
Transcriptome and Degradome Profiling Reveals a Role of miR530 in the Circadian Regulation of Gene Expression in Kalanchoë marnieriana

 , , ,
, , ,  and
and 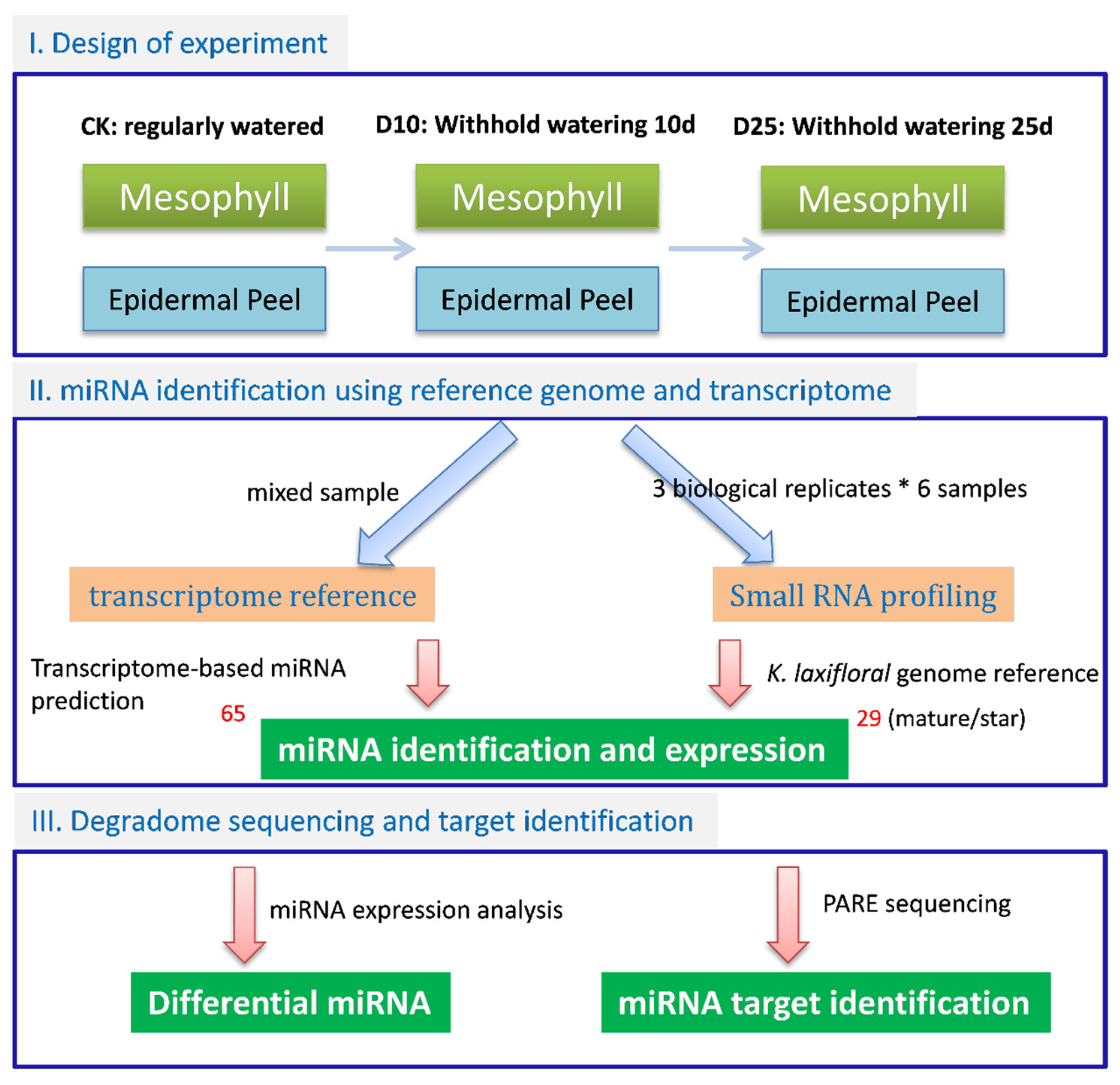{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Preparation
2.3. RNA Sequencing and Analysis
2.4. Dual-Fluorescence Assay
2.5. Real-Time Quantification of Gene Expression
3. Results
3.1. K. marnieriana Is an Obligate CAM Plant and Its CAM Expression Is Enhanced by Drought Treatment
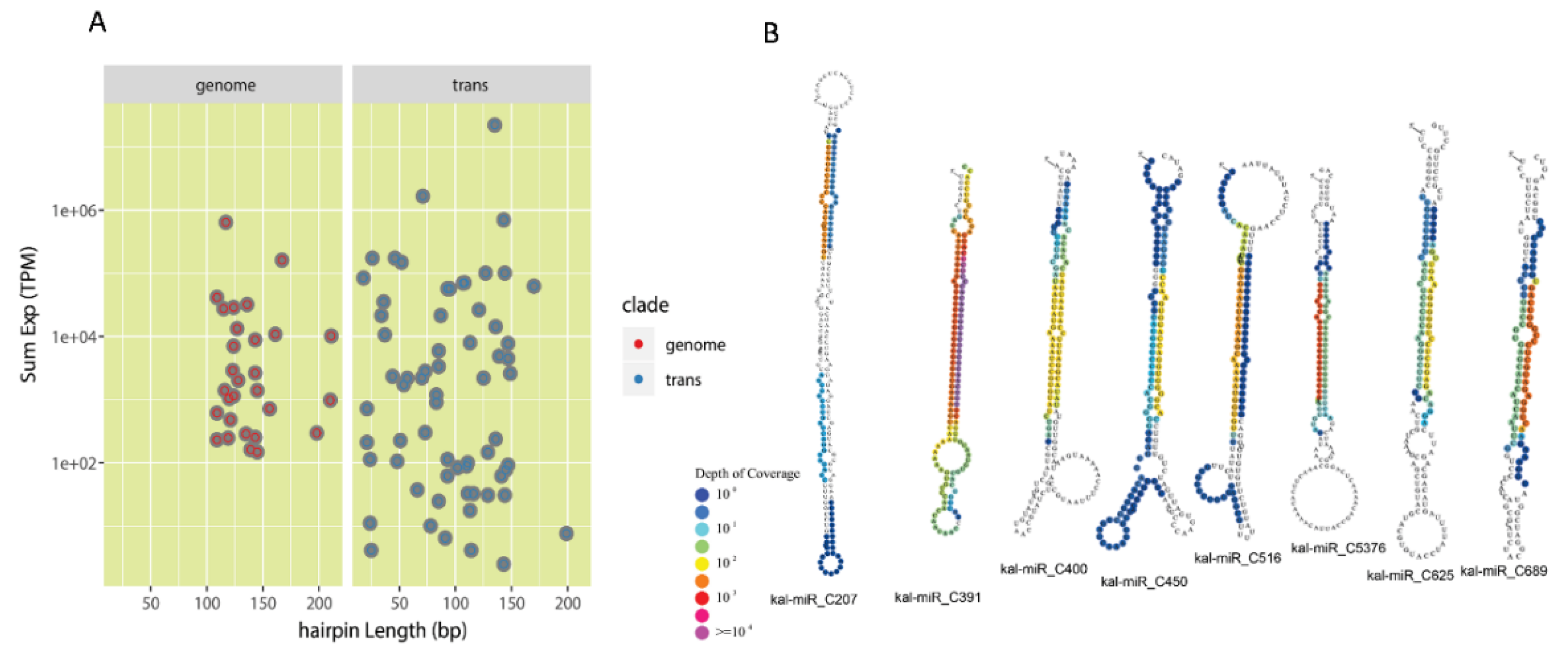
3.2. Identification of Conserved miRNAs and Lineage-Specific miRNAs in K. marnieriana
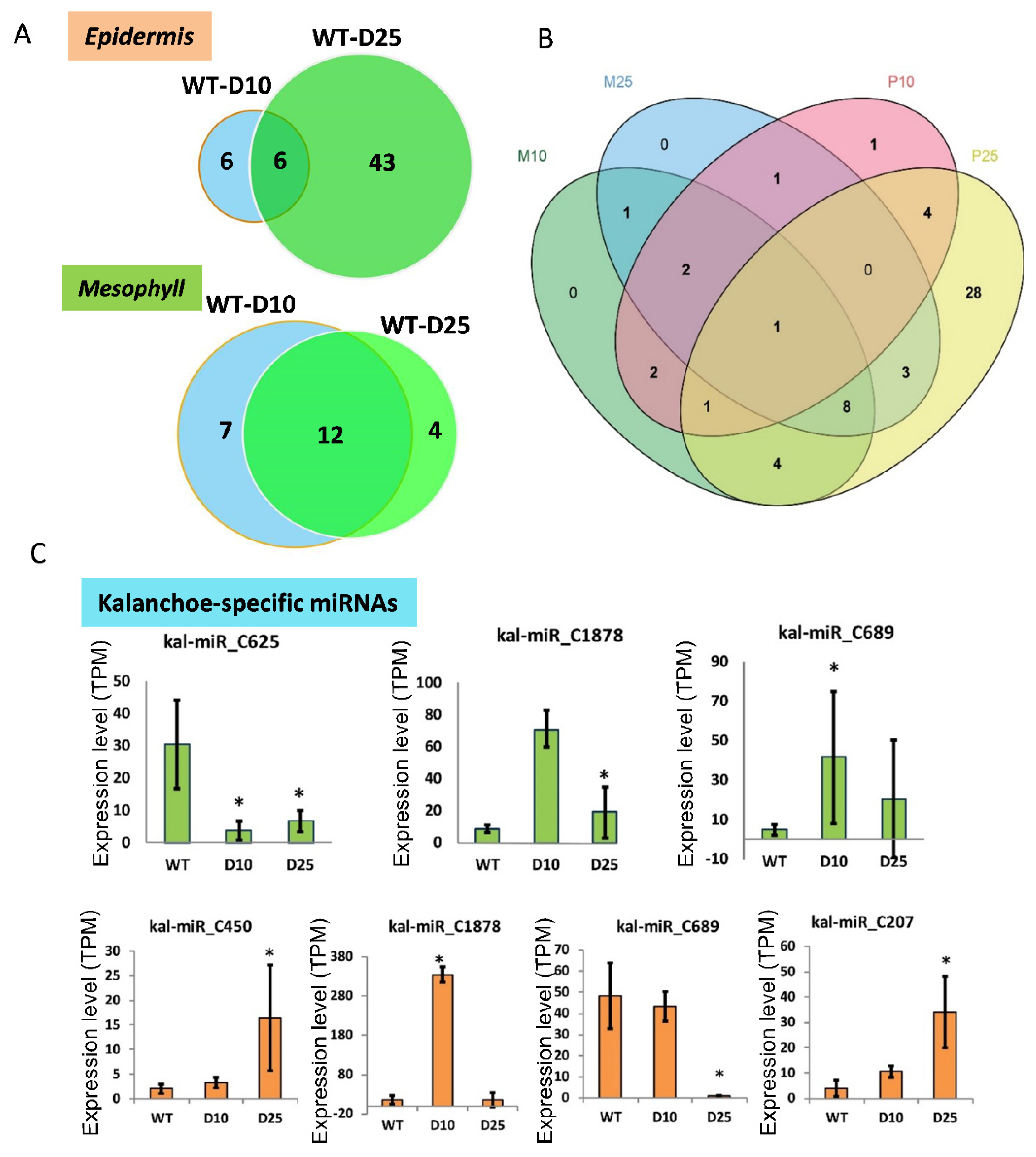
3.3. Differential Expression of miRNAs in Responsive to Drought Stress in Mesophyll and Epidermis
3.4. Diurnal Expression of the Targets of miRNAs in K. marnieriana Leaves
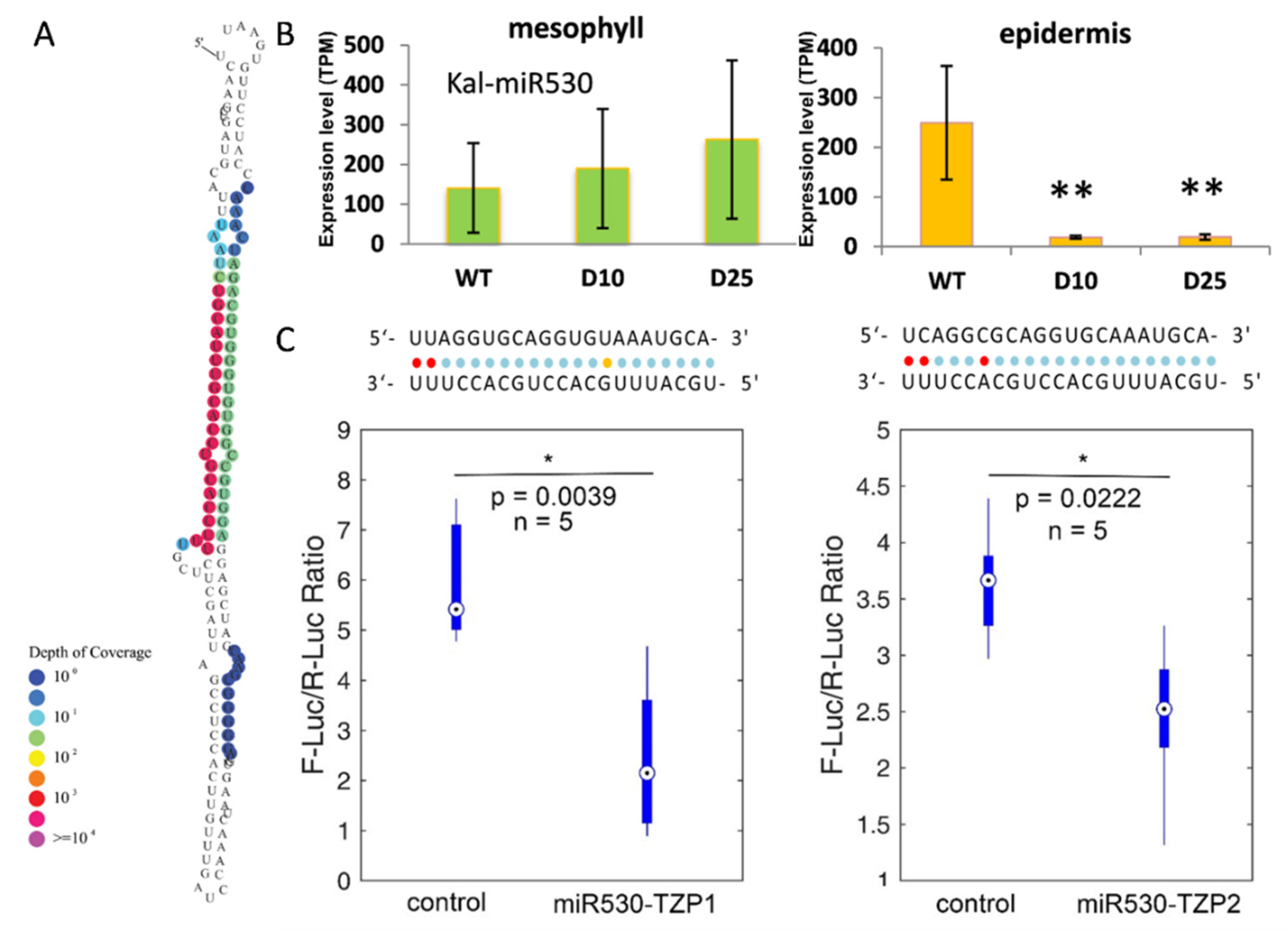
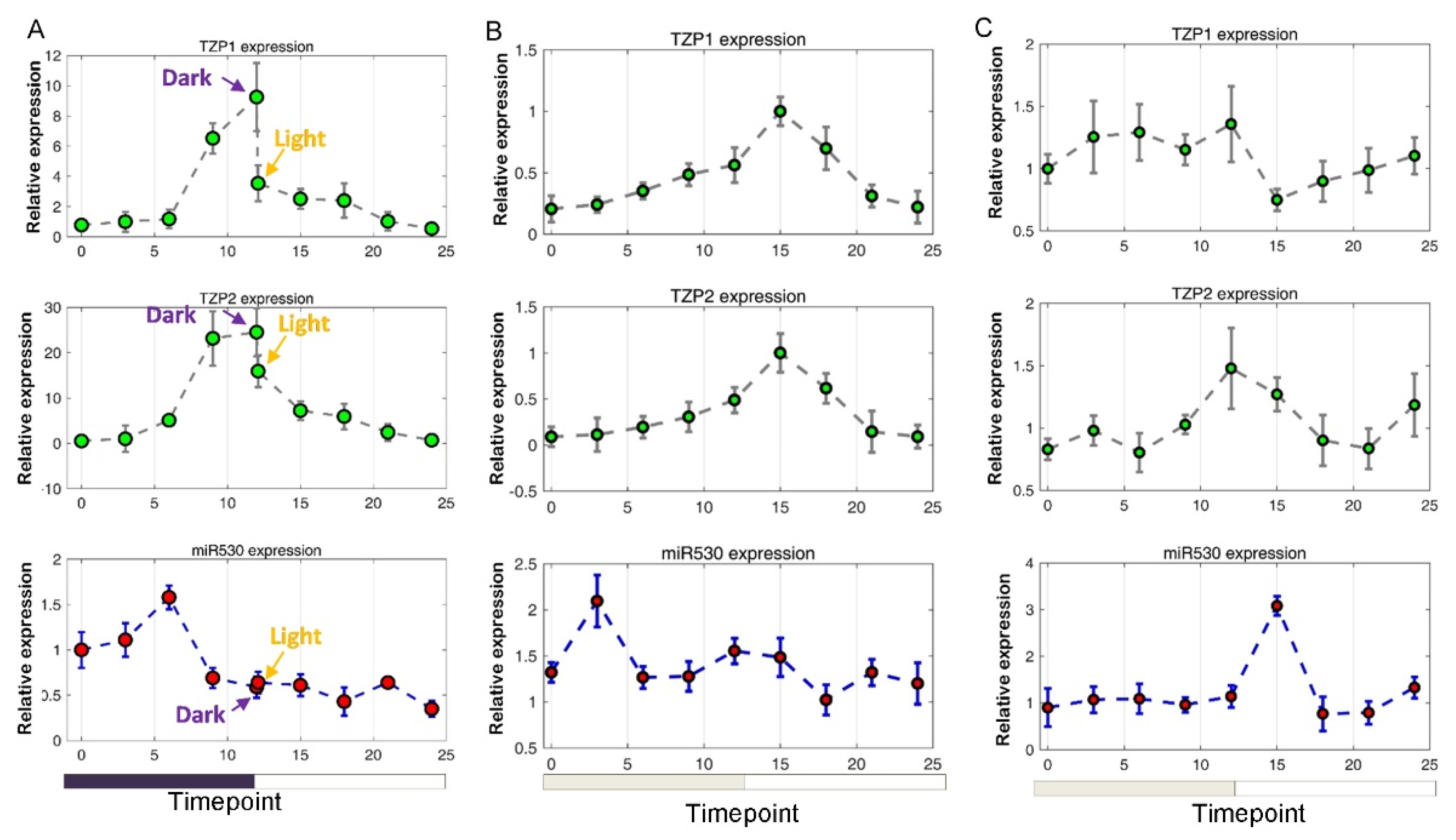
3.5. The Expression of miR530 and TZP Genes Is Affected by Circadian Clock and Light Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cushman, J.C.; Bohnert, H.J. Crassulacean acid metabolism: Molecular genetics. Annu. Rev. Plant Biol. 1999, 50, 305–332. [Google Scholar] [CrossRef] [PubMed]
- Cushman, J.C.; Bohnert, H.J. Molecular genetics of crassulacean acid metabolism. Plant Physiol. 1997, 113, 667–676. [Google Scholar] [CrossRef]
- Cushman, J.C. Crassulacean acid metabolism. A plastic photosynthetic adaptation to arid environments. Plant Physiol. 2001, 127, 1439–1448. [Google Scholar] [CrossRef]
- Cushman, J.; Borland, A. Induction of Crassulacean acid metabolism by water limitation. Plant Cell Environ. 2002, 25, 295–310. [Google Scholar] [CrossRef]
