
Lysophosphatidic Acid: Promoter of Cancer Progression and of Tumor Microenvironment Development. A Promising Target for Anticancer Therapies?

{kind=link}
Abstract
1. Introduction
2. Autotaxin, LPA and Its Receptors
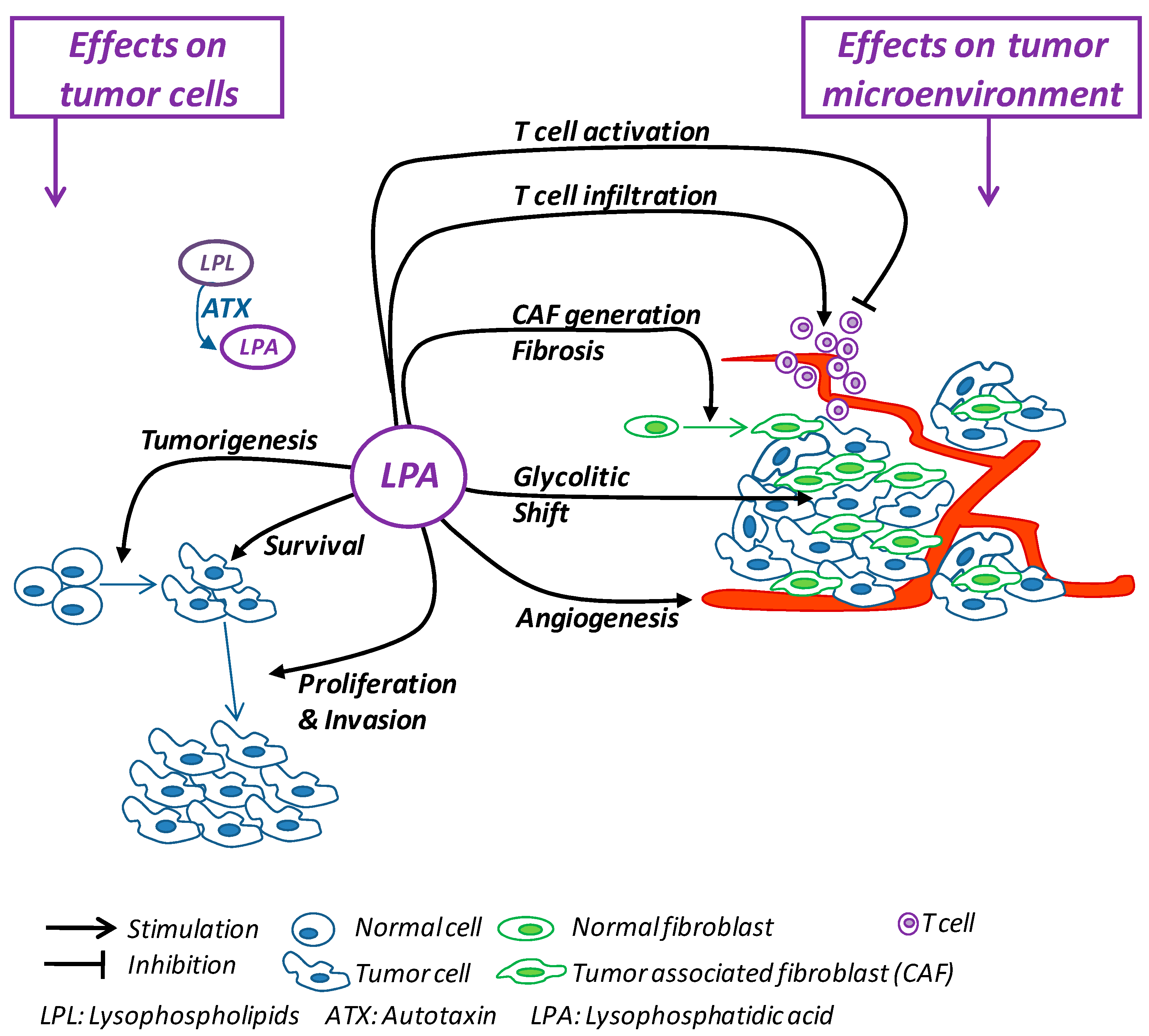
3. LPA: Direct Effects on Tumor Initiation and Progression
3.1. ATX/LPA Axis: A Promoter of Tumorigenesis, Tumor Invasion and Metastasis
3.2. ATX/LPA Axis: A Promoter of Tumor Cell Survival and Antitumor Therapy Resistance
4. LPA: Effects on the Tumor Microenvironment
4.1. ATX/LPA Axis: A Promoter of Angiogenesis and Lymphangiogenesis
4.2. ATX/LPA Axis: A Promoter of Tumor Glycolytic Shift
4.3. ATX/LPA Axis: A Promoter of CAF Generation and Fibrosis Development
4.4. ATX/LPA Axis: A Promoter of Tumor Escape from Immune Surveillance
5. Conclusions and Therapeutic Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef]
- Oikonomou, N.; Mouratis, M.-A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef]
- Sakai, N.; Chun, J.; Duffield, J.S.; Lagares, D.; Wada, T.; Luster, A.D.; Tager, A.M. Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor. Kidney Int. 2017, 91, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Bain, G.; Furuichi, K.; Iwata, Y.; Nakamura, M.; Hara, A.; Kitajima, S.; Sagara, A.; Miyake, T.; Toyama, T.; et al. The involvement of autotaxin in renal interstitial fibrosis through regulation of fibroblast functions and induction of vascular leakage. Sci. Rep. 2019, 9, 7414. [Google Scholar] [CrossRef]
- Bain, G.; Shannon, K.E.; Huang, F.; Darlington, J.; Goulet, L.; Prodanovich, P.; Ma, G.L.; Santini, A.M.; Stein, A.J.; Lonergan, D.; et al. Selective inhibition of autotaxin is efficacious in mouse models of liver fibrosis. J. Pharmacol. Exp. Ther. 2017, 360, 1–13. [Google Scholar] [CrossRef]
- Castelino, F.V.; Bain, G.; Pace, V.A.; Black, K.E.; George, L.; Probst, C.K.; Goulet, L.; Lafyatis, R.; Tager, A.M. An autotaxin/lysophosphatidic acid/interleukin-6 amplification loop drives scleroderma fibrosis. Arthritis Rheumatol. 2016, 68, 2964–2974. [Google Scholar] [CrossRef]
- Cortinovis, M.; Aiello, S.; Mister, M.; Conde-Knape, K.; Noris, M.; Novelli, R.; Solini, S.; Rodriguez Ordonez, P.Y.; Benigni, A.; Remuzzi, G. Autotaxin inhibitor protects from chronic allograft injury in rat kidney allotransplantation. Nephron 2020, 144, 38–48. [Google Scholar] [CrossRef]
- Cao, P.; Aoki, Y.; Badri, L.; Walker, N.M.; Manning, C.M.; Lagstein, A.; Fearon, E.R.; Lama, V.N. Autocrine lysophosphatidic acid signaling activates β-catenin and promotes lung allograft fibrosis. J. Clin. Investig. 2017, 127, 1517–1530. [Google Scholar] [CrossRef]
- Xu, Y. Targeting lysophosphatidic acid in cancer: The issues in moving from bench to bedside. Cancers 2019, 11, 1523. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Tang, X.; Brindley, D.N. Autotaxin and breast cancer: Towards overcoming treatment barriers and sequelae. Cancers 2020, 12, 374. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N. Lysophosphatidic acid signaling in cancer. Cancers 2020, 12, 3791. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a Lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest 2018, 154, 1061–1069. [Google Scholar] [CrossRef]
- Maher, T.M.; van der Aar, E.M.; Van de Steen, O.; Allamassey, L.; Desrivot, J.; Dupont, S.; Fagard, L.; Ford, P.; Fieuw, A.; Wuyts, W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): A phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 2018, 6, 627–635. [Google Scholar] [CrossRef]
