
Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The TME
Immunosuppressive and Inflammatory Properties of the TME
3. Phospholipases: Classification and General Properties
PLA2 in TME
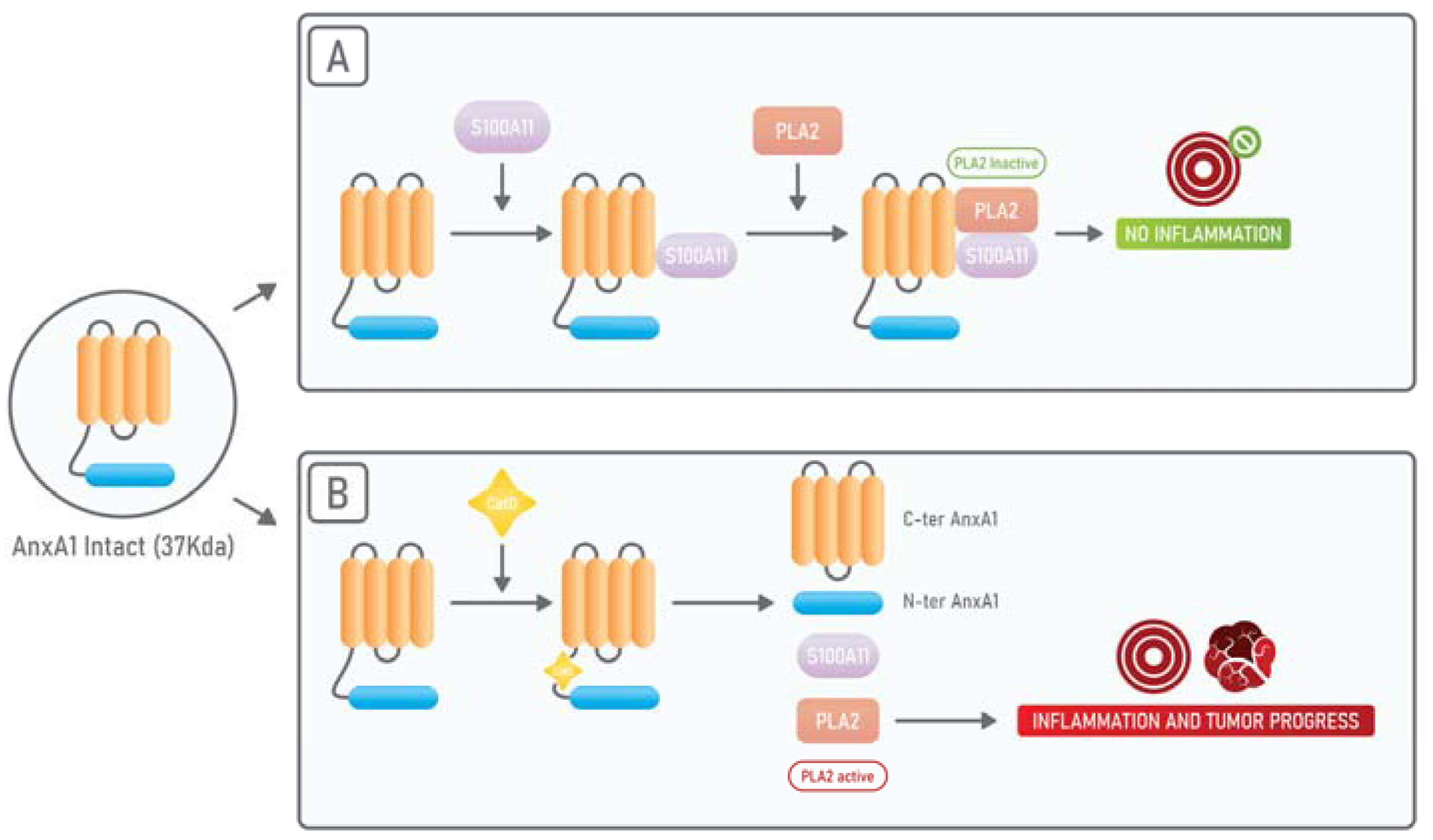
4. Annexin A1: An Endogenous PLA2 Inhibitor
AnxA1 in TME
5. PLA2 and Annexin A1 in Cancer-Derived Extracellular Vesicles
6. Use of PLA2 Inhibitors to Control Cancer Progression
7. Final Considerations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Breast Cancer: Prevention and Control. 2018. Available online: https://www.who.int/cancer/detection/breastcancer/en/ (accessed on 23 April 2021).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharm. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Cleary, A.S.; Leonard, T.L.; Gestl, S.A.; Gunther, E.J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014, 508, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Rodríguez-Ubreva, J.; Ballestar, E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin. Immunol. 2018, 196, 64–71. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Mohan, V.; Das, A.; Sagi, I. Emerging roles of ECM remodeling processes in cancer. Semin. Cancer Biol. 2020, 62, 192–200. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nistico, P.; Signore, A. Relevance of immune cell and tumor microenvironment imaging in the new era of immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 89. [Google Scholar] [CrossRef]
- Zhang, Y.; Ertl, H.C. Starved and Asphyxiated: How Can CD8(+) T Cells within a Tumor Microenvironment Prevent Tumor Progression. Front. Immunol. 2016, 7, 32. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Johnson, A.M.; Kleczko, E.K.; Nemenoff, R.A. Eicosanoids in Cancer: New Roles in Immunoregulation. Front. Pharm. 2020, 11, 595498. [Google Scholar] [CrossRef] [PubMed]
- Corn, K.C.; Windham, M.A.; Rafat, M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog. Lipid Res. 2020, 80, 101055. [Google Scholar] [CrossRef]
- Tang, S.; Ning, Q.; Yang, L.; Mo, Z.; Tang, S. Mechanisms of immune escape in the cancer immune cycle. Int. Immunopharmacol. 2020, 86, 106700. [Google Scholar] [CrossRef]
- Bretscher, P. On Analyzing How the Th1/Th2 Phenotype of an Immune Response Is Determined: Classical Observations Must Not Be Ignored. Front. Immunol. 2019, 10, 1024. [Google Scholar] [CrossRef] [PubMed]
- Zambrano-Zaragoza, J.F.; Romo-Martinez, E.J.; Duran-Avelar Mde, J.; Garcia-Magallanes, N.; Vibanco-Perez, N. Th17 cells in autoimmune and infectious diseases. Int. J. Inflam. 2014, 2014, 651503. [Google Scholar] [CrossRef]
- Kryczek, I.; Wei, S.; Szeliga, W.; Vatan, L.; Zou, W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood 2009, 114, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Restifo, N.P. T(H)17 cells in tumour immunity and immunotherapy. Nat. Rev. Immunol. 2010, 10, 248–256. [Google Scholar] [CrossRef]
- Williams, M.A.; Bevan, M.J. Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007, 25, 171–192. [Google Scholar] [CrossRef] [PubMed]
- Jost, S.; Altfeld, M. Control of human viral infections by natural killer cells. Annu. Rev. Immunol. 2013, 31, 163–194. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zheng, S.G. Hall of Fame among Pro-inflammatory Cytokines: Interleukin-6 Gene and Its Transcriptional Regulation Mechanisms. Front. Immunol. 2016, 7, 604. [Google Scholar] [CrossRef]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J. Cancer immunotherapy by dendritic cells. Immunity 2008, 29, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Budhu, S.; Schaer, D.A.; Li, Y.; Toledo-Crow, R.; Panageas, K.; Yang, X.; Zhong, H.; Houghton, A.N.; Silverstein, S.C.; Merghoub, T.; et al. Blockade of surface-bound TGF-beta on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hanabuchi, S.; Perng, O.A.; Gilliet, M.; Qin, F.X.; Liu, Y.J. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13138–13143. [Google Scholar] [CrossRef]
- Weber, F.; Byrne, S.N.; Le, S.; Brown, A.; Breit, S.N.; Scolyer, R.A.; Halliday, G.M. Transforming growth factor-beta1 immobilises dendritic cells within skin tumours and facilitates tumour escape from the immune system. Cancer Immunol. Immunother. 2005, 54, 898–906. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Lin, Y.C.; Yao, P.L.; Yuan, A.; Chen, H.Y.; Shun, C.T.; Tsai, M.F.; Chen, C.H.; Yang, P.C. Tumor-associated macrophages: The double-edged sword in cancer progression. J. Clin. Oncol. 2005, 23, 953–964. [Google Scholar] [CrossRef]