- Dodd, A.N.; Borland, A.M.; Haslam, R.P.; Griffiths, H.; Maxwell, K. Crassulacean acid metabolism: Plastic, fantastic. J. Exp. Bot. 2002, 53, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Silvera, K.; Neubig, K.M.; Whitten, W.M.; Williams, N.H.; Winter, K.; Cushman, J.C. Evolution along the crassulacean acid metabolism continuum. Funct. Plant Biol. 2010, 37, 995–1010. [Google Scholar] [CrossRef]
- Winter, K.; Holtum, J.A.; Smith, J.A.C. Crassulacean acid metabolism: A continuous or discrete trait? New Phytol. 2015, 208, 73–78. [Google Scholar] [CrossRef]
- Heyduk, K.; Moreno-Villena, J.J.; Gilman, I.S.; Christin, P.-A.; Edwards, E.J. The genetics of convergent evolution: Insights from plant photosynthesis. Nat. Rev. Genet. 2019, 20, 485–493. [Google Scholar] [CrossRef]
- Bräutigam, A.; Schlüter, U.; Eisenhut, M.; Gowik, U. On the evolutionary origin of CAM photosynthesis. Plant Physiol. 2017, 174, 473–477. [Google Scholar] [CrossRef]
- Borland, A.M.; Hartwell, J.; Weston, D.J.; Schlauch, K.A.; Tschaplinski, T.J.; Tuskan, G.A.; Yang, X.; Cushman, J.C. Engineering crassulacean acid metabolism to improve water-use efficiency. Trends Plant Sci. 2014, 19, 327–338. [Google Scholar] [CrossRef]
- Borland, A.M.; Wullschleger, S.D.; Weston, D.J.; Hartwell, J.; Tuskan, G.A.; Yang, X.; Cushman, J.C. Climate-resilient agroforestry: Physiological responses to climate change and engineering of crassulacean acid metabolism (CAM) as a mitigation strategy. Plant Cell Environ. 2015, 38, 1833–1849. [Google Scholar] [CrossRef]
