
Cell Reprogramming to Model Huntington’s Disease: A Comprehensive Review

Abstract
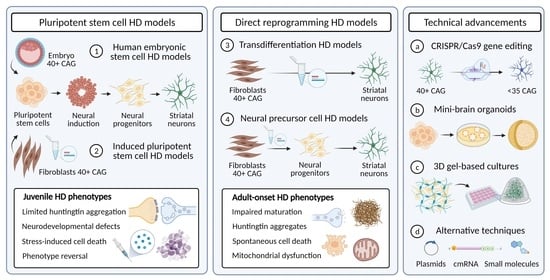
:
1. Introduction
2. Pluripotent Stem Cell Models of Huntington’s Disease
2.1. Human Embryonic Stem Cell Models of Huntington’s Disease
2.2. Induced Pluripotent Stem Cell Models of Huntington’s Disease
2.2.1. Phenotypes Observed in Huntington’s Disease induced Pluripotent Stem Cells
2.2.2. Phenotypes Observed in Huntington’s Disease Neural Precursor Cells Derived from Induced Pluripotent Stem Cells
2.2.3. Neurodevelopmental and Neuronal Maturation Phenotypes Observed in Huntington’s Disease Neurons Derived from Induced Pluripotent Stem Cells
2.2.4. Cell Stress and Degenerative Phenotypes Observed in Huntington’s Disease Neurons Derived from Induced Pluripotent Stem Cells
2.2.5. Genetic Correction and the Validation of Huntington’s Disease Phenotypes Using Neurons Derived from Induced Pluripotent Stem Cells Using Isogenic Controls
2.3. Challenges of Pluripotent Stem Cell Models of Huntington’s Disease
3. Direct Cell Reprogramming to Model Huntington’s Disease
3.1. Induced Neuron Reprogramming as an Alternative to Induced Pluripotent Stem Cells to Study Huntington’s Disease Neuropathogensis
3.2. Considerations for the Use of Directly Induced Neuron Models of Huntington’s Disease
3.3. Induced Neural Precursor Cells as an Alternative Method of Cell Reprogramming
3.4. Induced Neural Precursor Cell-Derived Neuronal Models of Huntington’s Disease
4. The Use of Cell Reprogramming for Disease Modelling and Recent Advancements in Cell Reprogramming Technologies
4.1. The Use of Cell Reprogramming to Study Cell-Autonomous and Non-Cell-Autonomous Mechanisms of Huntington’s Disease Neuropathogenesis
4.2. The Generation of Mixed Neuronal and Glial Cell Cultured from Directly Reprogrammed Induced Neural Precursor Cells for Modelling Huntington’s Disease
4.3. The Challenge of Using Small Molecules and Small Molecule Inhibitors to Enhance Nuronal Yield and Functionality for Huntington’s Disease Modelling Applications
4.4. The Generation of Multiple Neuronal Subtypes from Directly Reprogrammed Induced Neural Precursor Cells for Modelling Huntington’s Disease
4.5. The Inclusion of Three-Dimensional Culture Systems to Generate Functionally Mature Striatal Neurons from Reprogrammed Cells
4.6. The Generation of Huntington’s Disease Organoids for Modelling the Non-Cell-Autonomous Mechanisms Driving Medium Spiny Neuron Dysfunction
4.7. The Combination of Cell Reprogramming and Animal Models of Huntington’s Disease to Model Pathologies that May Be Dependent on the Specific In Vivo Environment
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verlinsky, Y.; Strelchenko, N.; Kukharenko, V.; Rechitsky, S.; Verlinsky, O.; Galat, V.; Kuliev, A. Human embryonic stem cell lines with genetic disorders. Reprod. Biomed. Online 2005, 10, 105–110. [Google Scholar] [CrossRef]
- Mateizel, I.; De Temmerman, N.; Ullmann, U.; Cauffman, G.; Sermon, K.; Van de Velde, H.; De Rycke, M.; Degreef, E.; Devroey, P.; Liebaers, I.; et al. Derivation of human embryonic stem cell lines from embryos obtained after IVF and after PGD for monogenic disorders. Hum. Reprod. 2006, 21, 503–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niclis, J.; Trounson, A.O.; Dottori, M.; Ellisdon, A.; Bottomley, S.P.; Verlinsky, Y.; Cram, D. Human embryonic stem cell models of Huntington disease. Reprod. Biomed. Online 2009, 19, 106–113. [Google Scholar] [CrossRef]
- Jacquet, L.; Neueder, A.; Foldes, G.; Karagiannis, P.; Hobbs, C.; Jolinon, N.; Mioulane, M.; Sakai, T.; Harding, S.E.; Ilic, D. Three Huntington’s Disease Specific Mutation-Carrying Human Embryonic Stem Cell Lines Have Stable Number of CAG Repeats upon In Vitro Differentiation into Cardiomyocytes. PLoS ONE 2015, 10, e0126860. [Google Scholar] [CrossRef]
- Bradley, C.K.; Scott, H.A.; Chami, O.; Peura, T.T.; Dumevska, B.; Schmidt, U.; Stojanov, T. Derivation of Huntington’s disease-affected human embryonic stem cell lines. Stem Cells Dev. 2011, 20, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Dumevska, B.; Chami, O.; McKernan, R.; Goel, D.; Schmidt, U. Derivation of Huntington Disease affected Genea046 human embryonic stem cell line. Stem Cell Res. 2016, 16, 446–448. [Google Scholar] [CrossRef] [Green Version]
- Dumevska, B.; Main, H.; McKernan, R.; Goel, D.; Schmidt, U.; Peura, T. Derivation of Huntington Disease affected Genea018 human embryonic stem cell line. Stem Cell Res. 2016, 16, 423–426. [Google Scholar] [CrossRef] [Green Version]
- Dumevska, B.; McKernan, R.; Goel, D.; Schmidt, U.; Peura, T. Derivation of Huntington Disease affected Genea017 human embryonic stem cell line. Stem Cell Res. 2016, 16, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Dumevska, B.; McKernan, R.; Hu, J.; Schmidt, U. Derivation of Huntington Disease affected Genea089 human embryonic stem cell line. Stem Cell Res. 2016, 16, 434–436. [Google Scholar] [CrossRef] [Green Version]
- Dumevska, B.; Peura, T.; McKernan, R.; Goel, D.; Schmidt, U. Derivation of Huntington disease affected Genea020 human embryonic stem cell line. Stem Cell Res. 2016, 16, 430–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumevska, B.; Schaft, J.; McKernan, R.; Hu, J.; Schmidt, U. Derivation of Huntington Disease affected Genea090 human embryonic stem cell line. Stem Cell Res. 2016, 16, 519–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumevska, B.; Schaft, J.; McKernan, R.; Hu, J.; Schmidt, U. Derivation of Huntington Disease affected Genea091 human embryonic stem cell line. Stem Cell Res. 2016, 16, 449–451. [Google Scholar] [CrossRef] [Green Version]
- Ruzo, A.; Croft, G.F.; Metzger, J.J.; Galgoczi, S.; Gerber, L.J.; Pellegrini, C.; Wang, H., Jr.; Fenner, M.; Tse, S.; Marks, A.; et al. Chromosomal instability during neurogenesis in Huntington’s disease. Development 2018, 145, dev156844. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 1820–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niclis, J.C.; Pinar, A.; Haynes, J.M.; Alsanie, W.; Jenny, R.; Dottori, M.; Cram, D.S. Characterization of forebrain neurons derived from late-onset Huntington’s disease human embryonic stem cell lines. Front. Cell. Neurosci. 2013, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- McQuade, L.R.; Balachandran, A.; Scott, H.A.; Khaira, S.; Baker, M.S.; Schmidt, U. Proteomics of Huntington’s disease-affected human embryonic stem cells reveals an evolving pathology involving mitochondrial dysfunction and metabolic disturbances. J. Proteome Res. 2014, 13, 5648–5659. [Google Scholar] [CrossRef] [PubMed]
- Feyeux, M.; Bourgois-Rocha, F.; Redfern, A.; Giles, P.; Lefort, N.; Aubert, S.; Bonnefond, C.; Bugi, A.; Ruiz, M.; Deglon, N. Early transcriptional changes linked to naturally occurring Huntington’s disease mutations in neural derivatives of human embryonic stem cells. Hum. Mol. Genet. 2012, 21, 3883–3895. [Google Scholar] [CrossRef] [Green Version]
