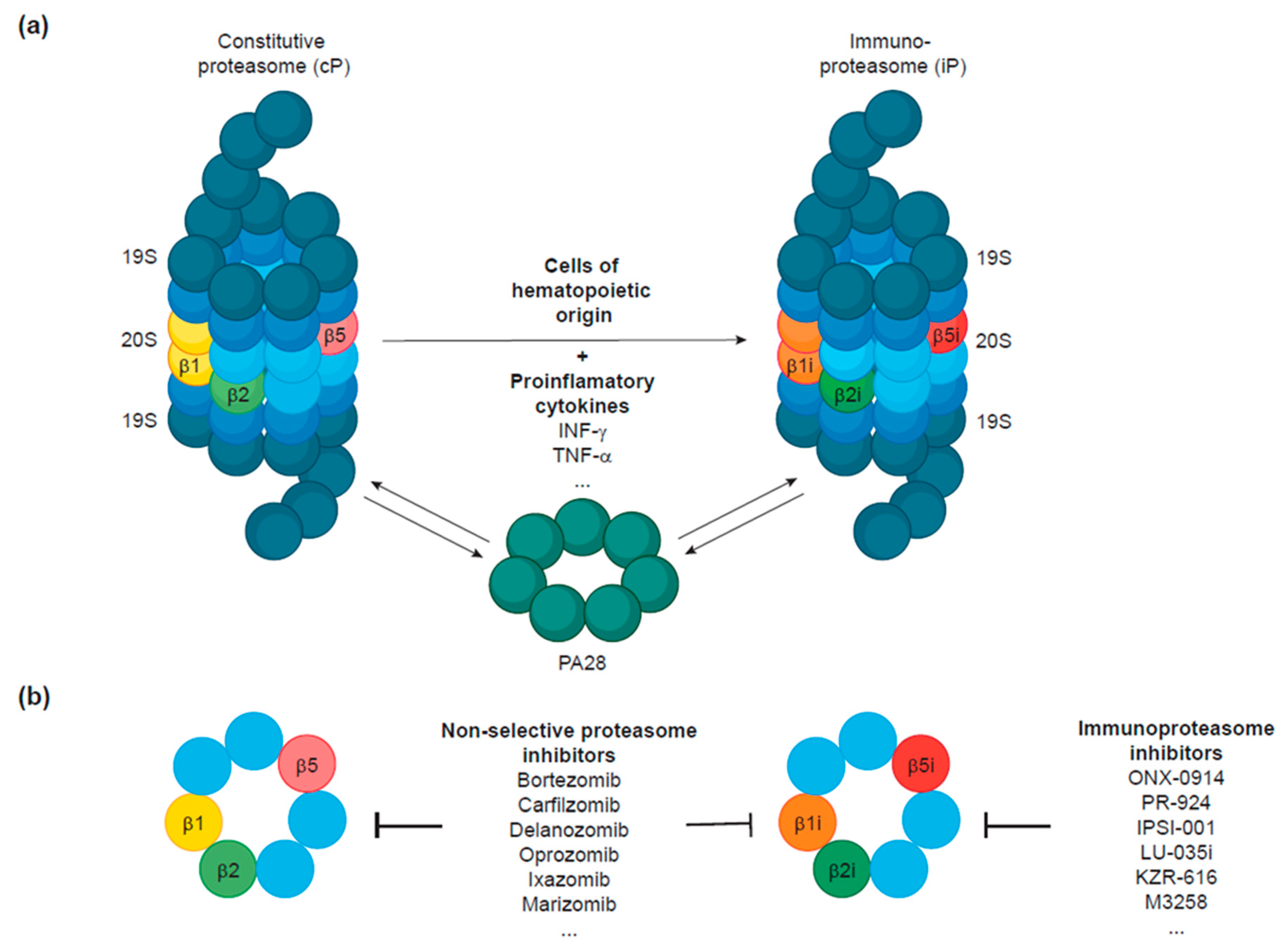
2. The Immunoproteasome: A Proteasomal Variant Linked to the Hematopoietic System
2.1. Immunoproteasome Structure
2.2. Immune and Non-Immune Functions of the Immunoproteasome
2.3. Expression Patterns of the Immunoproteasome
2.4. Genetic Variants of the Immunoproteasome
3. Pro- and Anti-Tumoral Properties of the Immunoproteasome
4. Development of Immunoproteasome Inhibitors to Target Hematologic Malignancies

{kind=link}
{kind=link}
| Inhibitor | Developed by | Backbone | Target | Binding Kinetics |
|---|---|---|---|---|
| UK-101 | [148] | Peptidyl epoxyketone | β1i subunit (144-fold more selective than β1, but only 10-fold to β5) | Covalent irreversible |
| ONX-0914 (PR-957) | [45] | Peptidyl epoxyketone | β5i subunit (20- to 40-fold more selective than β5 or β1i) | Covalent irreversible |
| IPSI-001 | [44] | Peptidyl aldehyde | β1i subunit (100-fold more selective than β1) | Covalent reversible |
| PR-924 | [41] | Peptidyl epoxyketone | β5i subunit (130-fold more selective than β5) | Covalent irreversible |
| LU-001i | [154] | Peptidyl epoxyketone | β1i subunit (925-fold more selective than β1) | Covalent irreversible |
| LU-015i | [154] | Peptidyl epoxyketone | β5i subunit (553-fold more selective than β5) | Covalent irreversible |
| LU-035i | [154] | Peptidyl epoxyketone | β5i subunit (500-fold more selective than β5) | Covalent irreversible |
| HT2210 and HT2106 | [155] | Oxathiazole | β5i subunit (>4700-fold more selective than β5c) | Covalent irreversible |
| 1-CA and 4-CA | [156] | Peptidyl epoxyketone | β5i subunit (75- to 150-fold more selective than β5) | Covalent irreversible |
| PKS2279 and PKS2252 | [157] | N,C-capped dipeptide | β5i subunit (5600 and 13,600-fold more selective than β5) | Non-covalent |
| KZR-504 | [158] | Peptidyl epoxyketone | β1i subunit (925-fold more selective than β1) | Covalent irreversible |
| KZR-616 | [158] | Peptidyl epoxyketone | β5i and β1i subunits (18- and 81-fold more selective than β5 and β1c) | Covalent irreversible |
| LU-002i | [159] | Peptidyl epoxyketone | β2i subunit | Covalent irreversible |
| M3258 | [160] | Boronic acid | β5i subunit (>500-fold more selective than β5) | Covalent reversible |
| Inhibitor | Effective against | In Vitro/In Vivo Experiments | References |
|---|---|---|---|
| UK-101 | MM | Patient samples | [150] |
| ONX-0914 (PR-957) | MM and MLLr-AML | Human cell lines Synergism with BTZ in a MM murine model | [99,127,136] |
| IPSI-001 | MM, NHL, CLL, and AML | Human cell lines and patient samples | [44] |
| PR-924 | MM, T-ALL, and AML | Human cell lines and patient samples MM xenograft model | [79,145] |
| LU-035i | MM | Human cell lines in conjugation with cytotoxic drug | [161] |
| HT2210 and HT2106 | NHL | Human cell lines | [155] |
| M3258 | MM, AML, and lymphoma | Human cell lines MM xenograft model Phase I clinical trial | [160,162,163] |
5. Pathways Affected by Immunoproteasome Inhibition
6. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Ardley, H.C.; Robinson, P.A. E3 ubiquitin ligases. Essays Biochem. 2005, 41, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Suryadinata, R.; Tan, A.R.; Roesley, S.N.A.; Sarcevic, B. Protein Monoubiquitination and Polyubiquitination Generate Structural Diversity to Control Distinct Biological Processes. IUBMB Life 2012, 64, 136–142. [Google Scholar] [CrossRef]
- Baumeister, W.; Walz, J.; Zuhl, F.; Seemüller, E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell 1998, 92, 367–380. [Google Scholar] [CrossRef]
- Coux, O.; Tanaka, K.; Goldberg, A.L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef]
- Liu, Y.C.; Penninger, J.; Karin, M. Immunity by ubiquitylation: A reversible process of modification. Nat. Rev. Immunol. 2005, 5, 941–952. [Google Scholar] [CrossRef]
- Kniepert, A.; Groettrup, M. The unique functions of tissue-specific proteasomes. Trends Biochem. Sci. 2014, 39, 17–24. [Google Scholar] [CrossRef]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef]
- Groettrup, M.; Kraft, R.; Kostka, S.; Standera, S.; Stohwasser, R.; Kloetzel, P.M. A third interferon-gamma-induced subunit exchange in the 20S proteasome. Eur. J. Immunol. 1996, 26, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Navarrete, V.; Seelig, A.; Gernold, M.; Frentzel, S.; Kloetzel, P.M.; Hämmerling, G.J. Subunit of the ‘20S’ proteasome (multicatalytic proteinase) encoded by the major histocompatibility complex. Nature 1991, 353, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Waters, J.B.; Früh, K.; Peterson, P.A. Proteasomes are regulated by interferon gamma: Implications for antigen processing. Proc. Natl. Acad. Sci. USA 1992, 89, 4928–4932. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, F.; Kloetzel, P.M.; Kruger, E.; Seifert, U. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol. Life Sci. 2012, 69, 2543–2558. [Google Scholar] [CrossRef] [PubMed]
- Groettrup, M.; Ruppert, T.; Kuehn, L.; Seeger, M.; Standera, S.; Koszinowski, U.; Kloetzel, P.M. The interferon-gamma-inducible 11 S regulator (PA28) and the LMP2/LMP7 subunits govern the peptide production by the 20 S proteasome in vitro. J. Biol. Chem. 1995, 270, 23808–23815. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; van Den Broek, M.; Kostka, S.; Kraft, R.; Soza, A.; Schmidtke, G.; Kloetzel, P.M.; Groettrup, M. Overexpression of the proteasome subunits LMP2, LMP7, and MECL-1, but not PA28 alpha/beta, enhances the presentation of an immunodominant lymphocytic choriomeningitis virus T cell epitope. J. Immunol. 2000, 165, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Moebius, J.; van den Broek, M.; Groettrup, M.; Basler, M. Immunoproteasomes are essential for survival and expansion of T cells in virus-infected mice. Eur. J. Immunol. 2010, 40, 3439–3449. [Google Scholar] [CrossRef]