- Moolenaar, W.H. Development of our current understanding of bioactive lysophospholipids. Ann. N. Y. Acad. Sci. 2000, 905, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bandoh, K.; Aoki, J.; Taira, A.; Tsujimoto, M.; Arai, H.; Inoue, K. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-Activity relationship of cloned LPA receptors. FEBS Lett. 2000, 478, 159–165. [Google Scholar] [CrossRef]
- Benesch, M.G.K.; Ko, Y.M.; McMullen, T.P.W.; Brindley, D.N. Autotaxin in the crosshairs: Taking aim at cancer and other inflammatory conditions. FEBS Lett. 2014, 588, 2712–2727. [Google Scholar] [CrossRef]
- Kawagoe, H.; Soma, O.; Goji, J.; Nishimura, N.; Narita, M.; Inazawa, J.; Nakamura, H.; Sano, K. Molecular cloning and chromosomal assignment of the human brain-type phosphodiesterase I/Nucleotide pyrophosphatase gene (PDNP2). Genomics 1995, 30, 380–384. [Google Scholar] [CrossRef]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 2008, 1781, 513–518. [Google Scholar] [CrossRef]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradère, J.P.; Pettit, T.R.; Wakelam, M.J.O.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell. Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef]
- Tang, X.; Zhao, Y.Y.; Dewald, J.; Curtis, J.M.; Brindley, D.N. Tetracyclines increase lipid phosphate phosphatase expression on plasma membranes and turnover of plasma lysophosphatidate. J. Lipid Res. 2016, 57, 597–606. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.K.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Zhao, Y.Y.; Curtis, J.M.; McMullen, T.P.W.; Brindley, D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and Sphingosine 1-Phosphate. J. Lipid Res. 2015, 56, 1134–1144. [Google Scholar] [CrossRef]
- Moolenaar, W.H.; van Meeteren, L.A.; Giepmans, B.N.G. The ins and outs of lysophosphatidic acid signaling. Bioessays 2004, 26, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A. Metabolic pathways and physiological and pathological significances of lysolipid phosphate mediators. J. Cell. Biochem. 2004, 92, 869–881. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/jcb.20147 (accessed on 8 April 2021). [CrossRef] [PubMed]
- Geraldo, L.H.M.; de Sampaio Spohr, T.C.L.; Amaral, R.F.D.; Fonseca, A.C.C.D.; Garcia, C.; Mendes, F.D.A.; Freitas, C.; DosSantos, M.F.; Lima, F.R.S. Role of lysophosphatidic acid and its receptors in health and disease: Novel therapeutic strategies. Signal. Transduct. Target. Ther. 2021, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Okudaira, S.; Kishi, Y.; Ohkawa, R.; Iseki, S.; Ota, M.; Noji, S.; Yatomi, Y.; Aoki, J.; Arai, H. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 2006, 281, 25822–25830. [Google Scholar] [CrossRef] [PubMed]
- Yukiura, H.; Kano, K.; Kise, R.; Inoue, A.; Aoki, J. Autotaxin overexpression causes embryonic lethality and vascular defects. PLoS ONE 2015, 10, e0126734. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, N.C.; Chun, J. Promising pharmacological directions in the world of lysophosphatidic acid signaling. Biomol. Ther. 2015, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.-W.; Mutoh, T.; Lin, M.-E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef]