- Yang, X.; Cushman, J.C.; Borland, A.M.; Edwards, E.J.; Wullschleger, S.D.; Tuskan, G.A.; Owen, N.A.; Griffiths, H.; Smith, J.A.C.; De Paolim, H.C. A roadmap for research on crassulacean acid metabolism (CAM) to enhance sustainable food and bioenergy production in a hotter, drier world. New Phytol. 2015, 207, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Medford, J.I.; Markel, K.; Shih, P.M.; De Paoli, H.C.; Trinh, C.T.; McCormick, A.J.; Ployet, R.; Hussey, S.G.; Myburg, A.A.; et al. Plant biosystems design research roadmap 1.0. BioDes. Res. 2020, 2020, 8051764. [Google Scholar] [CrossRef]
- Yuan, G.; Hassan, M.M.; Liu, D.; Lim, S.D.; Yim, W.C.; Cushman, J.C.; Markel, K.; Shih, P.M.; Lu, H.; Weston, D.J.; et al. Biosystems design to accelerate C3-to-CAM progression. BioDes. Res. 2020, 2020, 3686791. [Google Scholar] [CrossRef]
- Davis, S.C.; Simpson, J.; Gil-Vega, K.d.C.; Niechayev, N.A.; Tongerlo, E.v.; Castano, N.H.; Dever, L.V.; Búrquez, A. Undervalued potential of crassulacean acid metabolism for current and future agricultural production. J. Exp. Bot. 2019, 70, 6521–6537. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Palla, K.J.; Hu, R.; Moseley, R.C.; Mendoza, C.; Chen, M.; Abraham, P.E.; Labbé, J.L.; Kalluri, U.C.; Tschaplinski, T.J. Perspectives on the basic and applied aspects of crassulacean acid metabolism (CAM) research. Plant Sci. 2018, 274, 394–401. [Google Scholar] [CrossRef]
- Hartwell, J.; Dever, L.V.; Boxall, S.F. Emerging model systems for functional genomics analysis of crassulacean acid metabolism. Curr. Opin. Plant Biol. 2016, 31, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Abraham, P.E.; Yin, H.; Borland, A.M.; Weighill, D.; Lim, S.D.; De Paoli, H.C.; Engle, N.; Jones, P.C.; Agh, R.; Weston, D.J. Transcript, protein and metabolite temporal dynamics in the CAM plant Agave. Nat. Plants 2016, 2, 1–10. [Google Scholar] [CrossRef]