- Sica, A.; Allavena, P.; Mantovani, A. Cancer related inflammation: The macrophage connection. Cancer Lett. 2008, 267, 204–215. [Google Scholar] [CrossRef]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis. Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Birner, P.; Stockl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef]
- Iijima, J.; Konno, K.; Itano, N. Inflammatory alterations of the extracellular matrix in the tumor microenvironment. Cancers 2011, 3, 3189–3205. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 2009, 114, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, Y.; Odunsi, K.; Chen, W.; Peng, G.; Matsuzaki, J.; Wang, R.F. Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 15505–15510. [Google Scholar] [CrossRef]
- Guery, L.; Hugues, S. Th17 Cell Plasticity and Functions in Cancer Immunity. BioMed Res. Int. 2015, 2015, 314620. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 2006, 176, 284–290. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- Zippelius, A.; Batard, P.; Rubio-Godoy, V.; Bioley, G.; Lienard, D.; Lejeune, F.; Rimoldi, D.; Guillaume, P.; Meidenbauer, N.; Mackensen, A.; et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: A state of local functional tolerance. Cancer Res. 2004, 64, 2865–2873. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Driessens, G.; Kline, J.; Gajewski, T.F. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol. Rev. 2009, 229, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Murata, H.; Sonegawa, H.; Sakaguchi, Y.; Futami, J.; Kitazoe, M.; Yamada, H.; Huh, N.H. Truncation of annexin A1 is a regulatory lever for linking epidermal growth factor signaling with cytosolic phospholipase A2 in normal and malignant squamous epithelial cells. J. Biol. Chem. 2007, 282, 35679–35686. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A. Introduction to Thematic Review Series: Phospholipases: Central Role in Lipid Signaling and Disease. J. Lipid Res. 2015, 56, 1245–1247. [Google Scholar] [CrossRef] [PubMed]
- Wilton, D.C. CHAPTER 11—Phospholipases. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed.; Vance, D.E., Vance, J.E., Eds.; Elsevier: San Diego, CA, USA, 2008; pp. 305–329. [Google Scholar]
- Aloulou, A.; Rahier, R.; Arhab, Y.; Noiriel, A.; Abousalham, A. Phospholipases: An Overview. Methods Mol. Biol. 2018, 1835, 69–105. [Google Scholar]
- Brown, W.J.; Chambers, K.; Doody, A. Phospholipase A2 (PLA2) enzymes in membrane trafficking: Mediators of membrane shape and function. Traffic 2003, 4, 214–221. [Google Scholar] [CrossRef]
- Azevedo, F.V.; Lopes, D.S.; Cirilo Gimenes, S.N.; Ache, D.C.; Vecchi, L.; Alves, P.T.; Guimaraes Dde, O.; Rodrigues, R.S.; Goulart, L.R.; Rodrigues, V.d.M.; et al. Human breast cancer cell death induced by BnSP-6, a Lys-49 PLA(2) homologue from Bothrops pauloensis venom. Int. J. Biol. Macromol. 2016, 82, 671–677. [Google Scholar] [CrossRef] [PubMed]
- de Vasconcelos Azevedo, F.V.P.; Zoia, M.A.P.; Lopes, D.S.; Gimenes, S.N.; Vecchi, L.; Alves, P.T.; Rodrigues, R.S.; Silva, A.C.A.; Yoneyama, K.A.G.; Goulart, L.R.; et al. Antitumor and antimetastatic effects of PLA2-BthTX-II from Bothrops jararacussu venom on human breast cancer cells. Int. J. Biol. Macromol. 2019, 135, 261–273. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 biochemistry. Cardiovasc. Drugs Ther. 2009, 23, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Ilic, D.; Bollinger, J.M.; Gelb, M.; Mauro, T.M. sPLA2 and the epidermal barrier. Biochim. Biophys. Acta 2014, 1841, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating Phospholipase A2 Biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Slatter, D.A.; Aldrovandi, M.; O’Connor, A.; Allen, S.M.; Brasher, C.J.; Murphy, R.C.; Mecklemann, S.; Ravi, S.; Darley-Usmar, V.; O’Donnell, V.B. Mapping the Human Platelet Lipidome Reveals Cytosolic Phospholipase A2 as a Regulator of Mitochondrial Bioenergetics during Activation. Cell Metab. 2016, 23, 930–944. [Google Scholar] [CrossRef] [PubMed]
- Ishii, I.; Fukushima, N.; Ye, X.; Chun, J. Lysophospholipid receptors: Signaling and biology. Annu. Rev. Biochem. 2004, 73, 321–354. [Google Scholar] [CrossRef] [PubMed]