- Nikitopoulou, I.; Oikonomou, N.; Karouzakis, E.; Sevastou, I.; Nikolaidou-Katsaridou, N.; Zhao, Z.; Mersinias, V.; Armaka, M.; Xu, Y.; Masu, M.; et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925–933. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hecht, J.H.; Weiner, J.A.; Post, S.R.; Chun, J. Ventricular zone gene-1 (Vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell Biol. 1996, 135, 1071–1083. [Google Scholar] [CrossRef]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef]
- Contos, J.J.; Ishii, I.; Chun, J. Lysophosphatidic acid receptors. Mol. Pharmacol. 2000, 58, 1188–1196. [Google Scholar] [CrossRef]
- Hama, K.; Aoki, J.; Fukaya, M.; Kishi, Y.; Sakai, T.; Suzuki, R.; Ohta, H.; Yamori, T.; Watanabe, M.; Chun, J.; et al. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J. Biol. Chem. 2004, 279, 17634–17639. [Google Scholar] [CrossRef]
- Hayashi, M.; Okabe, K.; Kato, K.; Okumura, M.; Fukui, R.; Fukushima, N.; Tsujiuchi, T. Differential function of lysophosphatidic acid receptors in cell proliferation and migration of neuroblastoma cells. Cancer Lett. 2012, 316, 91–96. [Google Scholar] [CrossRef]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, Purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar] [CrossRef]
- Kawagoe, H.; Stracke, M.L.; Nakamura, H.; Sano, K. Expression and transcriptional regulation of the PD-Ialpha/Autotaxin gene in neuroblastoma. Cancer Res. 1997, 57, 2516–2521. [Google Scholar]
- Hoelzinger, D.B.; Mariani, L.; Weis, J.; Woyke, T.; Berens, T.J.; McDonough, W.S.; Sloan, A.; Coons, S.W.; Berens, M.E. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia 2005, 7, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Stassar, M.J.; Devitt, G.; Brosius, M.; Rinnab, L.; Prang, J.; Schradin, T.; Simon, J.; Petersen, S.; Kopp-Schneider, A.; Zöller, M. Identification of human renal cell carcinoma associated genes by suppression subtractive hybridization. Br. J. Cancer 2001, 85, 1372–1382. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kehlen, A.; Englert, N.; Seifert, A.; Klonisch, T.; Dralle, H.; Langner, J.; Hoang-Vu, C. Expression, regulation and function of autotaxin in thyroid carcinomas. Int. J. Cancer 2004, 109, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Lee, J.; Park, C.G.; Kim, S.; Hong, S.; Chung, H.C.; Min, S.K.; Han, J.W.; Lee, H.W.; Lee, H.Y. Expression of autotaxin (NPP-2) is closely linked to invasiveness of breast cancer cells. Clin. Exp. Metastasis 2002, 19, 603–608. [Google Scholar] [CrossRef]
- Yang, Y.; Mou, L.; Liu, N.; Tsao, M.S. Autotaxin expression in non-small-cell lung cancer. Am. J. Respir. Cell Mol. Biol. 1999, 21, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-M.; Xu, Y.; Skill, N.J.; Sheng, H.; Zhao, Z.; Yu, M.; Saxena, R.; Maluccio, M.A. Autotaxin expression and its connection with the TNF-α-NF-κB Axis in human hepatocellular carcinoma. Mol. Cancer 2010, 9, 71. [Google Scholar] [CrossRef]
- Mills, G.B.; Moolenaar, W.H. The Emerging Role of Lysophosphatidic Acid in Cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef]
- Leblanc, R.; Peyruchaud, O. New insights into the autotaxin/LPA axis in cancer development and metastasis. Exp. Cell Res. 2015, 333, 183–189. [Google Scholar] [CrossRef]
- Xu, Y.; Fang, X.J.; Casey, G.; Mills, G.B. Lysophospholipids activate ovarian and breast cancer cells. Biochem. J. 1995, 309, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Yoshikawa, K.; Tanabe, E.; Kitayoshi, M.; Fukui, R.; Fukushima, N.; Tsujiuchi, T. Opposite roles of LPA1 and LPA3 on cell motile and invasive activities of pancreatic cancer cells. Tumour Biol. 2012, 33, 1739–1744. [Google Scholar] [CrossRef]