- Murata, S.; Sasaki, K.; Kishimoto, T.; Niwa, S.; Hayashi, H.; Takahama, Y.; Tanaka, K. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science 2007, 316, 1349–1353. [Google Scholar] [CrossRef]
- Gomez, H.L.; Felipe-Medina, N.; Condezo, Y.B.; Garcia-Valiente, R.; Ramos, I.; Suja, J.A.; Barbero, J.L.; Roig, I.; Sanchez-Martin, M.; de Rooij, D.G.; et al. The PSMA8 subunit of the spermatoproteasome is essential for proper meiotic exit and mouse fertility. PLoS Genet. 2019, 15, e1008316. [Google Scholar] [CrossRef]
- Ustrell, V.; Hoffman, L.; Pratt, G.; Rechsteiner, M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002, 21, 3516–3525. [Google Scholar] [CrossRef]
- Qian, M.X.; Pang, Y.; Liu, C.H.; Haratake, K.; Du, B.Y.; Ji, D.Y.; Wang, G.F.; Zhu, Q.Q.; Song, W.; Yu, Y.; et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 2013, 153, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Mandemaker, I.K.; Geijer, M.E.; Kik, I.; Bezstarosti, K.; Rijkers, E.; Raams, A.; Janssens, R.C.; Lans, H.; Hoeijmakers, J.H.; Demmers, J.A.; et al. DNA damage-induced replication stress results in PA200-proteasome-mediated degradation of acetylated histones. EMBO Rep. 2018, 19, e45566. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Sousa-Victor, P.; Munoz-Cánoves, P. Proteostatic and Metabolic Control of Stemness. Cell Stem Cell 2017, 20, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.B.; Aifantis, I. Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 2012, 33, 357–363. [Google Scholar] [CrossRef][Green Version]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef]
- Kohli, L.; Passegue, E. Surviving change: The metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014, 24, 479–487. [Google Scholar] [CrossRef]
- Wilson, A.; Laurenti, E.; Oser, G.; Wath, R.C.V.D.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef]
- Signer, R.A.J.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004, 5, 739–751. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Schmidt, M.H.; Dikic, I. The Cbl interactome and ist functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 907–919. [Google Scholar] [CrossRef]
- Bedford, L.; Lowe, J.; Dick, L.R.; Mayer, R.J.; Brownell, J.E. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat. Rev. Drug Discov. 2011, 10, 29–46. [Google Scholar] [CrossRef]
- Micel, L.N.; Tentler, J.J.; Smith, P.G.; Eckhardt, G.S. Role of ubiquitin ligases and the proteasome in oncogenesis: Novel targets for anticancer therapies. J. Clin. Oncol. 2013, 31, 1231–1238. [Google Scholar] [CrossRef]
- Fisher, R.I.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; de Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2006, 24, 4867–4874. [Google Scholar] [CrossRef]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef]
- Chauhan, D.; Hideshima, T.; Mitsiades, C.; Richardson, P.; Anderson, K.C. Proteasome inhibitor therapy in multiple myeloma. Mol. Cancer Ther. 2005, 4, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef]
- Ruschak, A.M.; Slassi, M.; Kay, L.E.; Schimmer, A.D. Novel Proteasome Inhibitors to Overcome Bortezomib Resistance. J. Natl. Cancer Inst. 2011, 103, 1007–1017. [Google Scholar] [CrossRef]
- Florea, B.I.; Verdoes, M.; Li, N.; van der Linden, W.A.; Geurink, P.P.; van den Elst, H.; Hofmann, T.; de Ru, A.; van Veelen, P.A.; Tanaka, K. Activity-based profiling reveals reactivity of the murine thymoproteasome-specific subunit beta5t. Chem. Biol. 2010, 17, 795–801. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Hunsucker, S.A.; Chen, Q.; Voorhees, P.M.; Orlowski, M.; Orlowski, R.Z. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009, 113, 4667–4676. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef]
- Glickman, M.H.; Rubin, D.M.; Fried, V.A.; Finley, D. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell Biol. 1998, 18, 3149–3162. [Google Scholar] [CrossRef]
- Deveraux, Q.; van Nocker, S.; Mahaffey, D.; Vierstra, R.; Rechsteiner, M. Inhibition of ubiquitin-mediated proteolysis by the Arabidopsis 26 S protease subunit S5a. J. Biol. Chem. 1995, 270, 29660–29663. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chen, X.; Elsasser, S.; Stocks, B.B.; Tian, G.; Lee, B.H.; Shi, Y.; Zhang, N.; de Poot, S.A.; Tuebing, F.; et al. Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 2016, 351. [Google Scholar] [CrossRef]
- Früh, K.; Gossen, M.; Wang, K.; Bujard, H.; Peterson, P.A.; Yang, Y. Displacement of housekeeping proteasome subunits by MHC-encoded LMPs: A newly discovered mechanism for modulating the multicatalytic proteinase complex. EMBO J. 1994, 13, 3236–3244. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Yokota, K.; Kagawa, S.; Shimbara, N.; Tamura, T.; Akioka, H.; Nothwang, H.G.; Noda, C.; Tanaka, K.; Ichihara, A. cDNA cloning and interferon gamma down-regulation of proteasomal subunits X and Y. Science 1994, 265, 1231–1234. [Google Scholar] [CrossRef]