- Seriola, A.; Spits, C.; Simard, J.P.; Hilven, P.; Haentjens, P.; Pearson, C.E.; Sermon, K. Huntington’s and myotonic dystrophy hESCs: Down-regulated trinucleotide repeat instability and mismatch repair machinery expression upon differentiation. Hum. Mol. Genet. 2011, 20, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Carmon, D.; Sorek, M.; Lerner, V.; Divya, M.S.; Nissim-Rafinia, M.; Yarom, Y.; Meshorer, E. Progerin-Induced Transcriptional Changes in Huntington’s Disease Human Pluripotent Stem Cell-Derived Neurons. Mol. Neurobiol. 2020, 57, 1768–1777. [Google Scholar] [CrossRef]
- Quarrell, O.W.J.; Nance, M.A.; Nopoulos, P.; Paulsen, J.S.; Smith, J.A.; Squitieri, F. Managing juvenile Huntington’s disease. Neurodegener. Dis. Manag. 2013, 3, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Camnasio, S.; Carri, A.D.; Lombardo, A.; Grad, I.; Mariotti, C.; Castucci, A.; Rozell, B.; Riso, P.L.; Castiglioni, V.; Zuccato, C. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 2012, 46, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nekrasov, E.D.; Kiselev, S.L. Mitochondrial distribution violation and nuclear indentations in neurons differentiated from iPSCs of Huntington’s disease patients. J. Stem Cells Regen. Med. 2018, 14, 80–85. [Google Scholar]
- Vigont, V.; Nekrasov, E.; Shalygin, A.; Gusev, K.; Klushnikov, S.; Illarioshkin, S.; Lagarkova, M.; Kiselev, S.L.; Kaznacheyeva, E. Patient-Specific iPSC-Based Models of Huntington’s Disease as a Tool to Study Store-Operated Calcium Entry Drug Targeting. Front. Pharmacol. 2018, 9, 696–696. [Google Scholar] [CrossRef] [PubMed]
- Vigont, V.A.; Grekhnev, D.A.; Lebedeva, O.S.; Gusev, K.O.; Volovikov, E.A.; Skopin, A.Y.; Bogomazova, A.N.; Shuvalova, L.D.; Zubkova, O.A.; Khomyakova, E.A.; et al. STIM2 Mediates Excessive Store-Operated Calcium Entry in Patient-Specific iPSC-Derived Neurons Modeling a Juvenile Form of Huntington’s Disease. Front. Cell Dev. Biol. 2021, 9, 625231. [Google Scholar] [CrossRef] [PubMed]
- Mollica, P.A.; Zamponi, M.; Reid, J.A.; Sharma, D.K.; White, A.E.; Ogle, R.C.; Bruno, R.D.; Sachs, P.C. Epigenetic alterations mediate iPSC-induced normalization of DNA repair gene expression and TNR stability in Huntington’s disease cells. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Chiu, F.L.; Lin, J.T.; Chuang, C.Y.; Chien, T.; Chen, C.M.; Chen, K.H.; Hsiao, H.Y.; Lin, Y.S.; Chern, Y.; Kuo, H.C. Elucidating the role of the A2A adenosine receptor in neurodegeneration using neurons derived from Huntington’s disease iPSCs. Hum. Mol. Genet. 2015, 24, 6066–6079. [Google Scholar] [CrossRef] [Green Version]
- Machiela, E.; Jeloka, R.; Caron, N.S.; Mehta, S.; Schmidt, M.E.; Baddeley, H.J.E.; Tom, C.M.; Polturi, N.; Xie, Y.; Mattis, V.B.; et al. The Interaction of Aging and Cellular Stress Contributes to Pathogenesis in Mouse and Human Huntington Disease Neurons. Front. Aging Neurosci. 2020, 12, 524369. [Google Scholar] [CrossRef]
- Liu, Y.; Qiao, F.; Leiferman, P.C.; Ross, A.; Schlenker, E.H.; Wang, H. FOXOs modulate proteasome activity in human-induced pluripotent stem cells of Huntington’s disease and their derived neural cells. Hum. Mol. Genet. 2017, 26, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Geater, C.; Hernandez, S.J.; Lim, R.G.; Adam, M.; Wu, J.; Stocksdale, J.T.; Wassie, B.T.; Gold, M.P.; Wang, K.Q.; Miramontes, R.; et al. Aberrant Development Corrected in Adult-Onset Huntington’s Disease iPSC-Derived Neuronal Cultures via WNT Signaling Modulation. Stem Cell Rep. 2020, 14, 406–419. [Google Scholar] [CrossRef] [Green Version]
- Malankhanova, T.; Suldina, L.; Grigor’eva, E.; Medvedev, S.; Minina, J.; Morozova, K.; Kiseleva, E.; Zakian, S.; Malakhova, A. A Human Induced Pluripotent Stem Cell-Derived Isogenic Model of Huntington’s Disease Based on Neuronal Cells Has Several Relevant Phenotypic Abnormalities. J. Pers. Med. 2020, 10, 215. [Google Scholar] [CrossRef]
- Yao, Y.; Cui, X.; Al-Ramahi, I.; Sun, X.; Li, B.; Hou, J.; Difiglia, M.; Palacino, J.; Wu, Z.-Y.; Ma, L. A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. eLife 2015, 4, e05449. [Google Scholar] [CrossRef]
- The HD iPSC Consortium. Bioenergetic deficits in Huntington’s disease iPSC-derived neural cells and rescue with glycolytic metabolites. Hum. Mol. Genet. 2019, 29, 1757–1771. [Google Scholar] [CrossRef]
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.C.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 2017, 94, 93–107.e6. [Google Scholar] [CrossRef] [Green Version]
- Tidball, A.M.; Neely, M.D.; Chamberlin, R.; Aboud, A.A.; Kumar, K.K.; Han, B.; Bryan, M.R.; Aschner, M.; Ess, K.C.; Bowman, A.B. Genomic Instability Associated with p53 Knockdown in the Generation of Huntington’s Disease Human Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0150372. [Google Scholar] [CrossRef]
- Koyuncu, S.; Saez, I.; Lee, H.J.; Gutierrez-Garcia, R.; Pokrzywa, W.; Fatima, A.; Hoppe, T.; Vilchez, D. The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat. Commun. 2018, 9, 2886. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Bodnya, C.; Ilieva, I.; Neely, M.D.; Aschner, M.; Bowman, A.B. Huntington’s disease associated resistance to Mn neurotoxicity is neurodevelopmental stage and neuronal lineage dependent. Neurotoxicology 2019, 75, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Mattis, V.B.; Tom, C.; Akimov, S.; Saeedian, J.; Ostergaard, M.E.; Southwell, A.L.; Doty, C.N.; Ornelas, L.; Sahabian, A.; Lenaeus, L.; et al. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum. Mol. Genet. 2015, 24, 3257–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickey, A.S.; Sanchez, D.N.; Arreola, M.; Sampat, K.R.; Fan, W.; Arbez, N.; Akimov, S.; Van Kanegan, M.J.; Ohnishi, K.; Gilmore-Hall, S.K.; et al. PPARdelta activation by bexarotene promotes neuroprotection by restoring bioenergetic and quality control homeostasis. Sci. Transl. Med. 2017, 9, eaal2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The HD iPSC Consortium, Developmental alterations in Huntington’s disease neural cells and pharmacological rescue in cells and mice. Nat. Neurosci. 2017, 20, 648–660. [CrossRef] [PubMed] [Green Version]
- Mathkar, P.P.; Suresh, D.; Dunn, J.; Tom, C.M.; Mattis, V.B. Characterization of Neurodevelopmental Abnormalities in iPSC-Derived Striatal Cultures from Patients with Huntington’s Disease. J. Huntingt. Dis. 2019, 8, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The HD iPSC Consortium, Induced Pluripotent Stem Cells from Patients with Huntington’s Disease Show CAG-Repeat-Expansion-Associated Phenotypes. Cell Stem Cell 2012, 11, 264–278. [CrossRef] [Green Version]
- Martin-Flores, N.; Romani-Aumedes, J.; Rue, L.; Canal, M.; Sanders, P.; Straccia, M.; Allen, N.D.; Alberch, J.; Canals, J.M.; Perez-Navarro, E.; et al. RTP801 Is Involved in Mutant Huntingtin-Induced Cell Death. Mol. Neurobiol. 2016, 53, 2857–2868. [Google Scholar] [CrossRef] [PubMed]