- Nam, S.W.; Clair, T.; Campo, C.K.; Lee, H.Y.; Liotta, L.A.; Stracke, M.L. Autotaxin (ATX), a Potent tumor motogen, augments invasive and metastatic potential of ras-transformed cells. Oncogene 2000, 19, 241–247. [Google Scholar] [CrossRef]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, X.; Gajewiak, J.; Tsukahara, R.; Fujiwara, Y.; Liu, J.; Fells, J.I.; Perygin, D.; Parrill, A.L.; Tigyi, G.; et al. Dual activity lysophosphatidic acid receptor pan-antagonist/autotaxin inhibitor reduces breast cancer cell migration in vitro and causes tumor regression in vivo. Cancer Res. 2009, 69, 5441–5449. [Google Scholar] [CrossRef]
- Yu, S.; Murph, M.M.; Lu, Y.; Liu, S.; Hall, H.S.; Liu, J.; Stephens, C.; Fang, X.; Mills, G.B. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. J. Natl. Cancer Inst. 2008, 100, 1630–1642. [Google Scholar] [CrossRef]
- Peng, W.-T.; Sun, W.-Y.; Li, X.-R.; Sun, J.-C.; Du, J.-J.; Wei, W. Emerging roles of G protein-coupled receptors in hepatocellular carcinoma. Int. J. Mol. Sci. 2018, 19, 1366. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeong, K.J.; Panupinthu, N.; Yu, S.; Lee, J.; Han, J.W.; Kim, J.M.; Lee, J.-S.; Kang, J.; Park, C.G.; et al. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene 2011, 30, 1351–1359. [Google Scholar] [CrossRef]
- Popnikolov, N.K.; Dalwadi, B.H.; Thomas, J.D.; Johannes, G.J.; Imagawa, W.T. Association of autotaxin and lysophosphatidic acid receptor 3 with aggressiveness of human breast carcinoma. Tumour Biol. 2012, 33, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Ribeiro, J.; Descotes, F.; Serre, C.-M.; Barbier, M.; Murone, M.; Clézardin, P.; Peyruchaud, O. Targeting lysophosphatidic acid receptor type 1 with Debio 0719 inhibits spontaneous metastasis dissemination of breast cancer cells independently of cell proliferation and angiogenesis. Int. J. Oncol. 2012, 40, 1133–1141. [Google Scholar] [CrossRef]
- Mazzocca, A.; Dituri, F.; Lupo, L.; Quaranta, M.; Antonaci, S.; Giannelli, G. Tumor-Secreted lysophostatidic acid accelerates hepatocellular carcinoma progression by promoting differentiation of peritumoral fibroblasts in myofibroblasts. Hepatology 2011, 54, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Hofseth, L.J.; Hussain, S.P.; Harris, C.C. P53: 25 years after its discovery. Trends Pharmacol. Sci. 2004, 25, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Murph, M.M.; Hurst-Kennedy, J.; Newton, V.; Brindley, D.N.; Radhakrishna, H. Lysophosphatidic acid decreases the nuclear localization and cellular abundance of the P53 tumor suppressor in A549 lung carcinoma cells. Mol. Cancer Res. 2007, 5, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Furui, T.; LaPushin, R.; Mao, M.; Khan, H.; Watt, S.R.; Watt, M.A.; Lu, Y.; Fang, X.; Tsutsui, S.; Siddik, Z.H.; et al. Overexpression of Edg-2/Vzg-1 induces apoptosis and anoikis in ovarian cancer cells in a lysophosphatidic acid-independent manner. Clin. Cancer Res. 1999, 5, 4308–4318. [Google Scholar] [PubMed]
- Fukui, R.; Kato, K.; Okabe, K.; Kitayoshi, M.; Tanabe, E.; Fukushima, N.; Tsujiuchi, T. Enhancement of drug resistance by lysophosphatidic acid receptor-3 in mouse mammary tumor FM3A cells. J. Toxicol. Pathol. 2012, 25, 225–228. [Google Scholar] [CrossRef]
- Samadi, N.; Gaetano, C.; Goping, I.S.; Brindley, D.N. Autotaxin protects MCF-7 breast cancer and MDA-MB-435 melanoma cells against taxol-induced apoptosis. Oncogene 2009, 28, 1028–1039. [Google Scholar] [CrossRef]