- Yang, X.; Hu, R.; Yin, H.; Jenkins, J.; Shu, S.; Tang, H.; Liu, D.; Weighill, D.A.; Yim, W.C.; Ha, J. The Kalanchoë genome provides insights into convergent evolution and building blocks of crassulacean acid metabolism. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Yin, H.; Guo, H.-B.; Weston, D.J.; Borland, A.M.; Ranjan, P.; Abraham, P.E.; Jawdy, S.S.; Wachira, J.; Tuskan, G.A.; Tschaplinski, T.J. Diel rewiring and positive selection of ancient plant proteins enabled evolution of CAM photosynthesis in Agave. BMC Genom. 2018, 19, 588. [Google Scholar] [CrossRef]
- Moseley, R.C.; Mewalal, R.; Motta, F.; Tuskan, G.A.; Haase, S.; Yang, X. Conservation and diversification of circadian rhythmicity between a model crassulacean acid metabolism plant Kalanchoë fedtschenkoi and a model C3 photosynthesis plant Arabidopsis thaliana. Front. Plant Sci. 2018, 9, 1757. [Google Scholar] [CrossRef]
- Liu, D.; Chen, M.; Mendoza, B.; Cheng, H.; Hu, R.; Li, L.; Trinh, C.T.; Tuskan, G.A.; Yang, X. CRISPR/Cas9-mediated targeted mutagenesis for functional genomics research of crassulacean acid metabolism plants. J. Exp. Bot. 2019, 70, 6621–6629. [Google Scholar] [CrossRef] [PubMed]
- Boxall, S.F.; Dever, L.V.; Kneřová, J.; Gould, P.D.; Hartwell, J. Phosphorylation of phosphoenolpyruvate carboxylase is essential for maximal and sustained dark CO2 fixation and core circadian clock operation in the obligate crassulacean acid metabolism species Kalanchoë fedtschenkoi. Plant. Cell 2017, 29, 2519–2536. [Google Scholar] [CrossRef]