- Heink, S.; Ludwig, D.; Kloetzel, P.M.; Kruger, E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc. Natl. Acad. Sci. USA 2005, 102, 9241–9246. [Google Scholar] [CrossRef]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Van Holle, B.; Parvizi, G.; Bousquet-Dubouch, M.P.; Theate, I.; Parmentier, N.; Van den Eynde, B.J. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 18599–18604. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B. Mammalian proteasome subtypes: Their diversity in structure and function. Arch. Biochem. Biophys. 2016, 591, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, G.; Schregle, R.; Alvarez, G.; Huber, E.M.; Groettrup, M. The 20S immunoproteasome and constitutive proteasome bind with the same affinity to PA28α β and equally degrade FAT10. Mol. Immunol. 2017, 113, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, N.; Murakami, Y.; Minami, Y.; Shimbarai, N.; Hendil, K.B.; Tanaka, K. Hybrid Proteasomes: Induction by Interferon-g and contribution to ATP-dependent proteolysis. J. Biol. Chem. 2000, 275, 14336–14345. [Google Scholar] [CrossRef]
- Li, J.; Powell, S.R.; Wang, X. Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J. 2011, 25, 883–893. [Google Scholar] [CrossRef]
- Pickering, A.M.; Koop, A.L.; Teoh, C.Y.; Ermak, G.; Grune, T.; Davies, K.J. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010, 432, 585–594. [Google Scholar] [CrossRef]
- Dick, T.P.; Ruppert, T.; Groettrup, M.; Kloetzel, P.M.; Kuehn, L.; Koszinowski, U.H.; Stevanovic, S.; Schild, H.; Rammensee, H.G. Coordinated dual cleavages induced by the proteasome regulator PA28 lead to dominant MHC ligands. Cell 1996, 86, 253–262. [Google Scholar] [CrossRef]
- Respondek, D.; Voss, M.; Kuhlewindt, I.; Klingel, K.; Kruger, E.; Beling, A. PA28 modulates antigen processing and viral replication during coxsackievirus B3 infection. PLoS ONE 2017, 12, e0173259. [Google Scholar] [CrossRef]
- Sijts, A.; Sun, Y.; Janek, K.; Kral, S.; Paschen, A.; Schadendorf, D.; Kloetzel, P.M. The role of the proteasome activator PA28 in MHC class I antigen processing. Mol. Immunol. 2002, 39, 165–169. [Google Scholar] [CrossRef]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Mishto, M.; Liepe, J.; Textoris-Taube, K.; Keller, C.; Henklein, P.; Weberruss, M.; Dahlmann, B.; Enenkel, C.; Voigt, A.; Kuckelkorn, U.; et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur. J. Immunol. 2014, 44, 3508–3521. [Google Scholar] [CrossRef]
- Chapatte, L.; Ayyoub, M.; Morel, S.; Peitrequin, A.L.; Levy, N.; Servis, C.; Van den Eynde, B.J.; Valmori, D.; Levy, F. Processing of tumor-associated antigen by the proteasomes of dendritic cells controls in vivo T-cell responses. Cancer Res. 2006, 66, 5461–5468. [Google Scholar] [CrossRef] [PubMed]
- Chapiro, J.; Claverol, S.; Piette, F.; Ma, W.; Stroobant, V.; Guillaume, B.; Gairin, J.E.; Morel, S.; Burlet-Schiltz, O.; Monsarrat, B.; et al. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J. Immunol. 2006, 176, 1053–1061. [Google Scholar] [CrossRef]
- Ma, W.; Vigneron, N.; Chapiro, J.; Stroobant, V.; Germeau, C.; Boon, T.; Coulie, P.G.; Van den Eynde, B.J. A MAGE-C2 antigenic peptide processed by the immunoproteasome is recognized by cytolytic T cells isolated from a melanoma patient after successful immunotherapy. Int. J. Cancer 2011, 129, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Schultz, E.S.; Chapiro, J.; Lurquin, C.; Claverol, S.; Burlet-Schiltz, O.; Warnier, G.; Russo, V.; Morel, S.; Levy, F.; Boon, T.; et al. The production of a new MAGE-3 peptide presented to cytolytic T lymphocytes by HLA-B40 requires the immunoproteasome. J. Exp. Med. 2002, 195, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.; Knights, A.J.; Anaka, M.; Schittenhelm, R.B.; Purcell, A.W.; Behren, A.; Cebon, J. Mismatch in epitope specificities between IFNgamma inflamed and uninflamed conditions leads to escape from T lymphocyte killing in melanoma. J. Immunother. Cancer 2016, 4, 10. [Google Scholar] [CrossRef]
- Guillaume, B.; Stroobant, V.; Bousquet-Dubouch, M.P.; Colau, D.; Chapiro, J.; Parmentier, N.; Dalet, A.; Van den Eynde, B.J. Analysis of the processing of seven human tumor antigens by intermediate proteasomes. J. Immunol. 2012, 189, 3538–3547. [Google Scholar] [CrossRef]
- Fehling, H.J.; Swat, W.; Laplace, C.; Kuhn, R.; Rajewsky, K.; Muller, U.; von Boehmer, H. MHC class I expression in mice lacking the proteasome subunit LMP-7. Science 1994, 265, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Ashton-Rickardt, P.G.; Eichelberger, M.; Gaczynska, M.; Nagashima, K.; Rock, K.L.; Goldberg, A.L.; Doherty, P.C.; Tonegawa, S. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity 1994, 1, 533–541. [Google Scholar] [CrossRef]
- Basler, M.; Moebius, J.; Elenich, L.; Groettrup, M.; Monaco, J.J. An altered T cell repertoire in MECL-1-deficient mice. J. Immunol. 2006, 176, 6665–6672. [Google Scholar] [CrossRef]
- Kincaid, E.Z.; Che, J.W.; York, I.; Escobar, H.; Reyes-Vargas, E.; Delgado, J.C.; Welsh, R.M.; Karow, M.L.; Murphy, A.J.; Valenzuela, D.M.; et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 2011, 13, 129–135. [Google Scholar] [CrossRef]
- Zaiss, D.M.; de Graaf, N.; Sijts, A.J. The proteasome immunosubunit multicatalytic endopeptidase complex-like 1 is a T-cell-intrinsic factor influencing homeostatic expansion. Infect. Immun. 2008, 76, 1207–1213. [Google Scholar] [CrossRef]
- Bockstahler, M.; Fischer, A.; Goetzke, C.C.; Neumaier, H.L.; Sauter, M.; Kespohl, M.; Muller, A.M.; Meckes, C.; Salbach, C.; Schenk, M.; et al. Heart-Specific Immune Responses in an Animal Model of Autoimmune-Related Myocarditis Mitigated by an Immunoproteasome Inhibitor and Genetic Ablation. Circulation 2020, 141, 1885–1902. [Google Scholar] [CrossRef]
- Goetzke, C.C.; Althof, N.; Neumaier, H.L.; Heuser, A.; Kaya, Z.; Kespohl, M.; Klingel, K.; Beling, A. Mitigated viral myocarditis in A/J mice by the immunoproteasome inhibitor ONX 0914 depends on inhibition of systemic inflammatory responses in CoxsackievirusB3 infection. Basic Res. Cardiol. 2021, 116, 7. [Google Scholar] [CrossRef]