- Brindley, D.N.; Lin, F.-T.; Tigyi, G.J. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim. Biophys. Acta 2013, 1831, 74–85. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Cassis, L.; Aiello, S.; Noris, M. Natural versus adaptive regulatory T cells. Contrib. Nephrol. 2005, 146, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Du, Y.; Lin, X.; Qian, Y.; Zhou, T.; Huang, Z. CD4+CD25+ regulatory T cells in tumor immunity. Int. Immunopharmacol. 2016, 34, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Budhwar, S.; Verma, P.; Verma, R.; Rai, S.; Singh, K. The yin and yang of myeloid derived suppressor cells. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.C.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P.W.; et al. Inhibition of Autotaxin Delays Breast Tumor Growth and Lung Metastasis in Mice. FASEB J. 2014, 28, 2655–2666. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.W.; Clair, T.; Kim, Y.S.; McMarlin, A.; Schiffmann, E.; Liotta, L.A.; Stracke, M.L. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. 2001, 61, 6938–6944. [Google Scholar]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef]
- Hu, Y.L.; Tee, M.K.; Goetzl, E.J.; Auersperg, N.; Mills, G.B.; Ferrara, N.; Jaffe, R.B. Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells. J. Natl. Cancer Inst. 2001, 93, 762–768. [Google Scholar] [CrossRef]
- Fujita, T.; Miyamoto, S.; Onoyama, I.; Sonoda, K.; Mekada, E.; Nakano, H. Expression of lysophosphatidic acid receptors and vascular endothelial growth factor mediating lysophosphatidic acid in the development of human ovarian cancer. Cancer Lett. 2003, 192, 161–169. [Google Scholar] [CrossRef]
- Bachelier, R.; Confavreux, C.B.; Peyruchaud, O.; Croset, M.; Goehrig, D.; van der Pluijm, G.; Clézardin, P. Combination of anti-angiogenic therapies reduces osteolysis and tumor burden in experimental breast cancer bone metastasis. Int. J. Cancer 2014, 135, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Boucharaba, A.; Guillet, B.; Menaa, F.; Hneino, M.; van Wijnen, A.J.; Clézardin, P.; Philippe, C.; Peyruchaud, O.; Oliver, P. Bioactive lipids lysophosphatidic acid and sphingosine 1-Phosphate mediate breast cancer cell biological functions through distinct mechanisms. Oncol. Res. 2009, 18, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Jeon, E.S.; Heo, S.C.; Lee, I.H.; Choi, Y.J.; Park, J.H.; Choi, K.U.; Park, D.Y.; Suh, D.S.; Yoon, M.S.; Kim, J.H. Ovarian cancer-derived lysophosphatidic acid stimulates secretion of VEGF and stromal cell-derived factor-1 α from human mesenchymal stem cells. Exp. Mol. Med. 2010, 42, 280–293. [Google Scholar] [CrossRef]
- Su, J.-L.; Yen, C.-J.; Chen, P.-S.; Chuang, S.-E.; Hong, C.-C.; Kuo, I.-H.; Chen, H.-Y.; Hung, M.-C.; Kuo, M.-L. The role of the VEGF-C/VEGFR-3 axis in cancer progression. Br. J. Cancer 2007, 96, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.B.; Priceman, S.J.; Sung, J.L.; Brakenhielm, E.; An, D.S.; Pytowski, B.; Alitalo, K.; Wu, L. Suppression of prostate cancer nodal and systemic metastasis by blockade of the lymphangiogenic axis. Cancer Res. 2008, 68, 7828–7837. [Google Scholar] [CrossRef]