- Dever, L.V.; Boxall, S.F.; Kneřová, J.; Hartwell, J. Transgenic perturbation of the decarboxylation phase of Crassulacean acid metabolism alters physiology and metabolism but has only a small effect on growth. Plant Physiol. 2015, 167, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Carrington, J.C.; Ambros, V. Role of microRNAs in plant and animal development. Science 2003, 301, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Sunkar, R.; Li, Y.-F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef]
- Zhang, B. MicroRNA: A new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 2015, 66, 1749–1761. [Google Scholar] [CrossRef]
- Siré, C.; Moreno, A.B.; Garcia-Chapa, M.; López-Moya, J.J.; San Segundo, B. Diurnal oscillation in the accumulation of Arabidopsis microRNAs, miR167, miR168, miR171 and miR398. FEBS Lett. 2009, 583, 1039–1044. [Google Scholar] [CrossRef]
- Wai, C.M.; VanBuren, R.; Zhang, J.; Huang, L.; Miao, W.; Edger, P.P.; Yim, W.C.; Priest, H.D.; Meyers, B.C.; Mockler, T. Temporal and spatial transcriptomic and micro RNA dynamics of CAM photosynthesis in pineapple. Plant. J. 2017, 92, 19–30. [Google Scholar] [CrossRef]
- Liu, Q.; Axtell, M.J. Quantitating plant microRNA-mediated target repression using a dual-luciferase transient expression system. In Plant Functional Genomics; Springer: Berlin, Germany, 2015; pp. 287–303. [Google Scholar]
- Han, X.; Yin, H.; Song, X.; Zhang, Y.; Liu, M.; Sang, J.; Jiang, J.; Li, J.; Zhuo, R. Integration of small RNA s, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol. J. 2016, 14, 1470–1483. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Fan, Z.; Li, X.; Wang, J.; Liu, W.; Wu, B.; Ying, Z.; Liu, L.; Liu, Z.; Li, J. Phylogenetic tree-informed microRNAome analysis uncovers conserved and lineage-specific miRNAs in Camellia during floral organ development. J. Exp. Bot. 2016, 67, 2641–2653. [Google Scholar] [CrossRef]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. Rna 2013, 19, 740–751. [Google Scholar] [CrossRef]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, F.; Axtell, M.J. Analysis of complementarity requirements for plant microRNA targeting using a Nicotiana benthamiana quantitative transient assay. Plant Cell 2014, 26, 741–753. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, H.; Gaußmann, O.; Marx, H.; Schwarzott, D.; Kluge, M. Molecular phylogeny of the genus Kalanchoe (Crassulaceae) inferred from nucleotide sequences of the ITS-1 and ITS-2 regions. Plant Sci. 2001, 160, 827–835. [Google Scholar] [CrossRef]
- Loudet, O.; Michael, T.P.; Burger, B.T.; Le Metté, C.; Mockler, T.C.; Weigel, D.; Chory, J. A zinc knuckle protein that negatively controls morning-specific growth in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2008, 105, 17193–17198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, C.; Zhou, Y.; Wang, X.; Li, H.; Feng, Z.; Chen, H.; Qin, G.; Jin, D.; Terzaghi, W. TANDEM ZINC-FINGER/PLUS3 is a key component of phytochrome A signaling. Plant Cell 2018, 30, 835–852. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.E.; Hogenesch, J.B.; Kornacker, K. JTK_CYCLE: An Efficient Nonparametric Algorithm for Detecting Rhythmic Components in Genome-Scale Data Sets. J. Biol. Rhythm. 2010, 25, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and their regulatory roles in plant–environment interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef]