- Meyer zu Heringdorf, D.; Jakobs, K.H. Lysophospholipid receptors: Signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim. Biophys. Acta 2007, 1768, 923–940. [Google Scholar] [CrossRef]
- Law, S.H.; Chan, M.L.; Marathe, G.K.; Parveen, F.; Chen, C.H.; Ke, L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.; Ko, Y.M.; McMullen, T.P.; Brindley, D.N. Autotaxin in the crosshairs: Taking aim at cancer and other inflammatory conditions. FEBS Lett. 2014, 588, 2712–2727. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal. Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Six, D.A.; Dennis, E.A. The expanding superfamily of phospholipase A(2) enzymes: Classification and characterization. Biochim. Biophys. Acta 2000, 1488, 1–19. [Google Scholar] [CrossRef]
- Schaloske, R.H.; Dennis, E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta 2006, 1761, 1246–1259. [Google Scholar] [CrossRef]
- Leslie, C.C. Cytosolic phospholipase A(2): Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef]
- Brglez, V.; Lambeau, G.; Petan, T. Secreted phospholipases A2 in cancer: Diverse mechanisms of action. Biochimie 2014, 107, 114–123. [Google Scholar] [CrossRef]
- Cummings, B.S.; McHowat, J.; Schnellmann, R.G. Phospholipase A(2)s in cell injury and death. J. Pharmacol. Exp. Ther. 2000, 294, 793–799. [Google Scholar]
- Valentin, E.; Lambeau, G. Increasing molecular diversity of secreted phospholipases A(2) and their receptors and binding proteins. Biochim. Biophys. Acta 2000, 1488, 59–70. [Google Scholar] [CrossRef]
- Hunter, K.W.; Crawford, N.P.; Alsarraj, J. Mechanisms of metastasis. Breast Cancer Res. BCR 2008, 10, S2. [Google Scholar] [CrossRef]
- Scott, K.F.; Sajinovic, M.; Hein, J.; Nixdorf, S.; Galettis, P.; Liauw, W.; de Souza, P.; Dong, Q.; Graham, G.G.; Russell, P.J. Emerging roles for phospholipase A2 enzymes in cancer. Biochimie 2010, 92, 601–610. [Google Scholar] [CrossRef]
- Peng, Z.; Chang, Y.; Fan, J.; Ji, W.; Su, C. Phospholipase A2 superfamily in cancer. Cancer Lett. 2021, 497, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Neubauer, B.L.; Graff, J.R.; Chedid, M.; Thomas, J.E.; Roehm, N.W.; Zhang, S.; Eckert, G.J.; Koch, M.O.; Eble, J.N.; et al. Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. Am. J. Pathol. 2002, 160, 667–671. [Google Scholar] [CrossRef]
- Lu, S.; Dong, Z. Overexpression of secretory phospholipase A2-IIa supports cancer stem cell phenotype via HER/ERBB-elicited signaling in lung and prostate cancer cells. Int. J. Oncol. 2017, 50, 2113–2122. [Google Scholar] [CrossRef]
- Sved, P.; Scott, K.F.; McLeod, D.; King, N.J.; Singh, J.; Tsatralis, T.; Nikolov, B.; Boulas, J.; Nallan, L.; Gelb, M.H.; et al. Oncogenic action of secreted phospholipase A2 in prostate cancer. Cancer Res. 2004, 64, 6934–6940. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lu, B.; Zhang, X.; Zhang, J.; Lai, L.; Li, D.; Wu, Y.; Song, Y.; Luo, J.; Pang, X.; et al. Cucurbitacin E, a tetracyclic triterpenes compound from Chinese medicine, inhibits tumor angiogenesis through VEGFR2-mediated Jak2-STAT3 signaling pathway. Carcinogenesis 2010, 31, 2097–2104. [Google Scholar] [CrossRef]
- Oleksowicz, L.; Liu, Y.; Bracken, R.B.; Gaitonde, K.; Burke, B.; Succop, P.; Levin, L.; Dong, Z.; Lu, S. Secretory phospholipase A2-IIa is a target gene of the HER/HER2-elicited pathway and a potential plasma biomarker for poor prognosis of prostate cancer. Prostate 2012, 72, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Kidoguchi, Y.; Sato, M.; Taketomi, Y.; Taya, C.; Muramatsu, K.; Gelb, M.H.; Yamamoto, K.; Murakami, M. Dual Roles of Group IID Phospholipase A2 in Inflammation and Cancer. J. Biol. Chem. 2016, 291, 15588–15601. [Google Scholar] [CrossRef]
- Caiazza, F.; McCarthy, N.S.; Young, L.; Hill, A.D.; Harvey, B.J.; Thomas, W. Cytosolic phospholipase A2-alpha expression in breast cancer is associated with EGFR expression and correlates with an adverse prognosis in luminal tumours. Br. J. Cancer 2011, 104, 338–344. [Google Scholar] [CrossRef]
- Wendum, D.; Svrcek, M.; Rigau, V.; Boelle, P.Y.; Sebbagh, N.; Parc, R.; Masliah, J.; Trugnan, G.; Flejou, J.F. COX-2, inflammatory secreted PLA2, and cytoplasmic PLA2 protein expression in small bowel adenocarcinomas compared with colorectal adenocarcinomas. Mod. Pathol. 2003, 16, 130–136. [Google Scholar] [CrossRef][Green Version]