- Lin, C.-E.; Chen, S.-U.; Lin, C.-C.; Chang, C.-H.; Lin, Y.-C.; Tai, Y.-L.; Shen, T.-L.; Lee, H. Lysophosphatidic acid enhances vascular endothelial growth factor-C expression in human prostate cancer PC-3 cells. PLoS ONE 2012, 7, e41096. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Chen, C.-C.; Chen, W.-M.; Lu, K.-Y.; Shen, T.-L.; Jou, Y.-C.; Shen, C.-H.; Ohbayashi, N.; Kanaho, Y.; Huang, Y.-L.; et al. LPA1/3 signaling mediates tumor lymphangiogenesis through promoting CRT expression in prostate cancer. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2018, 1863, 1305–1315. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal. Transduct. Target. Ther. 2020, 5, 1–16. [Google Scholar] [CrossRef]
- Qiu, G.-Z.; Jin, M.-Z.; Dai, J.-X.; Sun, W.; Feng, J.-H.; Jin, W.-L. Reprogramming of the tumor in the hypoxic niche: The emerging concept and associated therapeutic strategies. Trends Pharmacol. Sci. 2017, 38, 669–686. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Ha, J.H.; Radhakrishnan, R.; Jayaraman, M.; Yan, M.; Ward, J.D.; Fung, K.-M.; Moxley, K.; Sood, A.K.; Isidoro, C.; Mukherjee, P.; et al. LPA induces metabolic reprogramming in ovarian cancer via a pseudohypoxic response. Cancer Res. 2018, 78, 1923–1934. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-Associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Buechler, M.B.; Turley, S.J. A short field guide to fibroblast function in immunity. Semin. Immunol. 2018, 35, 48–58. [Google Scholar] [CrossRef]
- Bu, L.; Baba, H.; Yoshida, N.; Miyake, K.; Yasuda, T.; Uchihara, T.; Tan, P.; Ishimoto, T. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene 2019, 38, 4887–4901. [Google Scholar] [CrossRef]
- Eiro, N.; González, L.; Martínez-Ordoñez, A.; Fernandez-Garcia, B.; González, L.O.; Cid, S.; Dominguez, F.; Perez-Fernandez, R.; Vizoso, F.J. Cancer-Associated fibroblasts affect breast cancer cell gene expression, invasion and angiogenesis. Cell Oncol. 2018, 41, 369–378. [Google Scholar] [CrossRef]
- Benyahia, Z.; Dussault, N.; Cayol, M.; Sigaud, R.; Berenguer-Daizé, C.; Delfino, C.; Tounsi, A.; Garcia, S.; Martin, P.-M.; Mabrouk, K.; et al. Stromal fibroblasts present in breast carcinomas promote tumor growth and angiogenesis through adrenomedullin secretion. Oncotarget 2017, 8, 15744–15762. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.C.; Fingas, C.D.; Christensen, J.D.; Smoot, R.L.; Bronk, S.F.; Werneburg, N.W.; Gustafson, M.P.; Dietz, A.B.; Roberts, L.R.; Sirica, A.E.; et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 2013, 73, 897–907. [Google Scholar] [CrossRef]
- Cadamuro, M.; Nardo, G.; Indraccolo, S.; Dall’olmo, L.; Sambado, L.; Moserle, L.; Franceschet, I.; Colledan, M.; Massani, M.; Stecca, T.; et al. Platelet-Derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013, 58, 1042–1053. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef]
- Pankova, D.; Chen, Y.; Terajima, M.; Schliekelman, M.J.; Baird, B.N.; Fahrenholtz, M.; Sun, L.; Gill, B.J.; Vadakkan, T.J.; Kim, M.P.; et al. Cancer-Associated fibroblasts induce a collagen cross-link switch in tumor stroma. Mol. Cancer Res. 2016, 14, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The fibrotic tumor stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Bager, C.L.; Willumsen, N.; Leeming, D.J.; Smith, V.; Karsdal, M.A.; Dornan, D.; Bay-Jensen, A.C. Collagen degradation products measured in serum can separate ovarian and breast cancer patients from healthy controls: A preliminary study. Cancer Biomark. 2015, 15, 783–788. [Google Scholar] [CrossRef]