- Kalim, K.W.; Basler, M.; Kirk, C.J.; Groettrup, M. Immunoproteasome subunit LMP7 deficiency and inhibition suppresses Th1 and Th17 but enhances regulatory T cell differentiation. J. Immunol. 2012, 189, 4182–4193. [Google Scholar] [CrossRef] [PubMed]
- Kruger, E.; Kloetzel, P.M. Immunoproteasomes at the interface of innate and adaptive immune responses: Two faces of one enzyme. Curr. Opin. Immunol. 2012, 24, 77–83. [Google Scholar] [CrossRef]
- Niewerth, D.; van Meerloo, J.; Jansen, G.; Assaraf, Y.G.; Hendrickx, T.C.; Kirk, C.J.; Anderl, J.L.; Zweegman, S.; Kaspers, G.J.; Cloos, J. Anti-leukemic activity and mechanisms underlying resistance to the novel immunoproteasome inhibitor PR-924. Biochem. Pharmacol. 2014, 89, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Opitz, E.; Koch, A.; Klingel, K.; Schmidt, F.; Prokop, S.; Rahnefeld, A.; Sauter, M.; Heppner, F.L.; Volker, U.; Kandolf, R.; et al. Impairment of immunoproteasome function by beta5i/LMP7 subunit deficiency results in severe enterovirus myocarditis. PLoS Pathog. 2011, 7, e1002233. [Google Scholar] [CrossRef] [PubMed]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schroter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Sachs, Z.; Noble-Orcutt, K.E.; Moriarity, B.S.; Ai, T.; Ding, R.; Williams, J.; Chen, L.; et al. mTORC1 Coordinates Protein Synthesis and Immunoproteasome Formation via PRAS40 to Prevent Accumulation of Protein Stress. Mol. Cell 2016, 61, 625–639. [Google Scholar] [CrossRef]
- Davies, K.J. Degradation of oxidized proteins by the 20S proteasome. Biochimie 2001, 83, 301–310. [Google Scholar] [CrossRef]
- Nathan, J.A.; Spinnenhirn, V.; Schmidtke, G.; Basler, M.; Groettrup, M.; Goldberg, A.L. Immuno- and constitutive proteasomes do not differ in their abilities to degrade ubiquitinated proteins. Cell 2013, 152, 1184–1194. [Google Scholar] [CrossRef]
- Abi Habib, J.; De Plaen, E.; Stroobant, V.; Zivkovic, D.; Bousquet, M.P.; Guillaume, B.; Wahni, K.; Messens, J.; Busse, A.; Vigneron, N.; et al. Efficiency of the four proteasome subtypes to degrade ubiquitinated or oxidized proteins. Sci. Rep. 2020, 10, 15765. [Google Scholar] [CrossRef] [PubMed]
- Muratani, M.; Tansey, W.P. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 2003, 4, 192–201. [Google Scholar] [CrossRef] [PubMed]
- De Verteuil, D.A.; Rouette, A.; Hardy, M.P.; Lavallee, S.; Trofimov, A.; Gaucher, E.; Perreault, C. Immunoproteasomes shape the transcriptome and regulate the function of dendritic cells. J. Immunol. 2014, 193, 1121–1132. [Google Scholar] [CrossRef]
- Cetin, G.; Klafack, S.; Studencka-Turski, M.; Kruger, E.; Ebstein, F. The Ubiquitin-Proteasome System in Immune Cells. Biomolecules 2021, 11, 60. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Meiners, S.; Heyken, D.; Weller, A.; Ludwig, A.; Stangl, K.; Kloetzel, P.M.; Kruger, E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of Mammalian proteasomes. J. Biol. Chem. 2003, 278, 21517–21525. [Google Scholar] [CrossRef]
- Brucet, M.; Marques, L.; Sebastian, C.; Lloberas, J.; Celada, A. Regulation of murine Tap1 and Lmp2 genes in macrophages by interferon gamma is mediated by STAT1 and IRF-1. Genes Immun. 2004, 5, 26–35. [Google Scholar] [CrossRef]
- Foss, G.S.; Prydz, H. Interferon regulatory factor 1 mediates the interferon-gamma induction of the human immunoproteasome subunit multicatalytic endopeptidase complex-like 1. J. Biol. Chem. 1999, 274, 35196–35202. [Google Scholar] [CrossRef]
- Namiki, S.; Nakamura, T.; Oshima, S.; Yamazaki, M.; Sekine, Y.; Tsuchiya, K.; Okamoto, R.; Kanai, T.; Watanabe, M. IRF-1 mediates upregulation of LMP7 by IFN-gamma and concerted expression of immunosubunits of the proteasome. FEBS Lett. 2005, 579, 2781–2787. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, A.M.; Angeletti, M.; Lupidi, G.; Tacconi, R.; Bini, L.; Fioretti, E. Isolation and characterization of bovine thymus multicatalytic proteinase complex. Protein Expr. Purif. 2000, 18, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Noda, C.; Tanahashi, N.; Shimbara, N.; Hendil, K.B.; Tanaka, K. Tissue distribution of constitutive proteasomes, immunoproteasomes, and PA28 in rats. Biochem. Biophys. Res. Commun. 2000, 277, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Heilman, D.; Diekmann, C.; Osna, N.; Donohue, T.M., Jr.; Ghorpade, A.; Persidsky, Y. Alcohol and HIV decrease proteasome and immunoproteasome function in macrophages: Implications for impaired immune function during disease. Cell Immunol. 2004, 229, 139–148. [Google Scholar] [CrossRef]
- Frisan, T.; Levitsky, V.; Masucci, M.G. Variations in proteasome subunit composition and enzymatic activity in B-lymphoma lines and normal B cells. Int. J. Cancer 2000, 88, 881–888. [Google Scholar] [CrossRef]
- Niewerth, D.; Franke, N.E.; Jansen, G.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Degenhardt, J.; Anderl, J.; Schimmer, A.D.; Zweegman, S.; et al. Higher ratio immune versus constitutive proteasome level as novel indicator of sensitivity of pediatric acute leukemia cells to proteasome inhibitors. Haematologica 2013, 98, 1896–1904. [Google Scholar] [CrossRef]
- Niewerth, D.; Kaspers, G.J.; Jansen, G.; van Meerloo, J.; Zweegman, S.; Jenkins, G.; Whitlock, J.A.; Hunger, S.P.; Lu, X.; Alonzo, T.A.; et al. Proteasome subunit expression analysis and chemosensitivity in relapsed paediatric acute leukaemia patients receiving bortezomib-containing chemotherapy. J. Hematol. Oncol. 2016, 9, 82. [Google Scholar] [CrossRef]
- Di Rosa, M.; Giallongo, C.; Romano, A.; Tibullo, D.; Li Volti, G.; Musumeci, G.; Barbagallo, I.; Imbesi, R.; Castrogiovanni, P.; Palumbo, G.A. Immunoproteasome Genes Are Modulated in CD34(+) JAK2(V617F) Mutated Cells from Primary Myelofibrosis Patients. Int. J. Mol. Sci. 2020, 21, 2926. [Google Scholar] [CrossRef]