- Conforti, P.; Besusso, D.; Bocchi, V.D.; Faedo, A.; Cesana, E.; Rossetti, G.; Ranzani, V.; Svendsen, C.N.; Thompson, L.M.; Toselli, M.; et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc. Natl. Acad. Sci. USA 2018, 115, E762–E771. [Google Scholar] [CrossRef] [Green Version]
- Morozko, E.L.; Smith-Geater, C.; Monteys, A.M.; Pradhan, S.; Lim, R.G.; Langfelder, P.; Kachemov, M.; Hill, A.; Stocksdale, J.T.; Cullis, P.R.; et al. PIAS1 modulates striatal transcription, DNA damage repair, and SUMOylation with relevance to Huntington’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2021836118. [Google Scholar] [CrossRef] [PubMed]
- Tidball, A.M.; Bryan, M.R.; Uhouse, M.A.; Kumar, K.K.; Aboud, A.A.; Feist, J.E.; Ess, K.C.; Neely, M.D.; Aschner, M.; Bowman, A.B. A novel manganese-dependent ATM-p53 signaling pathway is selectively impaired in patient-based neuroprogenitor and murine striatal models of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 1929–1944. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, W.J.; Switonski, P.M.; Krzyzosiak, W.J.; Figlerowicz, M.; Figiel, M. Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis. Models Mech. 2015, 8, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Switonska, K.; Szlachcic, W.J.; Handschuh, L.; Wojciechowski, P.; Marczak, L.; Stelmaszczuk, M.; Figlerowicz, M.; Figiel, M. Identification of Altered Developmental Pathways in Human Juvenile HD iPSC With 71Q and 109Q Using Transcriptome Profiling. Front. Cell. Neurosci. 2018, 12, 528. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; An, M.C.; Montoro, D.; Ellerby, L.M. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010, 2, RRN1193. [Google Scholar] [CrossRef] [PubMed]
- An, M.C.; Zhang, N.; Scott, G.; Montoro, D.; Wittkop, T.; Mooney, S.; Melov, S.; Ellerby, L.M. Genetic correction of huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 2012, 11, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Chae, J.I.; Kim, D.W.; Lee, N.; Jeon, Y.J.; Jeon, I.; Kwon, J.; Kim, J.; Soh, Y.; Lee, D.S.; Seo, K.S.; et al. Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem. J. 2012, 446, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, I.; Lee, N.; Li, J.Y.; Park, I.H.; Park, K.S.; Moon, J.; Shim, S.H.; Choi, C.; Chang, D.J.; Kwon, J. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells 2012, 30, 2054–2062. [Google Scholar] [CrossRef]
- Charbord, J.; Poydenot, P.; Bonnefond, C.; Feyeux, M.; Casagrande, F.; Brinon, B.; Francelle, L.; Auregan, G.; Guillermier, M.; Cailleret, M.; et al. High throughput screening for inhibitors of REST in neural derivatives of human embryonic stem cells reveals a chemical compound that promotes expression of neuronal genes. Stem Cells 2013, 31, 1816–1828. [Google Scholar] [CrossRef]
- Cheng, P.H.; Li, C.L.; Chang, Y.F.; Tsai, S.J.; Lai, Y.Y.; Chan, A.W.; Chen, C.M.; Yang, S.H. miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am. J. Hum. Genet. 2013, 93, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Ring, K.L.; An, M.C.; Zhang, N.; O’Brien, R.N.; Ramos, E.M.; Gao, F.; Atwood, R.; Bailus, B.J.; Melov, S.; Mooney, S.D.; et al. Genomic Analysis Reveals Disruption of Striatal Neuronal Development and Therapeutic Targets in Human Huntington’s Disease Neural Stem Cells. Stem Cell Rep. 2015, 5, 1023–1038. [Google Scholar] [CrossRef] [Green Version]
- Naphade, S.; Embusch, A.; Madushani, K.L.; Ring, K.L.; Ellerby, L.M. Altered Expression of Matrix Metalloproteinases and Their Endogenous Inhibitors in a Human Isogenic Stem Cell Model of Huntington’s Disease. Front. Neurosci. 2017, 11, 736. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.; Tang, Y.; Anjo, S.I.; Manadas, B.; Onofre, I.; de Almeida, L.P.; Daley, G.Q.; Schlaeger, T.M.; Rego, A.C.C. Mitochondrial and Redox Modifications in Huntington Disease Induced Pluripotent Stem Cells Rescued by CRISPR/Cas9 CAGs Targeting. Front. Cell Dev. Biol. 2020, 8, 576592–576592. [Google Scholar] [CrossRef]
- Le Cann, K.; Foerster, A.; Rösseler, C.; Erickson, A.; Hautvast, P.; Giesselmann, S.; Pensold, D.; Kurth, I.; Rothermel, M.; Mattis, V.B.; et al. The difficulty to model Huntington’s disease in vitro using striatal medium spiny neurons differentiated from human induced pluripotent stem cells. Sci. Rep. 2021, 11, 6934–6934. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.R.; Tom, C.M.; Wang, Y.; Bresee, C.; Rushton, D.; Mathkar, P.P.; Tang, J.; Mattis, V.B. Human Huntington’s Disease iPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell Rep. 2018, 25, 1081–1096.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Disatnik, M.-H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease–associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371. [Google Scholar] [CrossRef]
- Lu, X.H.; Mattis, V.B.; Wang, N.; Al-Ramahi, I.; van den Berg, N.; Fratantoni, S.A.; Waldvogel, H.; Greiner, E.; Osmand, A.; Elzein, K.; et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 2014, 6, 268ra178. [Google Scholar] [CrossRef]
- Aron, R.; Pellegrini, P.; Green, E.W.; Maddison, D.C.; Opoku-Nsiah, K.; Wong, J.S.; Daub, A.C.; Giorgini, F.; Finkbeiner, S. Deubiquitinase Usp12 functions noncatalytically to induce autophagy and confer neuroprotection in models of Huntington’s disease. Nat. Commun. 2018, 9, 3191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, J.; Hayden, M.R.; Pouladi, M.A. Inhibition of Excessive Monoamine Oxidase A/B Activity Protects Against Stress-induced Neuronal Death in Huntington Disease. Mol. Neurobiol. 2015, 52, 1850–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 619–633. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Cheah, F.S.H.; Tan, A.S.C.; Lian, M.; Phang, G.P.; Agarwal, A.; Chong, S.S. Robust Preimplantation Genetic Testing of Huntington Disease by Combined Triplet-Primed PCR Analysis of the HTT CAG Repeat and Multi-Microsatellite Haplotyping. Sci. Rep. 2019, 9, 16481. [Google Scholar] [CrossRef]
- Evers, M.M.; Schut, M.H.; Pepers, B.A.; Atalar, M.; van Belzen, M.J.; Faull, R.L.M.; Roos, R.A.C.; van Roon-Mom, W.M.C. Making (anti-) sense out of huntingtin levels in Huntington disease. Mol. Neurodegener. 2015, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Belyaev, N.; Conforti, P.; Ooi, L.; Tartari, M.; Papadimou, E.; MacDonald, M.; Fossale, E.; Zeitlin, S.; Buckley, N.; et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 6972–6983. [Google Scholar] [CrossRef]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccato, C.; Liber, D.; Ramos, C.; Tarditi, A.; Rigamonti, D.; Tartari, M.; Valenza, M.; Cattaneo, E. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacol. Res. 2005, 52, 133–139. [Google Scholar] [CrossRef]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 2008, 18, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.-H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, H.Y.; Chiu, F.L.; Chen, C.M.; Wu, Y.R.; Chen, H.M.; Chen, Y.C.; Kuo, H.C.; Chern, Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4328–4344. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.J.; Rushton, D.J.; Tom, C.M.; Allen, N.D.; Kemp, P.J.; Svendsen, C.N.; Mattis, V.B. Huntington’s Disease Patient-Derived Astrocytes Display Electrophysiological Impairments and Reduced Neuronal Support. Front. Neurosci. 2019, 13, 669. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kegel, K.B.; Kim, M.; Sapp, E.; McIntyre, C.; Castano, J.G.; Aronin, N.; DiFiglia, M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 7268–7278. [Google Scholar] [CrossRef]