- Willumsen, N.; Bager, C.L.; Leeming, D.J.; Smith, V.; Christiansen, C.; Karsdal, M.A.; Dornan, D.; Bay-Jensen, A.-C. Serum biomarkers reflecting specific tumor tissue remodeling processes are valuable diagnostic tools for lung cancer. Cancer Med. 2014, 3, 1136–1145. [Google Scholar] [CrossRef]
- Kehlet, S.N.; Sanz-Pamplona, R.; Brix, S.; Leeming, D.J.; Karsdal, M.A.; Moreno, V. Excessive collagen turnover products are released during colorectal cancer progression and elevated in serum from metastatic colorectal cancer patients. Sci. Rep. 2016, 6, 30599. [Google Scholar] [CrossRef] [PubMed]
- Leeming, D.J.; Koizumi, M.; Qvist, P.; Barkholt, V.; Zhang, C.; Henriksen, K.; Byrjalsen, I.; Karsdal, M.A. Serum N-Terminal Propeptide of collagen type I is associated with the number of bone metastases in breast and prostate cancer and correlates to other bone related markers. Biomark. Cancer 2011, 3, 15–23. [Google Scholar] [CrossRef]
- Gudmann, N.; Luo, Y.; Sand, J.M.B.; Trujillo, G.; Murphy, B.J.; Cheng, P.T.; Karsdal, M.A.; Nielsen, M.J.; Leeming, D.J.; Jarai, G. Fibroblast activation triggered by LPA results in matrix synthesis/fibrogenesis different from that of TGF-β1. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- King, C.S.; Nathan, S.D. Idiopathic pulmonary fibrosis: Effects and optimal management of comorbidities. Lancet Respir. Med. 2017, 5, 72–84. [Google Scholar] [CrossRef]
- Condorelli, A.G.; Dellambra, E.; Logli, E.; Zambruno, G.; Castiglia, D. Epidermolysis bullosa-associated squamous cell carcinoma: From pathogenesis to therapeutic perspectives. Int. J. Mol. Sci. 2019, 20, 5707. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Reinartz, S.; Lieber, S.; Pesek, J.; Brandt, D.T.; Asafova, A.; Finkernagel, F.; Watzer, B.; Nockher, W.A.; Nist, A.; Stiewe, T.; et al. Cell type-selective pathways and clinical associations of lysophosphatidic acid biosynthesis and signaling in the ovarian cancer microenvironment. Mol. Oncol. 2019, 13, 185–201. [Google Scholar] [CrossRef]
- Ray, R.; Rai, V. Lysophosphatidic acid converts monocytes into macrophages in both mice and humans. Blood 2017, 129, 1177–1183. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef]
- Oda, S.K.; Strauch, P.; Fujiwara, Y.; Al-Shami, A.; Oravecz, T.; Tigyi, G.; Pelanda, R.; Torres, R.M. Lysophosphatidic acid inhibits CD8 T cell activation and control of tumor progression. Cancer Immunol. Res. 2013, 1, 245–255. [Google Scholar] [CrossRef]
- Mathew, D.; Kremer, K.N.; Strauch, P.; Tigyi, G.; Pelanda, R.; Torres, R.M. LPA5 is an inhibitory receptor that suppresses CD8 T-cell cytotoxic function via disruption of early TCR signaling. Front. Immunol. 2019, 10, 1159. [Google Scholar] [CrossRef]
- Zheng, Y.; Kong, Y.; Goetzl, E.J. Lysophosphatidic acid receptor-selective effects on jurkat T cell migration through a matrigel model basement membrane. J. Immunol. 2001, 166, 2317–2322. [Google Scholar] [CrossRef]
- Kanda, H.; Newton, R.; Klein, R.; Morita, Y.; Gunn, M.D.; Rosen, S.D. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat. Immunol. 2008, 9, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.-C.M.; Krummel, M.F.; Rosen, S.D. Autotaxin through lysophosphatidic acid stimulates polarization, motility, and transendothelial migration of naive T cells. J. Immunol. 2012, 189, 3914–3924. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Cai, L.; Umemoto, E.; Takeda, A.; Tohya, K.; Komai, Y.; Veeraveedu, P.T.; Hata, E.; Sugiura, Y.; Kubo, A.; et al. Constitutive lymphocyte transmigration across the basal lamina of high endothelial venules is regulated by the autotaxin/lysophosphatidic acid axis. J. Immunol. 2013, 190, 2036–2048. [Google Scholar] [CrossRef]