- Visekruna, A.; Slavova, N.; Dullat, S.; Grone, J.; Kroesen, A.J.; Ritz, J.P.; Buhr, H.J.; Steinhoff, U. Expression of catalytic proteasome subunits in the gut of patients with Crohn’s disease. Int. J. Colorectal. Dis. 2009, 24, 1133–1139. [Google Scholar] [CrossRef]
- Visekruna, A.; Joeris, T.; Seidel, D.; Kroesen, A.; Loddenkemper, C.; Zeitz, M.; Kaufmann, S.H.; Schmidt-Ullrich, R.; Steinhoff, U. Proteasome-mediated degradation of IkappaBalpha and processing of p105 in Crohn disease and ulcerative colitis. J. Clin. Invest. 2006, 116, 3195–3203. [Google Scholar] [CrossRef]
- Chen, M.; Tabaczewski, P.; Truscott, S.M.; Van Kaer, L.; Stroynowski, I. Hepatocytes express abundant surface class I MHC and efficiently use transporter associated with antigen processing, tapasin, and low molecular weight polypeptide proteasome subunit components of antigen processing and presentation pathway. J. Immunol. 2005, 175, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Vasuri, F.; Capizzi, E.; Bellavista, E.; Mishto, M.; Santoro, A.; Fiorentino, M.; Capri, M.; Cescon, M.; Grazi, G.L.; Grigioni, W.F.; et al. Studies on immunoproteasome in human liver. Part I: Absence in fetuses, presence in normal subjects, and increased levels in chronic active hepatitis and cirrhosis. Biochem. Biophys. Res. Commun. 2010, 397, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Loukissa, A.; Cardozo, C.; Altschuller-Felberg, C.; Nelson, J.E. Control of LMP7 expression in human endothelial cells by cytokines regulating cellular and humoral immunity. Cytokine 2000, 12, 1326–1330. [Google Scholar] [CrossRef] [PubMed]
- Roby, K.F.; Laham, N.; Kroning, H.; Terranova, P.F.; Hunt, J.S. Expression and localization of messenger RNA for tumor necrosis factor receptor (TNF-R) I and TNF-RII in pregnant mouse uterus and placenta. Endocrine 1995, 3, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, F.; Textoris-Taube, K.; Keller, C.; Golnik, R.; Vigneron, N.; Van den Eynde, B.J.; Schuler-Thurner, B.; Schadendorf, D.; Lorenz, F.K.; Uckert, W.; et al. Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non-spliced epitopes. Sci. Rep. 2016, 6, 24032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Schulz, R.; Edmunds, S.; Kruger, E.; Markert, E.; Gaedcke, J.; Cormet-Boyaka, E.; Ghadimi, M.; Beissbarth, T.; Levine, A.J.; et al. Suppresses Tumor Cell Proliferation by Acting as an Endogenous Proteasome Inhibitor via Targeting the Proteasome Assembly Factor POMP. Mol. Cell 2015, 59, 243–257. [Google Scholar] [CrossRef]
- Sharifi, H.; Jafari Najaf Abadi, M.H.; Razi, E.; Mousavi, N.; Morovati, H.; Sarvizadeh, M.; Taghizadeh, M. MicroRNAs and response to therapy in leukemia. J. Cell Biochem. 2019, 120, 14233–14246. [Google Scholar] [CrossRef]
- Wang, C.Z.; Deng, F.; Li, H.; Wang, D.D.; Zhang, W.; Ding, L.; Tang, J.H. MiR-101: A potential therapeutic target of cancers. Am. J. Transl. Res. 2018, 10, 3310–3321. [Google Scholar] [PubMed]
- Gomes, A.V. Genetics of proteasome diseases. Scientifica 2013, 2013, 637629. [Google Scholar] [CrossRef] [PubMed]
- Kroll-Hermi, A.; Ebstein, F.; Stoetzel, C.; Geoffroy, V.; Schaefer, E.; Scheidecker, S.; Bar, S.; Takamiya, M.; Kawakami, K.; Zieba, B.A.; et al. Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol. Med. 2020, 12, e11861. [Google Scholar] [CrossRef]
- Ansar, M.; Ebstein, F.; Ozkoc, H.; Paracha, S.A.; Iwaszkiewicz, J.; Gesemann, M.; Zoete, V.; Ranza, E.; Santoni, F.A.; Sarwar, M.T.; et al. Biallelic variants in PSMB1 encoding the proteasome subunit beta6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum. Mol. Genet. 2020, 29, 1132–1143. [Google Scholar] [CrossRef]
- Cao, B.; Tian, X.; Li, Y.; Jiang, P.; Ning, T.; Xing, H.; Zhao, Y.; Zhang, C.; Shi, X.; Chen, D.; et al. LMP7/TAP2 gene polymorphisms and HPV infection in esophageal carcinoma patients from a high incidence area in China. Carcinogenesis 2005, 26, 1280–1284. [Google Scholar] [CrossRef]
- Mehta, A.M.; Jordanova, E.S.; van Wezel, T.; Uh, H.W.; Corver, W.E.; Kwappenberg, K.M.; Verduijn, W.; Kenter, G.G.; van der Burg, S.H.; Fleuren, G.J. Genetic variation of antigen processing machinery components and association with cervical carcinoma. Genes Chromosomes Cancer 2007, 46, 577–586. [Google Scholar] [CrossRef]
- Tang, Q.; Zhang, J.; Qi, B.; Shen, C.; Xie, W. Downregulation of HLA class I molecules in primary oral squamous cell carcinomas and cell lines. Arch. Med. Res. 2009, 40, 256–263. [Google Scholar] [CrossRef]
- Seliger, B.; Stoehr, R.; Handke, D.; Mueller, A.; Ferrone, S.; Wullich, B.; Tannapfel, A.; Hofstaedter, F.; Hartmann, A. Association of HLA class I antigen abnormalities with disease progression and early recurrence in prostate cancer. Cancer Immunol. Immunother. 2010, 59, 529–540. [Google Scholar] [CrossRef]
- Fellerhoff, B.; Gu, S.; Laumbacher, B.; Nerlich, A.G.; Weiss, E.H.; Glas, J.; Kopp, R.; Johnson, J.P.; Wank, R. The LMP7-K allele of the immunoproteasome exhibits reduced transcript stability and predicts high risk of colon cancer. Cancer Res. 2011, 71, 7145–7154. [Google Scholar] [CrossRef]