- Jeong, H.; Then, F.; Melia, T.J., Jr.; Mazzulli, J.R.; Cui, L.; Savas, J.N.; Voisine, C.; Paganetti, P.; Tanese, N.; Hart, A.C.; et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009, 137, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, W.T.; Warren, C.R.; Cowan, C.A. Genome Editing in Human Pluripotent Stem Cells: Approaches, Pitfalls, and Solutions. Cell Stem Cell 2016, 18, 53–65. [Google Scholar] [CrossRef] [Green Version]
- An, M.C.; O’Brien, R.N.; Zhang, N.; Patra, B.N.; De La Cruz, M.; Ray, A.; Ellerby, L.M. Polyglutamine Disease Modeling: Epitope Based Screen for Homologous Recombination using CRISPR/Cas9 System. PLoS Curr. 2014, 6. [Google Scholar] [CrossRef]
- Dabrowska, M.; Ciolak, A.; Kozlowska, E.; Fiszer, A.; Olejniczak, M. Generation of New Isogenic Models of Huntington’s Disease Using CRISPR-Cas9 Technology. Int. J. Mol. Sci. 2020, 21, 1854. [Google Scholar] [CrossRef] [Green Version]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef]
- Narva, E.; Autio, R.; Rahkonen, N.; Kong, L.; Harrison, N.; Kitsberg, D.; Borghese, L.; Itskovitz-Eldor, J.; Rasool, O.; Dvorak, P.; et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat. Biotechnol. 2010, 28, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Amps, K.; Andrews, P.W.; Anyfantis, G.; Armstrong, L.; Avery, S.; Baharvand, H.; Baker, J.; Baker, D.; Munoz, M.B.; Beil, S.; et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar] [PubMed] [Green Version]
- Maitra, A.; Arking, D.E.; Shivapurkar, N.; Ikeda, M.; Stastny, V.; Kassauei, K.; Sui, G.; Cutler, D.J.; Liu, Y.; Brimble, S.N.; et al. Genomic alterations in cultured human embryonic stem cells. Nat. Genet. 2005, 37, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Garitaonandia, I.; Amir, H.; Boscolo, F.S.; Wambua, G.K.; Schultheisz, H.L.; Sabatini, K.; Morey, R.; Waltz, S.; Wang, Y.C.; Tran, H.; et al. Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS ONE 2015, 10, e0118307. [Google Scholar] [CrossRef] [PubMed]
- Bershteyn, M.; Hayashi, Y.; Desachy, G.; Hsiao, E.C.; Sami, S.; Tsang, K.M.; Weiss, L.A.; Kriegstein, A.R.; Yamanaka, S.; Wynshaw-Boris, A. Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature 2014, 507, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, L.; Vera, E.; Cornacchia, D. Programming and Reprogramming Cellular Age in the Era of Induced Pluripotency. Cell Stem Cell 2015, 16, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [Green Version]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, J.; Simonini, M.V.; Palejev, D.; Tomasini, L.; Coppola, G.; Szekely, A.M.; Horvath, T.L.; Vaccarino, F.M. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12770–12775. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Abyzov, A.; Mariani, J.; Palejev, D.; Zhang, Y.; Haney, M.S.; Tomasini, L.; Ferrandino, A.F.; Rosenberg Belmaker, L.A.; Szekely, A.; Wilson, M.; et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 2012, 492, 438–442. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Barton, D.E.; Davison, B.C.; Ferguson-Smith, M.A. Analysis of the huntingtin gene reveals a trinucleotide-length polymorphism in the region of the gene that contains two CCG-rich stretches and a correlation between decreased age of onset of Huntington’s disease and CAG repeat number. Hum. Mol. Genet. 1993, 2, 1713–1715. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Chiano, M.; Dodge, A.; Norbury, G.; Rosser, E.; Craufurd, D. Genotypes at the GluR6 kainate receptor locus are associated with variation in the age of onset of Huntington disease. Proc. Natl. Acad. Sci. USA 1997, 94, 3872–3876. [Google Scholar] [CrossRef] [Green Version]
- Snell, R.G.; MacMillan, J.C.; Cheadle, J.P.; Fenton, I.; Lazarou, L.P.; Davies, P.; MacDonald, M.E.; Gusella, J.F.; Harper, P.S.; Shaw, D.J. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 1993, 4, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Holbert, S.; Denghien, I.; Kiechle, T.; Rosenblatt, A.; Wellington, C.; Hayden, M.R.; Margolis, R.L.; Ross, C.A.; Dausset, J.; Ferrante, R.J.; et al. The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: Neuropathologic and genetic evidence for a role in Huntington’s disease pathogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1811–1816. [Google Scholar] [CrossRef]
- Mollica, P.A.; Reid, J.A.; Ogle, R.C.; Sachs, P.C.; Bruno, R.D. DNA Methylation Leads to DNA Repair Gene Down-Regulation and Trinucleotide Repeat Expansion in Patient-Derived Huntington Disease Cells. Am. J. Pathol. 2016, 186, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Zheng, X.; Ansari, Z.; Bunnell, M.C.; Herdy, J.R.; Traxler, L.; Lee, H.; Paquola, A.C.M.; Blithikioti, C.; Ku, M.; et al. Mitochondrial Aging Defects Emerge in Directly Reprogrammed Human Neurons due to Their Metabolic Profile. Cell Rep. 2018, 23, 2550–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Bohnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Drouin-Ouellet, J.; Lau, S.; Brattas, P.L.; Rylander Ottosson, D.; Pircs, K.; Grassi, D.A.; Collins, L.M.; Vuono, R.; Andersson Sjoland, A.; Westergren-Thorsson, G.; et al. REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and -independent pathways. EMBO Mol. Med. 2017, 9, 1117–1131. [Google Scholar] [CrossRef]
- Victor, M.B.; Richner, M.; Olsen, H.E.; Lee, S.W.; Monteys, A.M.; Ma, C.; Huh, C.J.; Zhang, B.; Davidson, B.L.; Yang, X.W.; et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 2018, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xue, Y.; Ridley, S.; Zhang, D.; Rezvani, K.; Fu, X.-D.; Wang, H. Direct reprogramming of Huntington’s disease patient fibroblasts into neuron-like cells leads to abnormal neurite outgrowth, increased cell death, and aggregate formation. PLoS ONE 2014, 9, e109621. [Google Scholar] [CrossRef]
- Milnerwood, A.J.; Cummings, D.M.; Dallerac, G.M.; Brown, J.Y.; Vatsavayai, S.C.; Hirst, M.C.; Rezaie, P.; Murphy, K.P. Early development of aberrant synaptic plasticity in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2006, 15, 1690–1703. [Google Scholar] [CrossRef] [Green Version]
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.R.; et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.S.; Klapstein, G.J.; Koppel, A.; Gruen, E.; Cepeda, C.; Vargas, M.E.; Jokel, E.S.; Carpenter, E.M.; Zanjani, H.; Hurst, R.S.; et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J. Neurosci. Res. 1999, 58, 515–532. [Google Scholar] [CrossRef]