- Imaizumi, T. Arabidopsis circadian clock and photoperiodism: Time to think about location. Curr. Opin. Plant Biol. 2010, 13, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Kaiserli, E.; Paldi, K.; O’Donnell, L.; Batalov, O.; Pedmale, U.V.; Nusinow, D.A.; Kay, S.A.; Chory, J. Integration of light and photoperiodic signaling in transcriptional nuclear foci. Dev. Cell 2015, 35, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xu, X.H.; Li, Y.; Xie, L.; He, Y.; Li, W.; Lu, X.; Sun, H.; Xie, X. OsmiR530 acts downstream of OsPIL15 to regulate grain yield in rice. New Phytol. 2020, 226, 823. [Google Scholar] [CrossRef] [PubMed]
- Boxall, S.F.; Foster, J.M.; Bohnert, H.J.; Cushman, J.C.; Nimmo, H.G.; Hartwell, J. Conservation and divergence of circadian clock operation in a stress-inducible Crassulacean acid metabolism species reveals clock compensation against stress. Plant Physiol. 2005, 137, 969–982. [Google Scholar] [CrossRef]
- Boxall, S.F.; Kadu, N.; Dever, L.V.; Kneřová, J.; Waller, J.L.; Gould, P.J.; Hartwell, J. Kalanchoë PPC1 is essential for crassulacean acid metabolism and the regulation of core circadian clock and guard cell signaling genes. Plant Cell 2020, 32, 1136–1160. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.H.; Barkla, B.J.; Vera-Estrella, R.; Pantoja, O.; Lee, S.Y.; Bohnert, H.J.; Dassanayake, M. Cell type-specific responses to salinity–the epidermal bladder cell transcriptome of Mesembryanthemum crystallinum. New Phytol. 2015, 207, 627–644. [Google Scholar] [CrossRef]