- Patel, M.I.; Singh, J.; Niknami, M.; Kurek, C.; Yao, M.; Lu, S.; Maclean, F.; King, N.J.; Gelb, M.H.; Scott, K.F.; et al. Cytosolic phospholipase A2-alpha: A potential therapeutic target for prostate cancer. Clin. Cancer Res. 2008, 14, 8070–8079. [Google Scholar] [CrossRef]
- Weiser-Evans, M.C.; Wang, X.Q.; Amin, J.; Van Putten, V.; Choudhary, R.; Winn, R.A.; Scheinman, R.; Simpson, P.; Geraci, M.W.; Nemenoff, R.A. Depletion of cytosolic phospholipase A2 in bone marrow-derived macrophages protects against lung cancer progression and metastasis. Cancer Res. 2009, 69, 1733–1738. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Z.; Wei, G.; Yan, L.; Wang, D.; Zhang, H.; Sandusky, G.E.; Turk, J.; Xu, Y. Group VIA phospholipase A2 in both host and tumor cells is involved in ovarian cancer development. FASEB J. 2010, 24, 4103–4116. [Google Scholar] [CrossRef]
- Xu, Y.; Xiao, Y.J.; Zhu, K.; Baudhuin, L.M.; Lu, J.; Hong, G.; Kim, K.S.; Cristina, K.L.; Song, L.; Williams, F.S.; et al. Unfolding the pathophysiological role of bioactive lysophospholipids. Endocr. Metabol. Disord. 2003, 3, 23–32. [Google Scholar]
- Cai, Q.; Zhao, Z.; Antalis, C.; Yan, L.; Del Priore, G.; Hamed, A.H.; Stehman, F.B.; Schilder, J.M.; Xu, Y. Elevated and secreted phospholipase A(2) activities as new potential therapeutic targets in human epithelial ovarian cancer. FASEB J. 2012, 26, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Rodriguez-Vita, J.; Lawrence, T. The resolution of inflammation and cancer. Cytokine Growth Factor Rev. 2010, 21, 61–65. [Google Scholar] [CrossRef]
- Holt, D.M.; Ma, X.; Kundu, N.; Collin, P.D.; Fulton, A.M. Modulation of host natural killer cell functions in breast cancer via prostaglandin E2 receptors EP2 and EP4. J. Immunother. 2012, 35, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Z.; Mao, K.; Zou, J.; Wang, Y.; Tao, Z.; Lin, G.; Tian, L.; Ji, Y.; Wu, X.; et al. Dec2 promotes Th2 cell differentiation by enhancing IL-2R signaling. J. Immunol. 2009, 183, 6320–6329. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Baratelli, F.; Lin, Y.; Zhu, L.; Yang, S.C.; Heuze-Vourc’h, N.; Zeng, G.; Reckamp, K.; Dohadwala, M.; Sharma, S.; Dubinett, S.M. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J. Immunol. 2005, 175, 1483–1490. [Google Scholar] [CrossRef]
- Liu, L.; Ge, D.; Ma, L.; Mei, J.; Liu, S.; Zhang, Q.; Ren, F.; Liao, H.; Pu, Q.; Wang, T.; et al. Interleukin-17 and prostaglandin E2 are involved in formation of an M2 macrophage-dominant microenvironment in lung cancer. J. Thorac. Oncol. 2012, 7, 1091–1100. [Google Scholar] [CrossRef]
- Mathew, D.; Kremer, K.N.; Strauch, P.; Tigyi, G.; Pelanda, R.; Torres, R.M. LPA5 Is an Inhibitory Receptor That Suppresses CD8 T-Cell Cytotoxic Function via Disruption of Early TCR Signaling. Front. Immunol. 2019, 10, 1159. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xiao, M.; Zhang, Z.; Cui, R.; Jiang, X.; Wang, S.; Bai, H.; Liu, C.; Zhang, Z. Potential interaction between lysophosphatidic acid and tumor-associated macrophages in ovarian carcinoma. J. Inflamm. 2020, 17, 23. [Google Scholar] [CrossRef]
- Kamal, A.M.; Flower, R.J.; Perretti, M. An overview of the effects of annexin 1 on cells involved in the inflammatory process. Mem. Inst. Oswaldo. Cruz. 2005, 100, 39–47. [Google Scholar] [CrossRef]
- Yang, Y.H.; Morand, E.; Leech, M. Annexin A1: Potential for glucocorticoid sparing in RA. Nat. Rev. Rheumatol. 2013, 9, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.H.; Solito, E. Annexin A1: Uncovering the Many Talents of an Old Protein. Int. J. Mol. Sci. 2018, 19, 1045. [Google Scholar] [CrossRef]
- Barbosa, C.M.V.; Fock, R.A.; Hastreiter, A.A.; Reutelingsperger, C.; Perretti, M.; Paredes-Gamero, E.J.; Farsky, S.H.P. Extracellular annexin-A1 promotes myeloid/granulocytic differentiation of hematopoietic stem/progenitor cells via the Ca(2+)/MAPK signalling transduction pathway. Cell Death Discov. 2019, 5, 135. [Google Scholar] [CrossRef]
- Bizzarro, V.; Fontanella, B.; Franceschelli, S.; Pirozzi, M.; Christian, H.; Parente, L.; Petrella, A. Role of Annexin A1 in mouse myoblast cell differentiation. J. Cell Physiol. 2010, 224, 757–765. [Google Scholar] [CrossRef]