- Fan, M.M.; Raymond, L.A. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog. Neurobiol. 2007, 81, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, A.; Sipola, I.; Larsson, K.; Akerman, K.E.; Stoilov, P.; Stamm, S.; Wong, G.; Castren, E. Regulation of TRKB surface expression by brain-derived neurotrophic factor and truncated TRKB isoforms. J. Biol. Chem. 2002, 277, 43160–43167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef]
- Gines, S.; Bosch, M.; Marco, S.; Gavalda, N.; Diaz-Hernandez, M.; Lucas, J.J.; Canals, J.M.; Alberch, J. Reduced expression of the TrkB receptor in Huntington’s disease mouse models and in human brain. Eur. J. Neurosci. 2006, 23, 649–658. [Google Scholar] [CrossRef]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P.; et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, J.L.; Surmeier, D.J. Impaired striatal function in Huntington’s disease is due to aberrant p75NTR signaling. Rare Dis. 2014, 2, e968482–e968482. [Google Scholar] [CrossRef] [Green Version]
- Brito, V.; Puigdellivol, M.; Giralt, A.; del Toro, D.; Alberch, J.; Gines, S. Imbalance of p75NTR/TrkB protein expression in Huntington/’s disease: Implication for neuroprotective therapies. Cell Death Dis. 2013, 4, e595. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [CrossRef]
- Difiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Wapinski, O.L.; Vierbuchen, T.; Qu, K.; Lee, Q.Y.; Chanda, S.; Fuentes, D.R.; Giresi, P.G.; Ng, Y.H.; Marro, S.; Neff, N.F.; et al. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 2013, 155, 621–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colasante, G.; Lignani, G.; Rubio, A.; Medrihan, L.; Yekhlef, L.; Sessa, A.; Massimino, L.; Giannelli, S.G.; Sacchetti, S.; Caiazzo, M.; et al. Rapid Conversion of Fibroblasts into Functional Forebrain GABAergic Interneurons by Direct Genetic Reprogramming. Cell Stem Cell 2015, 17, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Wiatr, K.; Szlachcic, W.J.; Trzeciak, M.; Figlerowicz, M.; Figiel, M. Huntington Disease as a Neurodevelopmental Disorder and Early Signs of the Disease in Stem Cells. Mol. Neurobiol. 2018, 55, 3351–3371. [Google Scholar] [CrossRef]
- Shimojo, M. Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin p150 Glued. J. Biol. Chem. 2008, 283, 34880–34886. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, L.T.; Starr, A.; Nelson, A.T.; Sattler, R. Representing Diversity in the Dish: Using Patient-Derived in Vitro Models to Recreate the Heterogeneity of Neurological Disease. Front. Neurosci. 2018, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Wexler, N.S.; Lorimer, J.; Porter, J.; Gomez, F.; Moskowitz, C.; Shackell, E.; Marder, K.; Penchaszadeh, G.; Roberts, S.A.; Gayan, J.; et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar]
- Metzger, S.; Bauer, P.; Tomiuk, J.; Laccone, F.; DiDonato, S.; Gellera, C.; Mariotti, C.; Lange, H.W.; Weirich-Schwaiger, H.; Wenning, G.K. Genetic analysis of candidate genes modifying the age-at-onset in Huntington’s disease. Hum. Genet. 2006, 120, 285–292. [Google Scholar] [CrossRef]
- Maucksch, C.; Firmin, E.; Butler-Munro, C.; Montgomery, J.M.; Dottori, M.; Connor, B. Non-Viral Generation of Neural Precursor-like Cells from Adult Human Fibroblasts. J. Stem Cells Regen. Med. 2012, 8, 162–170. [Google Scholar]
- Connor, B.; Firmin, E.; McCaughey-Chapman, A.; Monk, R.; Lee, K.; Liot, S.; Geiger, J.; Rudolph, C.; Jones, K. Conversion of adult human fibroblasts into neural precursor cells using chemically modified mRNA. Heliyon 2018, 4, e00918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.S.; Chuang, C.Y.; Yeh, C.H.; Chiang, W.; Liu, H.J.; Lin, T.N.; Kuo, H.C. Direct Conversion of Human Fibroblasts into Neural Progenitors Using Transcription Factors Enriched in Human ESC-Derived Neural Progenitors. Stem Cell Rep. 2017, 8, 54–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, R.; Lee, K.; Jones, K.S.; Connor, B. Directly reprogrammed Huntington’s disease neural precursor cells generate striatal neurons exhibiting aggregates and impaired neuronal maturation. Stem Cells Transl. Med. 2021. [Google Scholar] [CrossRef]
- Trottier, Y.; Lutz, Y.; Stevanin, G.; Imbert, G.; Devys, D.; Cancel, G.; Saudou, F.; Weber, C.; David, G.; Tora, L.; et al. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 1995, 378, 403–406. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef] [PubMed]
- Canals, J.M.; Pineda, J.R.; Torres-Peraza, J.F.; Bosch, M.; Martín-Ibañez, R.; Muñoz, M.T.; Mengod, G.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 2004, 24, 7727–7739. [Google Scholar] [CrossRef] [Green Version]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar] [CrossRef]
- Baquet, Z.C.; Gorski, J.A.; Jones, K.R. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 4250–4258. [Google Scholar] [CrossRef]
- Baydyuk, M.; Xie, Y.; Tessarollo, L.; Xu, B. Midbrain-Derived Neurotrophins Support Survival of Immature Striatal Projection Neurons. J. Neurosci. 2013, 33, 3363–3369. [Google Scholar] [CrossRef] [Green Version]
- Khakh, B.S. Astrocyte–Neuron Interactions in the Striatum: Insights on Identity, Form, and Function. Trends Neurosci. 2019, 42, 617–630. [Google Scholar] [CrossRef]
- Benraiss, A.; Wang, S.; Herrlinger, S.; Li, X.; Chandler-Militello, D.; Mauceri, J.; Burm, H.B.; Toner, M.; Osipovitch, M.; Jim Xu, Q.; et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 2016, 7, 11758. [Google Scholar] [CrossRef]
- Tydlacka, S.; Wang, C.E.; Wang, X.; Li, S.; Li, X.J. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 13285–13295. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.E.; Barry, J.; Yang, Z.; Cepeda, C.; Levine, M.S.; Gray, M. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet. 2019, 28, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P.; et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: A correlation with Huntington’s disease subjects. Hum. Mol. Genet. 2010, 19, 3053–3067. [Google Scholar] [CrossRef]
- Shin, J.Y.; Fang, Z.H.; Yu, Z.X.; Wang, C.E.; Li, S.H.; Li, X.J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain J. Neurol. 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef]
- Merienne, N.; Meunier, C.; Schneider, A.; Seguin, J.; Nair, S.S.; Rocher, A.B.; Le Gras, S.; Keime, C.; Faull, R.; Pellerin, L.; et al. Cell-Type-Specific Gene Expression Profiling in Adult Mouse Brain Reveals Normal and Disease-State Signatures. Cell Rep. 2019, 26, 2477–2493 e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, A.S.; Sun, A.X.; Li, L.; Shcheglovitov, A.; Portmann, T.; Li, Y.; Lee-Messer, C.; Dolmetsch, R.E.; Tsien, R.W.; Crabtree, G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011, 476, 228–231. [Google Scholar] [CrossRef]