- Cushman, J.C.; Tillett, R.L.; Wood, J.A.; Branco, J.M.; Schlauch, K.A. Large-scale mRNA expression profiling in the common ice plant, Mesembryanthemum crystallinum, performing C3 photosynthesis and Crassulacean acid metabolism (CAM). J. Exp. Bot. 2008, 59, 1875–1894. [Google Scholar] [CrossRef]
- Mallona, I.; Egea-Cortines, M.; Weiss, J. Conserved and divergent rhythms of crassulacean acid metabolism-related and core clock gene expression in the cactus Opuntia ficus-indica. Plant Physiol. 2011, 156, 1978–1989. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Nie, Z.; Yan, C.; Huang, H.; Ma, X.; Wang, Y.; Ye, N.; Tuskan, G.A.; Yang, X.; Yin, H. Transcriptome and Degradome Profiling Reveals a Role of miR530 in the Circadian Regulation of Gene Expression in Kalanchoë marnieriana. Cells 2021, 10, 1526. https://doi.org/10.3390/cells10061526
Hu Z, Nie Z, Yan C, Huang H, Ma X, Wang Y, Ye N, Tuskan GA, Yang X, Yin H. Transcriptome and Degradome Profiling Reveals a Role of miR530 in the Circadian Regulation of Gene Expression in Kalanchoë marnieriana. Cells. 2021; 10(6):1526. https://doi.org/10.3390/cells10061526
Chicago/Turabian StyleHu, Zhikang, Ziyan Nie, Chao Yan, Hu Huang, Xianjin Ma, Yupeng Wang, Ning Ye, Gerald A. Tuskan, Xiaohan Yang, and Hengfu Yin. 2021. "Transcriptome and Degradome Profiling Reveals a Role of miR530 in the Circadian Regulation of Gene Expression in Kalanchoë marnieriana" Cells 10, no. 6: 1526. https://doi.org/10.3390/cells10061526
APA StyleHu, Z., Nie, Z., Yan, C., Huang, H., Ma, X., Wang, Y., Ye, N., Tuskan, G. A., Yang, X., & Yin, H. (2021). Transcriptome and Degradome Profiling Reveals a Role of miR530 in the Circadian Regulation of Gene Expression in Kalanchoë marnieriana. Cells, 10(6), 1526. https://doi.org/10.3390/cells10061526