- Knowlden, S.; Georas, S.N. The Autotaxin-LPA Axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef]
- Hozumi, H.; Hokari, R.; Kurihara, C.; Narimatsu, K.; Sato, H.; Sato, S.; Ueda, T.; Higashiyama, M.; Okada, Y.; Watanabe, C.; et al. Involvement of autotaxin/lysophospholipase D expression in intestinal vessels in aggravation of intestinal damage through lymphocyte migration. Lab. Investig. 2013, 93, 508–519. [Google Scholar] [CrossRef]
- Takeda, A.; Kobayashi, D.; Aoi, K.; Sasaki, N.; Sugiura, Y.; Igarashi, H.; Tohya, K.; Inoue, A.; Hata, E.; Akahoshi, N.; et al. Fibroblastic reticular cell-derived lysophosphatidic acid regulates confined intranodal t-cell motility. eLife 2016, 5, e10561. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, C.; Guillerm, S.; Quero, L. Combination of chemotherapy and radiotherapy: A thirty years evolution. Cancer Radiother. 2019, 23, 662–665. [Google Scholar] [CrossRef]
- Pradère, J.-P.; Klein, J.; Grès, S.; Guigné, C.; Neau, E.; Valet, P.; Calise, D.; Chun, J.; Bascands, J.-L.; Saulnier-Blache, J.-S.; et al. LPA1 receptor activation promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2007, 18, 3110–3118. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Ledein, L.; Léger, B.; Dees, C.; Beyer, C.; Distler, A.; Vettori, S.; Boukaiba, R.; Bidouard, J.P.; Schaefer, M.; Pernerstorfer, J.; et al. Translational engagement of lysophosphatidic acid receptor 1 in skin fibrosis: From dermal fibroblasts of patients with scleroderma to tight skin 1 mouse. Br. J. Pharmacol. 2020, 177, 4296–4309. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Distler, O.; Jagerschmidt, A.; Illiano, S.; Ledein, L.; Boitier, E.; Agueusop, I.; Denton, C.P.; Khanna, D. Lysophosphatidic acid receptor 1 antagonist SAR100842 for patients with diffuse cutaneous systemic sclerosis: A double-blind, randomized, eight-week placebo-controlled study followed by a sixteen-week open-label extension study. Arthritis Rheumatol. 2018, 70, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Kreuter, M.; Lederer, D.J.; Brown, K.K.; Wuyts, W.; Verbruggen, N.; Stutvoet, S.; Fieuw, A.; Ford, P.; Abi-Saab, W.; et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir. Res. 2019, 6, e000422. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiello, S.; Casiraghi, F. Lysophosphatidic Acid: Promoter of Cancer Progression and of Tumor Microenvironment Development. A Promising Target for Anticancer Therapies? Cells 2021, 10, 1390. https://doi.org/10.3390/cells10061390
Aiello S, Casiraghi F. Lysophosphatidic Acid: Promoter of Cancer Progression and of Tumor Microenvironment Development. A Promising Target for Anticancer Therapies? Cells. 2021; 10(6):1390. https://doi.org/10.3390/cells10061390
Chicago/Turabian StyleAiello, Sistiana, and Federica Casiraghi. 2021. "Lysophosphatidic Acid: Promoter of Cancer Progression and of Tumor Microenvironment Development. A Promising Target for Anticancer Therapies?" Cells 10, no. 6: 1390. https://doi.org/10.3390/cells10061390
APA StyleAiello, S., & Casiraghi, F. (2021). Lysophosphatidic Acid: Promoter of Cancer Progression and of Tumor Microenvironment Development. A Promising Target for Anticancer Therapies? Cells, 10(6), 1390. https://doi.org/10.3390/cells10061390