- Liu, M.-L.; Zang, T.; Zou, Y.; Chang, J.C.; Gibson, J.R.; Huber, K.M.; Zhang, C.-L. Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat. Commun. 2013, 4, 2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladewig, J.; Mertens, J.; Kesavan, J.; Doerr, J.; Poppe, D.; Glaue, F.; Herms, S.; Wernet, P.; Kögler, G.; Müller, F.-J. Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat. Methods 2012, 9, 575. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, W.; Zhang, Y.; Wei, W.; Ambasudhan, R.; Xia, P.; Talantova, M.; Lin, T.; Kim, J.; Wang, X.; et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 8299–8304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Li, X.; Zuo, X.; Jing, J.; Ma, Y.; Wang, J.; Liu, D.; Zhu, J.; Du, X.; Xiong, L.; Du, Y.; et al. Small-Molecule-Driven Direct Reprogramming of Mouse Fibroblasts into Functional Neurons. Cell Stem Cell 2015, 17, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G. Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 2015, 17, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Hogarth, P.; Lovrecic, L.; Krainc, D. Sodium phenylbutyrate in Huntington’s disease: A dose-finding study. Mov. Disord. Off. J. Mov. Disord. Soc. 2007, 22, 1962–1964. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yuan, J.; Sander, B.; Golas, M.M. In Vitro Differentiation of Human Neural Progenitor Cells Into Striatal GABAergic Neurons. Stem Cells Transl. Med. 2015, 4, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pallares, J.; Rodriguez-Perez, A.I.; Munoz, A.; Parga, J.A.; Toledo-Aral, J.J.; Labandeira-Garcia, J.L. Effects of Rho Kinase Inhibitors on Grafts of Dopaminergic Cell Precursors in a Rat Model of Parkinson’s Disease. Stem Cells Transl. Med. 2016, 5, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Tejeda, G.S.; Diaz-Guerra, M. Integral Characterization of Defective BDNF/TrkB Signalling in Neurological and Psychiatric Disorders Leads the Way to New Therapies. Int. J. Mol. Sci. 2017, 18, 268. [Google Scholar] [CrossRef] [Green Version]
- Simmons, D.A. Modulating Neurotrophin Receptor Signaling as a Therapeutic Strategy for Huntington’s Disease. J. Huntingt. Dis. 2017, 6, 303–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazi, E.; Moradi, S.; Nemati, S.; Satarian, L.; Basiri, M.; Gourabi, H.; Mehrjardi, N.Z.; Günther, P.; Lampert, A.; Händler, K. Conversion of human fibroblasts to stably self-renewing neural stem cells with a single zinc-finger transcription factor. Stem Cell Rep. 2016, 6, 539–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gines, S.; Seong, I.S.; Fossale, E.; Ivanova, E.; Trettel, F.; Gusella, J.F.; Wheeler, V.C.; Persichetti, F.; MacDonald, M.E. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum. Mol. Genet. 2003, 12, 497–508. [Google Scholar] [CrossRef]
- Mehan, S.; Parveen, S.; Kalra, S. Adenyl cyclase activator forskolin protects against Huntington’s disease-like neurodegenerative disorders. Neural Regen. Res. 2017, 12, 290–300. [Google Scholar] [CrossRef]
- Kim, J.E.; O’Sullivan, M.L.; Sanchez, C.A.; Hwang, M.; Israel, M.A.; Brennand, K.; Deerinck, T.J.; Goldstein, L.S.; Gage, F.H.; Ellisman, M.H.; et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 3005–3010. [Google Scholar] [CrossRef] [Green Version]
- Zirra, A.; Wiethoff, S.; Patani, R. Neural Conversion and Patterning of Human Pluripotent Stem Cells: A Developmental Perspective. Stem Cells Int. 2016, 2016, 8291260. [Google Scholar] [CrossRef] [Green Version]
- Cudkowicz, M.; Kowall, N.W. Degeneration of pyramidal projection neurons in Huntington’s disease cortex. Ann. Neurol. 1990, 27, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Hedreen, J.C.; Peyser, C.E.; Folstein, S.E.; Ross, C.A. Neuronal loss in layers V and VI of cerebral cortex in Huntington’s disease. Neurosci. Lett. 1991, 133, 257–261. [Google Scholar] [CrossRef]
- Bodner, R.A.; Outeiro, T.F.; Altmann, S.; Maxwell, M.M.; Cho, S.H.; Hyman, B.T.; McLean, P.J.; Young, A.B.; Housman, D.E.; Kazantsev, A.G. Pharmacological promotion of inclusion formation: A therapeutic approach for Huntington’s and Parkinson’s diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 4246–4251. [Google Scholar] [CrossRef] [Green Version]
- Bohanna, I.; Georgiou-Karistianis, N.; Hannan, A.J.; Egan, G.F. Magnetic resonance imaging as an approach towards identifying neuropathological biomarkers for Huntington’s disease. Brain Res. Rev. 2008, 58, 209–225. [Google Scholar] [CrossRef]
- Her, L.S.; Goldstein, L.S. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 13662–13672. [Google Scholar] [CrossRef]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- Hassel, B.; Tessler, S.; Faull, R.L.; Emson, P.C. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 2008, 33, 232–237. [Google Scholar] [CrossRef]
- Arzberger, T.; Krampfl, K.; Leimgruber, S.; Weindl, A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease-an in situ hybridization study. J. Neuropathol. Exp. Neurol. 1997, 56, 440–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, M.B.; Richner, M.; Hermanstyne, T.O.; Ransdell, J.L.; Sobieski, C.; Deng, P.Y.; Klyachko, V.A.; Nerbonne, J.M.; Yoo, A.S. Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron 2014, 84, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Virlogeux, A.; Moutaux, E.; Christaller, W.; Genoux, A.; Bruyère, J.; Fino, E.; Charlot, B.; Cazorla, M.; Saudou, F. Reconstituting Corticostriatal Network on-a-Chip Reveals the Contribution of the Presynaptic Compartment to Huntington’s Disease. Cell Rep. 2018, 22, 110–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, E.Y.; Ichida, J.K.; Wainger, B.J.; Toma, J.S.; Rafuse, V.F.; Woolf, C.J.; Eggan, K. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 2011, 9, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takazawa, T.; Croft, G.F.; Amoroso, M.W.; Studer, L.; Wichterle, H.; Macdermott, A.B. Maturation of spinal motor neurons derived from human embryonic stem cells. PLoS ONE 2012, 7, e40154. [Google Scholar] [CrossRef]
- Kole, A.J.; Annis, R.P.; Deshmukh, M. Mature neurons: Equipped for survival. Cell Death Dis. 2013, 4, e689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Adil, M.M.; Gaj, T.; Rao, A.T.; Kulkarni, R.U.; Fuentes, C.M.; Ramadoss, G.N.; Ekman, F.K.; Miller, E.W.; Schaffer, D.V. hPSC-Derived Striatal Cells Generated Using a Scalable 3D Hydrogel Promote Recovery in a Huntington Disease Mouse Model. Stem Cell Rep. 2018, 10, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Lunyak, V.V.; Rosenfeld, M.G. Epigenetic regulation of stem cell fate. Hum. Mol. Genet. 2008, 17, R28–R36. [Google Scholar] [CrossRef]
- Qin, H.; Zhao, A.; Zhang, C.; Fu, X. Epigenetic Control of Reprogramming and Transdifferentiation by Histone Modifications. Stem Cell Rev. Rep. 2016, 12, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Eiraku, M.; Sasai, Y. Self-formation of layered neural structures in three-dimensional culture of ES cells. Curr. Opin. Neurobiol. 2012, 22, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Chiu, F.L.; Yeh, C.S.; Kuo, H.C. Opportunities and challenges for the use of induced pluripotent stem cells in modelling neurodegenerative disease. Open Biol. 2019, 9, 180177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepeda, C.; Wu, N.; André, V.M.; Cummings, D.M.; Levine, M.S. The corticostriatal pathway in Huntington’s disease. Prog. Neurobiol. 2007, 81, 253–271. [Google Scholar] [CrossRef] [Green Version]