- Williams, S.L.; Milne, I.R.; Bagley, C.J.; Gamble, J.R.; Vadas, M.A.; Pitson, S.M.; Khew-Goodall, Y. A proinflammatory role for proteolytically cleaved annexin A1 in neutrophil transendothelial migration. J. Immunol. 2010, 185, 3057–3063. [Google Scholar] [CrossRef]
- Blume, K.E.; Soeroes, S.; Keppeler, H.; Stevanovic, S.; Kretschmer, D.; Rautenberg, M.; Wesselborg, S.; Lauber, K. Cleavage of annexin A1 by ADAM10 during secondary necrosis generates a monocytic “find-me” signal. J. Immunol. 2012, 188, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Rescher, U.; Goebeler, V.; Wilbers, A.; Gerke, V. Proteolytic cleavage of annexin 1 by human leukocyte elastase. Biochim. Biophys. Acta 2006, 1763, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Gavins, F.N.; Yona, S.; Kamal, A.M.; Flower, R.J.; Perretti, M. Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood 2003, 101, 4140–4147. [Google Scholar] [CrossRef]
- Le, Y.; Murphy, P.M.; Wang, J.M. Formyl-peptide receptors revisited. Trends Immunol. 2002, 23, 541–548. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int. J. Mol. Sci. 2013, 14, 7193–7230. [Google Scholar] [CrossRef]
- Snapkov, I.; Oqvist, C.O.; Figenschau, Y.; Kogner, P.; Johnsen, J.I.; Sveinbjornsson, B. The role of formyl peptide receptor 1 (FPR1) in neuroblastoma tumorigenesis. BMC Cancer 2016, 16, 490. [Google Scholar] [CrossRef]
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-inflammatory role of the murine formyl-peptide receptor 2: Ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 2010, 184, 2611–2619. [Google Scholar] [CrossRef]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic Potential in Microvascular Disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, J.; Liu, L.; Li, X.; Liu, S.; Xia, Q.; Shi, J. Annexin A1 translocates to nucleus and promotes the expression of pro-inflammatory cytokines in a PKC-dependent manner after OGD/R. Sci. Rep. 2016, 6, 27028. [Google Scholar] [CrossRef] [PubMed]
- Tcherniuk, S.; Cenac, N.; Comte, M.; Frouard, J.; Errazuriz-Cerda, E.; Galabov, A.; Morange, P.E.; Vergnolle, N.; Si-Tahar, M.; Alessi, M.C.; et al. Formyl Peptide Receptor 2 Plays a Deleterious Role During Influenza A Virus Infections. J. Infect. Dis. 2016, 214, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.L.; Yap, G.; Cui, J.; Lim, L.H.K. Annexin-A1—A Blessing or a Curse in Cancer? Trends Mol. Med. 2019, 25, 315–327. [Google Scholar] [CrossRef]
- Araujo, T.G.; Marangoni, K.; Rocha, R.M.; Maia, Y.C.; Araujo, G.R.; Alcantar, T.M.; Alves, P.T.; Calabria, L.; Neves, A.F.; Soares, F.A.; et al. Dynamic dialog between cytokeratin 18 and annexin A1 in breast cancer: A transcriptional disequilibrium. Acta. Histochem. 2014, 116, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, L.D.; Vishwanatha, J.K. Prognostic impact of AnxA1 and AnxA2 gene expression in triple-negative breast cancer. Oncotarget 2018, 9, 2697–2704. [Google Scholar] [CrossRef]
- de Graauw, M.; van Miltenburg, M.H.; Schmidt, M.K.; Pont, C.; Lalai, R.; Kartopawiro, J.; Pardali, E.; Le Devedec, S.E.; Smit, V.T.; van der Wal, A.; et al. Annexin A1 regulates TGF-beta signaling and promotes metastasis formation of basal-like breast cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6340–6345. [Google Scholar] [CrossRef]
- Vecchi, L.; Alves Pereira Zoia, M.; Goss Santos, T.; de Oliveira Beserra, A.; Colaco Ramos, C.M.; Franca Matias Colombo, B.; Paiva Maia, Y.C.; Piana de Andrade, V.; Teixeira Soares Mota, S.; Goncalves de Araujo, T.; et al. Inhibition of the AnxA1/FPR1 autocrine axis reduces MDA-MB-231 breast cancer cell growth and aggressiveness in vitro and in vivo. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1368–1382. [Google Scholar] [CrossRef]
- Mota, S.T.S.; Vecchi, L.; Alves, D.A.; Cordeiro, A.O.; Guimaraes, G.S.; Campos-Fernandez, E.; Maia, Y.C.P.; Dornelas, B.C.; Bezerra, S.M.; de Andrade, V.P.; et al. Annexin A1 promotes the nuclear localization of the epidermal growth factor receptor in castration-resistant prostate cancer. Int. J. Biochem. Cell Biol. 2020, 127, 105838. [Google Scholar] [CrossRef]
- Boer, J.C.; Domanska, U.M.; Timmer-Bosscha, H.; Boer, I.G.; de Haas, C.J.; Joseph, J.V.; Kruyt, F.A.; de Vries, E.G.; den Dunnen, W.F.; van Strijp, J.A.; et al. Inhibition of formyl peptide receptor in high-grade astrocytoma by CHemotaxis Inhibitory Protein of S. aureus. Br. J. Cancer 2013, 108, 587–596. [Google Scholar] [CrossRef]