- Ozbas-Gerceker, F.; Bozman, N.; Kok, S.; Pehlivan, M.; Yilmaz, M.; Pehlivan, S.; Oguzkan-Balci, S. Association of an LMP2 polymorphism with acute myeloid leukemia and multiple myeloma. Asian Pac. J. Cancer Prev. 2013, 14, 6399–6402. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Kinoshita, A.; Mishima, H.; Kanazawa, N.; Kaneko, T.; Mizushima, T.; Ichinose, K.; Nakamura, H.; Tsujino, A.; Kawakami, A.; et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 14914–14919. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Maekawa, Y.; Uehara, H.; Izumi, K.; Kawachi, I.; Nishizawa, M.; Toyoshima, Y.; Takahashi, H.; Standley, D.M.; Tanaka, K.; et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Investig. 2011, 121, 4150–4160. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Kruger, E. Dysfunction in protein clearance by the proteasome: Impact on autoinflammatory diseases. Semin. Immunopathol. 2015, 37, 323–333. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, A.A.; Brehm, A.; VanTries, R.; Pillet, P.; Parentelli, A.S.; Montealegre Sanchez, G.A.; Deng, Z.; Paut, I.K.; Goldbach-Mansky, R.; Kruger, E. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J. Allergy Clin. Immunol. 2019, 143, 1939–1943. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ramot, Y.; Torrelo, A.; Paller, A.S.; Si, N.; Babay, S.; Kim, P.W.; Sheikh, A.; Lee, C.C.; Chen, Y.; et al. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012, 64, 895–907. [Google Scholar] [CrossRef]
- Poli, M.C.; Ebstein, F.; Nicholas, S.K.; de Guzman, M.M.; Forbes, L.R.; Chinn, I.K.; Mace, E.M.; Vogel, T.P.; Carisey, A.F.; Benavides, F.; et al. Heterozygous Truncating Variants in POMP Escape Nonsense-Mediated Decay and Cause a Unique Immune Dysregulatory Syndrome. Am. J. Hum. Genet. 2018, 102, 1126–1142. [Google Scholar] [CrossRef]
- Sarrabay, G.; Mechin, D.; Salhi, A.; Boursier, G.; Rittore, C.; Crow, Y.; Rice, G.; Tran, T.A.; Cezar, R.; Duffy, D.; et al. PSMB10, the last immunoproteasome gene missing for PRAAS. J. Allergy Clin. Immunol. 2020, 145, 1015–1017. [Google Scholar] [CrossRef]
- Garrido, F.; Cabrera, T.; Concha, A.; Glew, S.; Ruiz-Cabello, F.; Stern, P.L. Natural history of HLA expression during tumour development. Immunol. Today 1993, 14, 491–499. [Google Scholar] [CrossRef]
- Garrido, F.; Ruiz-Cabello, F.; Cabrera, T.; Perez-Villar, J.J.; Lopez-Botet, M.; Duggan-Keen, M.; Stern, P.L. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol. Today 1997, 18, 89–95. [Google Scholar] [CrossRef]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1664. [Google Scholar] [CrossRef]
- Azimi, F.; Scolyer, R.A.; Rumcheva, P.; Moncrieff, M.; Murali, R.; McCarthy, S.W.; Saw, R.P.; Thompson, J.F. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J. Clin. Oncol. 2012, 30, 2678–2683. [Google Scholar] [CrossRef]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.H.; Pages, F.; et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013, 73, 3499–3510. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Lee, J.S.; Barnea, E.; Levy, R.; Greenberg, P.; Alon, M.; Yagel, G.; Bar Eli, G.; Oren, R.; Peri, A.; et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat. Commun. 2020, 11, 896. [Google Scholar] [CrossRef]
- Lee, M.; Song, I.H.; Heo, S.H.; Kim, Y.A.; Park, I.A.; Bang, W.S.; Park, H.S.; Gong, G.; Lee, H.J. Expression of Immunoproteasome Subunit LMP7 in Breast Cancer and Its Association with Immune-Related Markers. Cancer Res. Treat. 2019, 51, 80–89. [Google Scholar] [CrossRef]
- Rouette, A.; Trofimov, A.; Haberl, D.; Boucher, G.; Lavallee, V.P.; D’Angelo, G.; Hebert, J.; Sauvageau, G.; Lemieux, S.; Perreault, C. Expression of immunoproteasome genes is regulated by cell-intrinsic and -extrinsic factors in human cancers. Sci. Rep. 2016, 6, 34019. [Google Scholar] [CrossRef]
- Basler, M.; Dajee, M.; Moll, C.; Groettrup, M.; Kirk, C.J. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J. Immunol. 2010, 185, 634–641. [Google Scholar] [CrossRef]
- Vachharajani, N.; Joeris, T.; Luu, M.; Hartmann, S.; Pautz, S.; Jenike, E.; Pantazis, G.; Prinz, I.; Hofer, M.J.; Steinhoff, U.; et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 2017, 8, 50447–50459. [Google Scholar] [CrossRef]
- Yang, X.W.; Wang, P.; Liu, J.Q.; Zhang, H.; Xi, W.D.; Jia, X.H.; Wang, K.K. Coordinated regulation of the immunoproteasome subunits by PML/RARalpha and PU.1 in acute promyelocytic leukemia. Oncogene 2014, 33, 2700–2708. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Merin, N.M.; Kelly, K.R. Clinical use of proteasome inhibitors in the treatment of multiple myeloma. Pharmaceuticals 2014, 8, 1–20. [Google Scholar] [CrossRef]
- Cloos, J.; Roeten, M.S.; Franke, N.E.; van Meerloo, J.; Zweegman, S.; Kaspers, G.J.; Jansen, G. (Immuno)proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev. 2017, 36, 599–615. [Google Scholar] [CrossRef]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Studencka-Turski, M.; Cetin, G.; Junker, H.; Ebstein, F.; Kruger, E. Molecular Insight Into the IRE1alpha-Mediated Type I Interferon Response Induced by Proteasome Impairment in Myeloid Cells of the Brain. Front. Immunol. 2019, 10, 2900. [Google Scholar] [CrossRef]
- Singh, A.V.; Bandi, M.; Aujay, M.A.; Kirk, C.J.; Hark, D.E.; Raje, N.; Chauhan, D.; Anderson, K.C. PR-924, a selective inhibitor of the immunoproteasome subunit LMP-7, blocks multiple myeloma cell growth both in vitro and in vivo. Br. J. Haematol. 2011, 152, 155–163. [Google Scholar] [CrossRef]
- Ettari, R.; Previti, S.; Bitto, A.; Grasso, S.; Zappala, M. Immunoproteasome-Selective Inhibitors: A Promising Strategy to Treat Hematologic Malignancies, Autoimmune and Inflammatory Diseases. Curr. Med. Chem. 2016, 23, 1217–1238. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, H.; Shao, J.; He, R.; Xi, J.; Zhuang, R.; Zhang, J. Immunoproteasome-selective inhibitors: The future of autoimmune diseases? Future Med. Chem. 2020, 12, 269–272. [Google Scholar] [CrossRef]