- Miura, Y.; Li, M.Y.; Birey, F.; Ikeda, K.; Revah, O.; Thete, M.V.; Park, J.Y.; Puno, A.; Lee, S.H.; Porteus, M.H.; et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.J. The molecular chaperone concept. Semin. Biol. 1990, 1, 1–9. [Google Scholar]
- van Roon-Mom, W.; Reid, S.; Jones, A.L.; MacDonald, M.; Faull, R.; Snell, R. Insoluble TATA-binding protein accumulation in Huntington’s disease cortex. Brain Res. Mol. Brain Res. 2003, 109, 1–10. [Google Scholar] [CrossRef]
- Schaffar, G.; Breuer, P.; Boteva, R.; Behrends, C.; Tzvetkov, N.; Strippel, N.; Sakahira, H.; Siegers, K.; Hayer-Hartl, M.; Hartl, F.U. Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol. Cell 2004, 15, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Suhr, S.T.; Senut, M.C.; Whitelegge, J.P.; Faull, K.F.; Cuizon, D.B.; Gage, F.H. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J. Cell Biol. 2001, 153, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Study | HD Line | Differentiation | Huntington’s Disease Phenotype |
|---|---|---|---|
| Niclis et al. (2009) [6] | 37 CAG 51 CAG | Subpopulation TUJ1+, MAP2+, GFAP+ |
|
| Bradley et al. (2011) [8] | 40 CAG 45 CAG 46 CAG 48 CAG | Subpopulation MAP2+ |
|
| Feyeux et al. (2012) [20] | 40 CAG 44 CAG 44 CAG 46 CAG 48 CAG 51 CAG | Subpopulation MAP2+ |
|
| Niclis et al. (2013) [18] | 37 CAG 51 CAG | Subpopulation GABA+, TUJ1+ |
|
| Lu et al.(2013) [17] | 73 CAG * 145 CAG * | Subpopulation TUJ1+ |
|
| McQuade at al. (2014) [19] | 40 CAG 41 CAG 42 CAG 45 CAG 46 CAG 46 CAG 48 CAG | 50% MAP2+ <30% GAD65/67+ (normal CAG) >40% GAD65/67+ (HD CAG) |
|
| Ruzo et al. (2018) [16] | 45 CAG * 50 CAG * 58 CAG * 67 CAG * 74 CAG * | Subpopulation MAP2+, Nestin+ |
|
| Cohen-Carmon et al. (2020) [22] | 41 CAG 45 CAG 46 CAG 48 CAG | 40% GABAergic |
|
| Adult HD | Age of Onset | Method | Reprogramming Factors | Studies |
|---|---|---|---|---|
| 44/42 CAG | ≤59 years | Lentivirus | OCT4, SOX2, KLF4, cMYC | Camnasio et al. (2012) [24] |
| 43/39 CAG | ≤44 years | Lentivirus | OCT4, SOX2, KLF4, cMYC | Camnasio et al. (2012) [24] |
| 40 CAG | Not reported | Lentiviral | OCT4, SOX2, KLF4, cMYC | Nekrasov et al. (2016) [25] Nekrasov et al. (2018) [26] Vigont et al. (2018) [27] Vigont et al. (2021) [28] |
| 42 CAG | Not reported | Lentiviral | OCT4, SOX2, KLF4, cMYC | Nekrasov et al. (2016) [25] Nekrasov et al. (2018) [26] Vigont et al. (2018) [27] Vigont et al. (2021) [28] |
| Sendai virus | OCT4, SOX2, KLF4, cMYC | Mollica et al. (2018) [29] | ||
| 43 CAG | Not reported | Episomal | OCT4, SOX2, KLF4, cMYC, NANOG | Chiu et al. (2015) [30] |
| 44 CAG | Not reported | Sendai virus | OCT4, SOX2, KLF4, cMYC | Mollica et al. (2018) [29] |
| OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Machiela et al. (2020) [31] | |||
| 45 CAG | ≤36 years | Lentivirus | OCT4, SOX2, KLF4 | Camnasio et al. (2012) [24] |
| 46 CAG | Adult-onset | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Liu et al. (2017) [32] Smith-Geater et al. (2020) [33] |
| 47 CAG | Not reported | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, Trp53 | Malankhanova et al. (2020) [34] |
| Lentiviral | OCT4, SOX2, KLF4, cMYC | Nekrasov et al. (2016) [25] Nekrasov et al. (2018) [26] Vigont et al. (2018) [27] Vigont et al. (2021) [28] | ||
| Retroviral | OCT4, SOX2, KLF4, cMYC | Yao et al. (2015) [35] | ||
| 50 CAG | Not reported | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | The HD iPSC Consortium (2019) [36] |
| 53 CAG | Adult-onset | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Grima et al. (2017) [37] Smith-Geater et al. (2020) [33] |
| 57 CAG | ≤19 years | Episomal | OCT4, SOX2, KLF4, LMYC, p53 shRNA | Tidball et al. (2016) [38] |
| 26 years | OCT4, SOX2, KLF4, LMYC, LIN28 | Koyuncu et al. (2018) [39] | ||
| 58 CAG | ≤20 years | Episomal | OCT4, SOX2, KLF4, LMYC, p53 shRNA | Tidball et al. (2016) [38] Joshi et al. (2019) [40] |
| Juvenile HD | Age of Onset | Method | Reprogramming Factors | Studies |
| 60 CAG | 18 years | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Mattis et al. (2015) [41] Dickey et al. (2016) [42] The HD iPSC Consortium (2017) [43] Mathkar et al. (2019) [44] The HD iPSC Consortium (2019) [36] |
| Lentiviral | OCT4, SOX2, KLF4, LMYC, LIN28, NANOG | The HD iPSC Consortium (2012) [45] Martin-Flores et al. (2015) [46] Conforti et al. (2018) [47] | ||
| 66 CAG | ≤20 years | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | The HD iPSC Consortium (2019) [36] Smith-Geater et al. (2020) [33] Morozko et al. (2021) [48] |
| 69 CAG | Not reported | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Liu et al. (2017) [32] |
| 70 CAG | ≤50 years | Retroviral | OCT4, SOX2, KLF4, cMYC | Yao et al. (2015) [35] Cohen-Carmon et al. (2020) [22] |
| Not reported | Episomal | OCT4, SOX2, KLF4, LMYC | Tidball et al. (2015) [49] | |
| OCT4, SOX2, KLF4, LMYC, p53 shRNA | Tidball et al. (2016) [38] Joshi et al. (2019) [40] | |||
| OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Liu et al. (2017) [32] | |||
| 71 CAG | 14 years | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Mattis et al. (2015) [41] Szlachcic et al. (2015) [50] Koyuncu et al. (2018) [39] Switonska et al. (2019) [51] Mathkar et al. (2019) [44] Smith-Geater et al. (2020) [33] Machiela et al. (2020) [31] Morozko et al. (2021) [48] |
| 72 CAG | 14 years | Retroviral | OCT4, SOX2, KLF4, cMYC | Park et al. (2008) [52] Zhang et al. (2010) [53] An et al. (2012) [54] Chae et al. (2012) [55] Jeon et al. (2012) [56] Charbord et al. (2013) [57] Cheng et al. (2013) [58] Ring et al. (2015) [59] Naphade et al. (2018) [60] Lopes et al. (2020) [61] Le Cann et al. (2021) [62] |
| 76 CAG | ≤17 years | Sendai virus | OCT4, SOX2, KLF4, LMYC | Vigont et al. (2021) [28] |
| Lentiviral | OCT4, SOX2, KLF4, LMYC | Vigont et al. (2021) [28] | ||
| 77 CAG | Juvenile | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Mehta et al. (2018) [63] Mathkar et al. (2019) [44] |
| 99 CAG | Not reported | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Liu et al. (2017) [32] |
| 100 CAG * | 2 years | Lentiviral | OCT4, SOX2, KLF4, cMYC | Guo et al. (2013) [64] |
| 109 CAG | 3 years | Episomal | OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Lu et al. (2014) [65] Mattis et al. (2015) [41] Szlachcic et al. (2015) [50] Grima et al. (2017) [37] The HD iPSC Consortium (2017) [43] Mehta et al. (2018) [63] Switonska et al. (2019) [51] Mathkar et al. (2019) [44] The HD iPSC Consortium (2019) [36] Smith-Geater et al. (2020) [33] Machiela et al. (2020) [31] Morozko et al. (2021) [48] Le Cann et al. (2021) [62] |
| Lentiviral | OCT4, SOX2, KLF4, LMYC, LIN28, NANOG | The HD iPSC Consortium (2012) [45] Conforti et al. (2018) [47] Aron et al. (2018) [66] | ||
| 180 CAG | 6 years | Retroviral | OCT4, SOX2, KLF4, cMYC | Cohen-Carmon et al. (2020) [22] |
| Lentiviral | OCT4, SOX2, KLF4, LMYC, LIN28, NANOG | The HD iPSC Consortium (2012) [45] Ooi et al. (2015) [67] Xu et al. (2017) [68] Conforti et al. (2018) [47] | ||