- Locatelli, I.; Sutti, S.; Jindal, A.; Vacchiano, M.; Bozzola, C.; Reutelingsperger, C.; Kusters, D.; Bena, S.; Parola, M.; Paternostro, C.; et al. Endogenous annexin A1 is a novel protective determinant in nonalcoholic steatohepatitis in mice. Hepatology 2014, 60, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M.; et al. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci. Rep. 2017, 7, 17925. [Google Scholar] [CrossRef]
- Ampomah, P.B.; Moraes, L.A.; Lukman, H.M.; Lim, L.H.K. Formyl peptide receptor 2 is regulated by RNA mimics and viruses through an IFN-beta-STAT3-dependent pathway. FASEB J. 2018, 32, 1468–1478. [Google Scholar] [CrossRef]
- Li, Y.; Cai, L.; Wang, H.; Wu, P.; Gu, W.; Chen, Y.; Hao, H.; Tang, K.; Yi, P.; Liu, M.; et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene 2011, 30, 3887–3899. [Google Scholar] [CrossRef]
- Bai, F.; Zhang, P.; Fu, Y.; Chen, H.; Zhang, M.; Huang, Q.; Li, D.; Li, B.; Wu, K. Targeting ANXA1 abrogates Treg-mediated immune suppression in triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000169. [Google Scholar] [CrossRef]
- Oggero, S.; Austin-Williams, S.; Norling, L.V. The Contrasting Role of Extracellular Vesicles in Vascular Inflammation and Tissue Repair. Front. Pharmacol. 2019, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Maresh, G.; Zhang, X.; Salomon, C.; Hooper, J.; Margolin, D.; Li, L. The Emerging Roles of Extracellular Vesicles As Communication Vehicles within the Tumor Microenvironment and Beyond. Front. Endocrinol. 2017, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Xiao, Y.T.; Wu, T.; Yao, M.; Du, L.; Ren, S.; Wang, J. Microvesicles and chemokines in tumor microenvironment: Mediators of intercellular communications in tumor progression. Mol. Cancer 2019, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Czystowska-Kuzmicz, M.; Whiteside, T.L. The potential role of tumor-derived exosomes in diagnosis, prognosis, and response to therapy in cancer. Expert. Opin. Biol. Ther. 2021, 21, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, B.; Rodriguez, M.; San Millan, C.; Garcia, V.; Fernandez-Perianez, R.; Gil-Calderon, B.; Martin, P.; Garcia-Grande, A.; Silva, J.; Bonilla, F.; et al. Tumor-derived exosomes are enriched in DeltaNp73, which promotes oncogenic potential in acceptor cells and correlates with patient survival. Hum. Mol. Genet. 2014, 23, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Rezaie, J. Tumor cells derived-exosomes as angiogenenic agents: Possible therapeutic implications. J. Transl. Med. 2020, 18, 249. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Abusamra, A.J.; Zhong, Z.; Zheng, X.; Li, M.; Ichim, T.E.; Chin, J.L.; Min, W.P. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol. Dis. 2005, 35, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mason, M.D. Exosomes in tumour immunity. Curr. Oncol. 2009, 16, 46–49. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Subra, C.; Grand, D.; Laulagnier, K.; Stella, A.; Lambeau, G.; Paillasse, M.; De Medina, P.; Monsarrat, B.; Perret, B.; Silvente-Poirot, S.; et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J. Lipid Res. 2010, 51, 2105–2120. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef]
- Boilard, E. Extracellular vesicles and their content in bioactive lipid mediators: More than a sack of microRNA. J. Lipid Res. 2018, 59, 2037–2046. [Google Scholar] [CrossRef]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Pessolano, E.; Belvedere, R.; Bizzarro, V.; Franco, P.; Marco, I.; Porta, A.; Tosco, A.; Parente, L.; Perretti, M.; Petrella, A. Annexin A1 May Induce Pancreatic Cancer Progression as a Key Player of Extracellular Vesicles Effects as Evidenced in the In Vitro MIA PaCa-2 Model System. Int. J. Mol. Sci. 2018, 19, 3878. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; Neumann, P.A.; Kamaly, N.; Quiros, M.; Nishio, H.; Jones, H.R.; Sumagin, R.; Hilgarth, R.S.; Alam, A.; Fredman, G.; et al. Annexin A1-containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J. Clin. Investig. 2015, 125, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e418. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Buffolo, F.; Schlotter, F.; Atkins, S.K.; Lee, L.H.; Halu, A.; Blaser, M.C.; Tsolaki, E.; Higashi, H.; Luther, K.; et al. Annexin A1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci. Adv. 2020, 6, eabb1244. [Google Scholar] [CrossRef]