- Ho, Y.K.; Bargagna-Mohan, P.; Wehenkel, M.; Mohan, R.; Kim, K.B. LMP2-specific inhibitors: Chemical genetic tools for proteasome biology. Chem. Biol. 2007, 14, 419–430. [Google Scholar] [CrossRef]
- Wehenkel, M.; Ban, J.O.; Ho, Y.K.; Carmony, K.C.; Hong, J.T.; Kim, K.B. A selective inhibitor of the immunoproteasome subunit LMP2 induces apoptosis in PC-3 cells and suppresses tumour growth in nude mice. Br. J. Cancer 2012, 107, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J. The Development of Novel Proteasome Inhibitors for the Treatment of Multiple Myeloma and Alzheimer’s Disease. Ph.D. Thesis, University of Kentucky, Lexington, KY, USA, April 2019. [Google Scholar]
- Basler, M.; Mundt, S.; Muchamuel, T.; Moll, C.; Jiang, J.; Groettrup, M.; Kirk, C.J. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol. Med. 2014, 6, 226–238. [Google Scholar] [CrossRef]
- Ichikawa, H.T.; Conley, T.; Muchamuel, T.; Jiang, J.; Lee, S.; Owen, T.; Barnard, J.; Nevarez, S.; Goldman, B.I.; Kirk, C.J.; et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012, 64, 493–503. [Google Scholar] [CrossRef]
- Downey-Kopyscinski, S.; Daily, E.W.; Gautier, M.; Bhatt, A.; Florea, B.I.; Mitsiades, C.S.; Richardson, P.G.; Driessen, C.; Overkleeft, H.S.; Kisselev, A.F. An inhibitor of proteasome beta2 sites sensitizes myeloma cells to immunoproteasome inhibitors. Blood Adv. 2018, 2, 2443–2451. [Google Scholar] [CrossRef]
- De Bruin, G.; Huber, E.M.; Xin, B.T.; van Rooden, E.J.; Al-Ayed, K.; Kim, K.B.; Kisselev, A.F.; Driessen, C.; van der Stelt, M.; van der Marel, G.A.; et al. Structure-based design of beta1i or beta5i specific inhibitors of human immunoproteasomes. J. Med. Chem. 2014, 57, 6197–6209. [Google Scholar] [CrossRef]
- Fan, H.; Angelo, N.G.; Warren, J.D.; Nathan, C.F.; Lin, G. Oxathiazolones Selectively Inhibit the Human Immunoproteasome over the Constitutive Proteasome. ACS Med. Chem. Lett. 2014, 5, 405–410. [Google Scholar] [CrossRef]
- Dubiella, C.; Baur, R.; Cui, H.; Huber, E.M.; Groll, M. Selective Inhibition of the Immunoproteasome by Structure-Based Targeting of a Non-catalytic Cysteine. Angew. Chem. Int. Ed. Engl. 2015, 54, 15888–15891. [Google Scholar] [CrossRef]
- Singh, P.K.; Fan, H.; Jiang, X.; Shi, L.; Nathan, C.F.; Lin, G. Immunoproteasome beta5i-Selective Dipeptidomimetic Inhibitors. ChemMedChem 2016, 11, 2127–2131. [Google Scholar] [CrossRef]
- Johnson, H.W.B.; Anderl, J.L.; Bradley, E.K.; Bui, J.; Jones, J.; Arastu-Kapur, S.; Kelly, L.M.; Lowe, E.; Moebius, D.C.; Muchamuel, T.; et al. Discovery of Highly Selective Inhibitors of the Immunoproteasome Low Molecular Mass Polypeptide 2 (LMP2) Subunit. ACS Med. Chem. Lett. 2017, 8, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.T.; Huber, E.M.; de Bruin, G.; Heinemeyer, W.; Maurits, E.; Espinal, C.; Du, Y.; Janssens, M.; Weyburne, E.S.; Kisselev, A.F.; et al. Structure-Based Design of Inhibitors Selective for Human Proteasome beta2c or beta2i Subunits. J. Med. Chem. 2019, 62, 1626–1642. [Google Scholar] [CrossRef]
- Klein, M.; Busch, M.; Esdar, C.; Friese-Hamim, M.; Krier, M.; Musil, D.; Rohdich, F.; Sanderson, M.; Walter, G.; Schadt, O.; et al. Abstract LB-054: Discovery and profiling of M3258, a potent and selective LMP7 inhibitor demonstrating high efficacy in multiple myeloma models. Cancer Res. 2019, 79 (Suppl. S13). [Google Scholar] [CrossRef]
- Maurits, E.; van de Graaff, M.J.; Maiorana, S.; Wander, D.P.A.; Dekker, P.M.; van der Zanden, S.Y.; Florea, B.I.; Neefjes, J.J.C.; Overkleeft, H.S.; van Kasteren, S.I. Immunoproteasome Inhibitor-Doxorubicin Conjugates Target Multiple Myeloma Cells and Release Doxorubicin upon Low-Dose Photon Irradiation. J. Am. Chem. Soc. 2020, 142, 7250–7253. [Google Scholar] [CrossRef]
- Downey-Kopyscinski, S.L.; De Matos Simoes, R.; Bariteau, M.; Borah, M.L.; Bender, S.; Foneska, J.; Roth, J.; Groen, R.; Walter, G.; Friese-Hamim, M.; et al. Pharmacological Perturbation of the Immunoproteasome in Hematologic Neoplasias: Therapeutic Implications. Blood 2019, 134 (Suppl. S1), 1291. [Google Scholar] [CrossRef]
- First in Human Dose Escalation of M3258 as a Single Agent and Expansion Study of M3258 in Combination With Dexamethasone. Available online: https://ClinicalTrials.gov/show/NCT04075721 (accessed on 20 March 2021).
- Britton, M.; Lucas, M.M.; Downey, S.L.; Screen, M.; Pletnev, A.A.; Verdoes, M.; Tokhunts, R.A.; Amir, O.; Goddard, A.L.; Pelphrey, P.M.; et al. Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem. Biol. 2009, 16, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, E.I.; Mezo, G.; Tzakos, A.G. On the design principles of peptide-drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein. J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef]
- Ashley, J.D.; Quinlan, C.J.; Schroeder, V.A.; Suckow, M.A.; Pizzuti, V.J.; Kiziltepe, T.; Bilgicer, B. Dual Carfilzomib and Doxorubicin-Loaded Liposomal Nanoparticles for Synergistic Efficacy in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2 S,3 R)- N-(( S)-3-(Cyclopent-1-en-1-yl)-1-(( R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-(( S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar]
- Lickliter, J.; Bomba, D.; Anderl, J.; Fan, A.; Kirk, C.J.; Wang, J. AB0509 Kzr-616, a selective inhibitor of the immunoproteasome, shows a promising safety and target inhibition profile in a phase i, double-blind, single (SAD) and multiple ascending dose (MAD) study in healthy volunteers. Ann. Rheum. Dis. 2018, 77 (Suppl. S2), 1413–1414. [Google Scholar]
- Open-Label Extension to the Phase 2 Crossover Study (PRESIDIO) Evaluating KZR-616 in Patients With PM and DM. Available online: https://ClinicalTrials.gov/show/NCT04628936 (accessed on 20 March 2021).