| Episomal | OCT4, SOX2, KLF4, LMYC | Tidball et al. (2015) [49] | ||
| OCT4, SOX2, KLF4, LMYC, LIN28, p53 shRNA | Lu et al. (2014) [65] Mattis et al. (2015) [41] Mehta et al. (2017) [63] Mathkar et al. (2019) [44] Smith-Geater et al. (2020) [33] Le Cann et al. (2021) [62] | |||
| OCT4, SOX2, KLF4, LMYC, p53 shRNA | Tidball et al. (2016) [38] Koyuncu et al. (2018) [39] |
| Study | HD Line | Huntington’s Disease Phenotype |
|---|---|---|
| Zhang et al. (2010) [53] | 72 CAG |
|
| An et al. (2012) [54] | 72 CAG |
|
| Chae et al. (2012) [55] | 72 CAG |
|
| Camnasio et al. (2012) [24] | 44/42 CAG 43/39 CAG 45 CAG |
|
| Tidball et al. (2015) [49] | 70 CAG 180 CAG |
|
| Szlachcic et al. (2015) [50] | 71 CAG 109 CAG |
|
| Tidball et al. (2016) [38] | 57 CAG 58 CAG 70 CAG 180 CAG |
|
| Liu et al. (2017) [32] | 46 CAG 69 CAG 70 CAG 99 CAG |
|
| Mollica et al. (2018) [29] | 42 CAG 44 CAG |
|
| Switonska et al. (2018) [51] | 71 CAG 109 CAG |
|
| The HD iPSC Consortium (2019) [36] | 50 CAG 60 CAG 66 CAG 109 CAG |
|
| Joshi et al. (2019) [40] | 58 CAG 70 CAG |
|
| Lopes et al. (2020) [61] | 72 CAG |
|
| Study | HD Line | Markers | Huntington’s Disease Phenotype |
|---|---|---|---|
| Zhang et al. (2010) [53] | 72 CAG | Nestin, SOX1, PAX6 |
|
| An et al. (2012) [54] | 72 CAG | Nestin |
|
| The HD iPSC Consortium (2012) [45] | 60 CAG 109 CAG 180 CAG | Nestin, PAX6, SOX1, SOX2 |
|
| Camnasio et al. (2012) [24] | 44/42 CAG 43/39 CAG 45 CAG | Not reported |
|
| Jeon et al. (2012) [56] | 72 CAG | Nestin |
|
| Charbord et al. (2013) [57] | 72 CAG | Not reported |
|
| Tidball et al. (2015) [49] | 70 CAG 180 CAG | ISLET, PAX6, FOXG1 |
|
| Ring et al. (2015) [59] | 72 CAG | Nestin |
|
| Ooi et al. (2015) [67] | 180 CAG | Nestin |
|
| Martin-Flores et al. (2016) [46] | 60 CAG | Not reported |
|
| Xu et al. (2017) [68] | 180 CAG | Nestin |
|
| Liu et al. (2017) [32] | 46 CAG 69 CAG 70 CAG 99 CAG | Nestin, SOX2 |
|
| Naphade et al. (2017) [60] | 72 CAG | Nestin |
|
| Mollica et al. (2018) [29] | 42 CAG 44 CAG | Nestin, SOX2, SOX1, PAX6 |
|
| Conforti et al. (2018) [47] | 60 CAG 109 CAG 180 CAG | Nestin, SOX2 |
|
| The HD iPSC Consortium (2019) [36] | 50 CAG 60 CAG 66 CAG 109 CAG | Not reported |
|
| Joshi et al. (2019) [40] | 58 CAG 70 CAG | PAX6, SOX1 or ISLET1 |
|
| Lopes et al. (2020) [61] | 72 CAG | Nestin, SOX2 |
|
| Malankhanova et al. (2020) [34] | 47 CAG 69 CAG * | PAX6, SOX1, OTX2 |
|
| Study | HD Line | Differentiation | Huntington’s Disease Phenotype |
|---|---|---|---|
| The HD iPSC Consortium (2012) [45] | 60 CAG 109 CAG 180 CAG | <10% TUJ1+, <10% MAP2+, <5% DARPP32+/TUJ1+ |
|
| Chae et al. (2012) [55] | 72 CAG | 36% MAP2+ (HD) 51% MAP2+ (normal) |
|
| Camnasio et al. (2012) [24] | 44/42 CAG 43/39 CAG 45 CAG | 12–34% TUJ1+, subpopulation GABA+, GAD65/67+, MAP2+ |
|
| Cheng et al. (2013) [58] | 72 CAG | Subpopulation TUJ1+ |
|
| Guo et al. (2013) [64] | 100 CAG | Subpopulation DARPP32+, GAD67+, TUJ1+, MAP2+ |
|
| Lu et al. (2014) [65] | 109 CAG 180 CAG | 5% DARPP32+, 10% TUJ1+ |
|
| Chiu et al. (2015) [30] | 43 CAG | 70% GABA+, GAD65+, DARPP32+, Calbindin+ of TUJ1+, MAP2+ |
|
| Yao et al. (2015) [35] | 47 CAG 70 CAG | Subpopulation TUJ1+, DARPP32+, GABA+ |
|
| Mattis et al. (2015) [41] | 60 CAG 109 CAG 180 CAG | 5% TUJ1+ (normal) 4% DARPP32+ (normal) 20% TUJ1+ (HD) 1–4% DARPP32+ (HD) |
|
| Nekrasov et al. (2016) [25] | 40 CAG 42 CAG 47 CAG | 93% TUJ1+, 79% DARPP32+/TUJ1+ |
|
| Dickey et al. (2017) [42] | 60 CAG | Subpopulation TUJ1+, DARPP32+ |
|
| Grima et al. (2017) [37] | 53 CAG 109 CAG | Subpopulation MAP2+ |
|
| The HD iPSC Consortium (2017) [43] | 60 CAG 109 CAG | 27% TUJ1+, 15% MAP2+, 14% DARPP32+ |
|
| Xu et al. (2017) [68] | 180 CAG | Subpopulation GABA+, GAD65+, MAP2+ |
|
| Liu et al. (2017) [32] | 46 CAG 69 CAG 70 CAG 99 CAG | Subpopulation DARPP32+, GABA+, TUJ1+ |
|
| Conforti et al. (2018) [47] | 60 CAG 109 CAG 180 CAG | Subpopulation DARPP32+, CTIP2+, TUJ1+, MAP2+ |
|
| Mehta et al. (2018) [63] | 77 CAG 109 CAG 180 CAG | 25–30% MAP2+, TUJ1+ 20% CTIP2+, TBR1+ of MAP2+, TUJ1+ |
|
| Nekrasov et al. (2018) [26] | 40 CAG 42 CAG 47 CAG | Subpopulation TUJ1+, striatal differentiation protocol |
|
| Vigont et al. (2018) [27] | 40 CAG 42 CAG 47 CAG | Subpopulation GABA+ |
|
| The HD iPSC Consortium (2019) [36] | 50 CAG 60 CAG 66 CAG 109 CAG | 90% MAP2+, 20–30% DARPP32+/MAP2+, 60–80% CTIP2+/MAP2+ |
|
| Mathkar et al. (2019) [44] | 60 CAG 71 CAG 77 CAG 109 CAG 180 CAG | 10–20% TUJ1+, MAP2+ |
|
| Joshi et al. (2019) [40] | 58 CAG 70 CAG | Subpopulation TUJ1+, Glutamate+, GFAP+, TH+ |
|
| Cohen-Carmon et al. (2020) [22] | 70 CAG 180 CAG | 40% GABAergic |
|
| Smith-Geater et al. (2020) [33] | 46 CAG 53 CAG 66 CAG 71 CAG 109 CAG 180 CAG | 79–98% MAP2+, 4–28% DARPP32+ |
|
| Malankhanova et al. (2020) [34] | 47 CAG 69 CAG * | Subpopulation of DARPP32+, GABA+, TUJ1+ |
|
| Machiela et al. (2020) [31] | 43 CAG 71 CAG 109 CAG | Subpopulation TUJ1+, striatal differentiation protocol |
|
| Le Cann et al. (2021) [62] | 72 CAG 109 CAG 180 CAG | 0–64% DARPP32+/GAD+ 0–40% GAD67+, 9–40% TUJ1+ |
|
| Morozko et al. (2021) [48] | 66 CAG 71 CAG 109 CAG | Subpopulation DARPP32+, CTIP2+, MAP2+ |
|
| Vigont et al. (2021) [28] | 40 CAG 42 CAG 47 CAG 76 CAG | 80% DARPP32, ≤100% MAP2+ |
|
| Study | HD Line | Differentiation | Huntington’s Disease Phenotype |
|---|---|---|---|
| Liu et al. (2014) [111] | 68 CAG (onset 14 years) 86 CAG * (onset 2 years) | 8% TUJ1+ (normal) <40% DARPP32+ (normal) 8% TUJ1+ (HD) 60% DARPP32+ (HD) |
|
| Drouin- Ouellet et al. (2017) [109] | 41 CAG (onset ≤ 58 years) | 25–62% MAP2+ |
|
| Victor et al. (2018) [110] | 40 CAG (onset 63 years) 42 CAG (onset ≤ 51 years) 43 CAG (onset 50 years) 44 CAG (onset ≤ 52 years) 46 CAG (onset ≤ 60 years) 47 CAG (onset ≤ 63 years) 50 CAG (onset 27 years) | 90% MAP2+, 70–80% GABA+, 70–80% DARPP32+ |
|
| Study | HD Line | Differentiation | Huntington’s Disease Phenotype |
|---|---|---|---|
| Hou et al. (2017) [133] | 41 CAG (onset not reported) 41 CAG (onset not reported) | Subpopulation TUJ1+, GFAP+, GALC+ |
|
| Monk et al. (2021) [134] | 41 CAG (onset 56 years) 43 CAG (onset 50 years) 44 CAG (onset 33 years) 57 CAG (onset 27 years) | 20% TUJ1+ (normal) 12% TUJ1+ (HD) 69% DARPP32+/TUJ1+ (normal) 54% DARPP32+/TUJ1+ (HD) |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monk, R.; Connor, B. Cell Reprogramming to Model Huntington’s Disease: A Comprehensive Review. Cells 2021, 10, 1565. https://doi.org/10.3390/cells10071565
Monk R, Connor B. Cell Reprogramming to Model Huntington’s Disease: A Comprehensive Review. Cells. 2021; 10(7):1565. https://doi.org/10.3390/cells10071565
Chicago/Turabian StyleMonk, Ruth, and Bronwen Connor. 2021. "Cell Reprogramming to Model Huntington’s Disease: A Comprehensive Review" Cells 10, no. 7: 1565. https://doi.org/10.3390/cells10071565
APA StyleMonk, R., & Connor, B. (2021). Cell Reprogramming to Model Huntington’s Disease: A Comprehensive Review. Cells, 10(7), 1565. https://doi.org/10.3390/cells10071565