- Draeger, A.; Wray, S.; Babiychuk, E.B. Domain architecture of the smooth-muscle plasma membrane: Regulation by annexins. Biochem. J. 2005, 387, 309–314. [Google Scholar] [CrossRef]
- Pessolano, E.; Belvedere, R.; Bizzarro, V.; Franco, P.; Marco, I.; Petrella, F.; Porta, A.; Tosco, A.; Parente, L.; Perretti, M.; et al. Annexin A1 Contained in Extracellular Vesicles Promotes the Activation of Keratinocytes by Mesoglycan Effects: An Autocrine Loop Through FPRs. Cells 2019, 8, 753. [Google Scholar] [CrossRef]
- Aalberts, M.; Stout, T.A.; Stoorvogel, W. Prostasomes: Extracellular vesicles from the prostate. Reproduction 2014, 147, R1-14. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.R.; Sanchez-Heras, E.; Tsapara, A.; Sobota, A.; Levine, T.P.; Futter, C.E. Annexin A1 Tethers Membrane Contact Sites that Mediate ER to Endosome Cholesterol Transport. Dev. Cell 2016, 37, 473–483. [Google Scholar] [CrossRef]
- Rentero, C.; Blanco-Munoz, P.; Meneses-Salas, E.; Grewal, T.; Enrich, C. Annexins-Coordinators of Cholesterol Homeostasis in Endocytic Pathways. Int. J. Mol. Sci. 2018, 19, 1444. [Google Scholar] [CrossRef]
- Bardou, M.; Barkun, A.N.; Ghosn, J.; Hudson, M.; Rahme, E. Effect of chronic intake of NSAIDs and cyclooxygenase 2-selective inhibitors on esophageal cancer incidence. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2004, 2, 880–887. [Google Scholar] [CrossRef]
- Rayburn, E.R.; Ezell, S.J.; Zhang, R. Anti-Inflammatory Agents for Cancer Therapy. Mol. Cell. Pharmacol. 2009, 1, 29–43. [Google Scholar] [CrossRef] [PubMed]
- de Gaetano, G.; Donati, M.B.; Cerletti, C. Prevention of thrombosis and vascular inflammation: Benefits and limitations of selective or combined COX-1, COX-2 and 5-LOX inhibitors. Trends Pharmacol. Sci. 2003, 24, 245–252. [Google Scholar] [CrossRef]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef]
- Yu, J.A.; Sadaria, M.R.; Meng, X.; Mitra, S.; Ao, L.; Fullerton, D.A.; Weyant, M.J. Lung cancer cell invasion and expression of intercellular adhesion molecule-1 (ICAM-1) are attenuated by secretory phospholipase A(2) inhibition. J. Thorac. Cardiovasc. Surg. 2012, 143, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Makrilia, N.; Kollias, A.; Manolopoulos, L.; Syrigos, K. Cell adhesion molecules: Role and clinical significance in cancer. Cancer Investig. 2009, 27, 1023–1037. [Google Scholar] [CrossRef]
- Sadaria, M.R.; Meng, X.; Fullerton, D.A.; Reece, T.B.; Shah, R.R.; Grover, F.L.; Weyant, M.J. Secretory phospholipase A2 inhibition attenuates intercellular adhesion molecule-1 expression in human esophageal adenocarcinoma cells. Ann. Thorac. Surg. 2011, 91, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Sadaria, M.R.; Yu, J.A.; Meng, X.; Fullerton, D.A.; Reece, T.B.; Weyant, M.J. Secretory phospholipase A2 mediates human esophageal adenocarcinoma cell growth and proliferation via ERK 1/2 pathway. Anticancer Res. 2013, 33, 1337–1342. [Google Scholar] [PubMed]
- Nikolaou, A.; Kokotou, M.G.; Vasilakaki, S.; Kokotos, G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 941–956. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Hislop, C.; Elliott, M.; Stasiv, Y.; Goulder, M.; Waters, D. Effects of varespladib methyl on biomarkers and major cardiovascular events in acute coronary syndrome patients. J. Am. Coll. Cardiol. 2010, 56, 1079–1088. [Google Scholar] [CrossRef]
- Suckling, K. Phospholipase A2s: Developing drug targets for atherosclerosis. Atherosclerosis 2010, 212, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Hislop, C.; Christie, R.M.; Rick, H.L.; Reidy, C.A.; Chouinard, M.L.; Eacho, P.I.; Gould, K.E.; Trias, J. Varespladib (A-002), a secretory phospholipase A2 inhibitor, reduces atherosclerosis and aneurysm formation in ApoE-/- mice. J. Cardiovasc. Pharmacol. 2009, 53, 60–65. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Kastelein, J.J.; Schwartz, G.G.; Bash, D.; Rosenson, R.S.; Cavender, M.A.; Brennan, D.M.; Koenig, W.; Jukema, J.W.; Nambi, V.; et al. Varespladib and cardiovascular events in patients with an acute coronary syndrome: The VISTA-16 randomized clinical trial. Jama 2014, 311, 252–262. [Google Scholar] [CrossRef]