- A Study of KZR-616 in Patients With SLE With and Without Lupus Nephritis. Available online: https://ClinicalTrials.gov/show/NCT03393013 (accessed on 20 March 2021).
- Sanderson, M.; Busch, M.; Esdar, C.; Friese-Hamim, M.; Krier, M.; Ma, J.; Musil, D.; Rohdich, F.; Sloot, W.; Walter, G.; et al. Abstract DDT02-01: First-time disclosure of M3258: A selective inhibitor of the immunoproteasome subunit LMP7 with potential for improved therapeutic utility in multiple myeloma compared to pan-proteasome inhibitors. Cancer Res. 2019, 79 (Suppl. S13). [Google Scholar] [CrossRef]
- Ebstein, F.; Poli Harlowe, M.C.; Studencka-Turski, M.; Kruger, E. Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front. Immunol. 2019, 10, 2756. [Google Scholar] [CrossRef]
- Hideshima, T.; Ikeda, H.; Chauhan, D.; Okawa, Y.; Raje, N.; Podar, K.; Mitsiades, C.; Munshi, N.C.; Richardson, P.G.; Carrasco, R.D.; et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009, 114, 1046–1052. [Google Scholar] [CrossRef]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Qin, J.Z.; Ziffra, J.; Stennett, L.; Bodner, B.; Bonish, B.K.; Chaturvedi, V.; Bennett, F.; Pollock, P.M.; Trent, J.M.; Hendrix, M.J.; et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005, 65, 6282–6293. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.R.; Ahmed, M.; Ahmed, S.O.; Al-Thari, S.; Khan, A.S.; Razack, S.; Platanias, L.C.; Al-Kuraya, K.S.; Uddin, S. Proteasome inhibitor MG-132 mediated expression of p27Kip1 via S-phase kinase protein 2 degradation induces cell cycle coupled apoptosis in primary effusion lymphoma cells. Leuk. Lymphoma 2009, 50, 1204–1213. [Google Scholar] [CrossRef]
- Zarfati, M.; Avivi, I.; Brenner, B.; Katz, T.; Aharon, A. Extracellular vesicles of multiple myeloma cells utilize the proteasome inhibitor mechanism to moderate endothelial angiogenesis. Angiogenesis 2019, 22, 185–196. [Google Scholar] [CrossRef]
- Motegi, A.; Murakawa, Y.; Takeda, S. The vital link between the ubiquitin-proteasome pathway and DNA repair: Impact on cancer therapy. Cancer Lett. 2009, 283, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Berger, T.; Groettrup, M.; Basler, M. Immunoproteasome Inhibition Impairs T and B Cell Activation by Restraining ERK Signaling and Proteostasis. Front. Immunol. 2018, 9, 2386. [Google Scholar] [CrossRef]
- Paeschke, A.; Possehl, A.; Klingel, K.; Voss, M.; Voss, K.; Kespohl, M.; Sauter, M.; Overkleeft, H.S.; Althof, N.; Garlanda, C.; et al. The immunoproteasome controls the availability of the cardioprotective pattern recognition molecule Pentraxin3. Eur. J. Immunol. 2016, 46, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Althof, N.; Goetzke, C.C.; Kespohl, M.; Voss, K.; Heuser, A.; Pinkert, S.; Kaya, Z.; Klingel, K.; Beling, A. The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. EMBO Mol. Med. 2018, 10, 200–218. [Google Scholar] [CrossRef]
- Hensley, S.E.; Zanker, D.; Dolan, B.P.; David, A.; Hickman, H.D.; Embry, A.C.; Skon, C.N.; Grebe, K.M.; Griffin, T.A.; Chen, W.; et al. Unexpected role for the immunoproteasome subunit LMP2 in antiviral humoral and innate immune responses. J. Immunol. 2010, 184, 4115–4122. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; Anderson, B.O.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar]
- Pulte, D.; Jansen, L.; Castro, F.A.; Brenner, H. Changes in the survival of older patients with hematologic malignancies in the early 21st century. Cancer 2016, 122, 2031–2040. [Google Scholar] [CrossRef]
- Xu, G.W.; Ali, M.; Wood, T.E.; Wong, D.; Maclean, N.; Wang, X.; Gronda, M.; Skrtic, M.; Li, X.; Hurren, R.; et al. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 2010, 115, 2251–2259. [Google Scholar] [CrossRef]
- Milhollen, M.A.; Traore, T.; Adams-Duffy, J.; Thomas, M.P.; Berger, A.J.; Dang, L.; Dick, L.R.; Garnsey, J.J.; Koenig, E.; Langston, S.P.; et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: Rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood 2010, 116, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; O’Dwyer, M.; Nawrocki, S.T.; et al. Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.M.; Okoye, I.; Chaleshtari, M.G.; Hazhirkarzar, B.; Mohamadnejad, J.; Azizi, G.; Hojjat-Farsangi, M.; Mohammadi, H.; Shotorbani, S.S.; Jadidi-Niaragh, F. E2 ubiquitin-conjugating enzymes in cancer: Implications for immunotherapeutic interventions. Clin. Chim. Acta 2019, 498, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Sperling, A.S.; Ebert, B.L. Cancer therapies based on targeted protein degradation—Lessons learned with lenalidomide. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Kronke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Besse, A.; Besse, L.; Kraus, M.; Mendez-Lopez, M.; Bader, J.; Xin, B.T.; de Bruin, G.; Maurits, E.; Overkleeft, H.S.; Driessen, C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26, 340–351. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tubío-Santamaría, N.; Ebstein, F.; Heidel, F.H.; Krüger, E. Immunoproteasome Function in Normal and Malignant Hematopoiesis. Cells 2021, 10, 1577. https://doi.org/10.3390/cells10071577
Tubío-Santamaría N, Ebstein F, Heidel FH, Krüger E. Immunoproteasome Function in Normal and Malignant Hematopoiesis. Cells. 2021; 10(7):1577. https://doi.org/10.3390/cells10071577
Chicago/Turabian StyleTubío-Santamaría, Nuria, Frédéric Ebstein, Florian H. Heidel, and Elke Krüger. 2021. "Immunoproteasome Function in Normal and Malignant Hematopoiesis" Cells 10, no. 7: 1577. https://doi.org/10.3390/cells10071577
APA StyleTubío-Santamaría, N., Ebstein, F., Heidel, F. H., & Krüger, E. (2021). Immunoproteasome Function in Normal and Malignant Hematopoiesis. Cells, 10(7), 1577. https://doi.org/10.3390/cells10071577