- Dong, Z.; Liu, Y.; Scott, K.F.; Levin, L.; Gaitonde, K.; Bracken, R.B.; Burke, B.; Zhai, Q.J.; Wang, J.; Oleksowicz, L.; et al. Secretory phospholipase A2-IIa is involved in prostate cancer progression and may potentially serve as a biomarker for prostate cancer. Carcinogenesis 2010, 31, 1948–1955. [Google Scholar] [CrossRef]
- Moon, T.C.; Hwang, H.S.; Quan, Z.; Son, K.H.; Kim, C.H.; Kim, H.P.; Kang, S.S.; Son, J.K.; Chang, H.W. Ochnaflavone, naturally occurring biflavonoid, inhibits phospholipase A2 dependent phosphatidylethanolamine degradation in a CCl4-induced rat liver microsome. Biol. Pharm. Bull. 2006, 29, 2359–2361. [Google Scholar] [CrossRef][Green Version]
- Suh, S.J.; Jin, U.H.; Kim, S.H.; Chang, H.W.; Son, J.K.; Lee, S.H.; Son, K.H.; Kim, C.H. Ochnaflavone inhibits TNF-alpha-induced human VSMC proliferation via regulation of cell cycle, ERK1/2, and MMP-9. J. Cell. Biochem. 2006, 99, 1298–1307. [Google Scholar] [CrossRef]
- Suh, S.J.; Chung, T.W.; Son, M.J.; Kim, S.H.; Moon, T.C.; Son, K.H.; Kim, H.P.; Chang, H.W.; Kim, C.H. The naturally occurring biflavonoid, ochnaflavone, inhibits LPS-induced iNOS expression, which is mediated by ERK1/2 via NF-kappaB regulation, in RAW264.7 cells. Arch. Biochem. Biophys. 2006, 447, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Elhady, S.S.; El-Halawany, A.M.; Alahdal, A.M.; Hassanean, H.A.; Ahmed, S.A. A New Bioactive Metabolite Isolated from the Red Sea Marine Sponge Hyrtios erectus. Molecules 2016, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- Yap, W.H.; Ahmed, N.; Lim, Y.M. Inhibition of Human Group IIA-Secreted Phospholipase A2 and THP-1 Monocyte Recruitment by Maslinic Acid. Lipids 2016, 51, 1153–1159. [Google Scholar] [CrossRef]
- Yap, W.H.; Ooi, B.K.; Ahmed, N.; Lim, Y.M. Maslinic acid modulates secreted phospholipase A2-IIA (sPLA2-IIA)-mediated inflammatory effects in macrophage foam cells formation. J. Biosci. 2018, 43, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhang, B.; Li, P.; Wen, X.; Yang, J. Maslinic Acid Inhibits Colon Tumorigenesis by the AMPK-mTOR Signaling Pathway. J. Agric. Food Chem. 2019, 67, 4259–4272. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Grover, A. Maslinic acid differentially exploits the MAPK pathway in estrogen-positive and triple-negative breast cancer to induce mitochondrion-mediated, caspase-independent apoptosis. Apoptosis 2020, 25, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Gimenes, S.N.C.; Lopes, D.S.; Alves, P.T.; Azevedo, F.; Vecchi, L.; Goulart, L.R.; Rodrigues, T.C.S.; Santos, A.L.Q.; Brites, V.L.C.; Teixeira, T.L.; et al. Antitumoral effects of gammaCdcPLI, a PLA2 inhibitor from Crotalus durissus collilineatus via PI3K/Akt pathway on MDA-MB-231 breast cancer cell. Sci. Rep. 2017, 7, 7077. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, J.S.; Yang, X.; Pang, X.; Li, L.; Wang, S.S.; Wu, J.B.; Tang, Y.J.; Liang, X.H.; Zheng, M.; et al. Overexpression Cathepsin D Contributes to Perineural Invasion of Salivary Adenoid Cystic Carcinoma. Front. Oncol. 2018, 8, 492. [Google Scholar] [CrossRef]
- Zoia, M.A.P.; Azevedo, F.V.P.; Vecchi, L.; Mota, S.T.S.; Rodovalho, V.R.; Cordeiro, A.O.; Correia, L.I.V.; Silva, A.C.A.; Avila, V.M.R.; Araujo, T.G.; et al. Inhibition of Triple-Negative Breast Cancer Cell Aggressiveness by Cathepsin D Blockage: Role of Annexin A1. Int. J. Mol. Sci. 2019, 20, 1337. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vecchi, L.; Araújo, T.G.; Azevedo, F.V.P.d.V.; Mota, S.T.S.; Ávila, V.d.M.R.; Ribeiro, M.A.; Goulart, L.R. Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1. Cells 2021, 10, 1472. https://doi.org/10.3390/cells10061472
Vecchi L, Araújo TG, Azevedo FVPdV, Mota STS, Ávila VdMR, Ribeiro MA, Goulart LR. Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1. Cells. 2021; 10(6):1472. https://doi.org/10.3390/cells10061472
Chicago/Turabian StyleVecchi, Lara, Thaise Gonçalves Araújo, Fernanda Van Petten de Vasconcelos Azevedo, Sara Teixeria Soares Mota, Veridiana de Melo Rodrigues Ávila, Matheus Alves Ribeiro, and Luiz Ricardo Goulart. 2021. "Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1" Cells 10, no. 6: 1472. https://doi.org/10.3390/cells10061472
APA StyleVecchi, L., Araújo, T. G., Azevedo, F. V. P. d. V., Mota, S. T. S., Ávila, V. d. M. R., Ribeiro, M. A., & Goulart, L. R. (2021). Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1. Cells, 10(6), 1472. https://doi.org/10.3390/cells10061472