
Oxidative Stress-Induced Unscheduled CDK1–Cyclin B1 Activity Impairs ER–Mitochondria-Mediated Bioenergetic Metabolism



Abstract
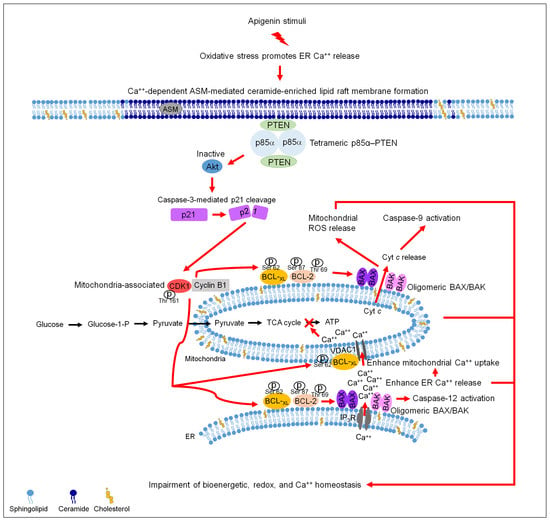
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Chemicals, Reagents, and Plasmids
2.3. Antibodies
2.4. Measurement of DNA Fragmentation
2.5. Subcellular Fractionation
2.6. Density Based Membrane Flotation Technique
2.7. Western Blot Analysis and Co-Immunoprecipitation
2.8. Cell Surface Biotinylation
2.9. Measurement of Cytosolic Ca++
2.10. Detection of Reactive Oxygen Species (ROS)
2.11. Plasmid and siRNA Transfection
2.12. Rac1 Activation Assay
2.13. Determination of Cholesterol, Sphingomyelin, and Ceramide
2.14. Measurement of Intracellular Glucose
2.15. Measurement of Cellular ATP
2.16. Statistical Analysis
3. Results
3.1. BCL-2 (Thr 69 and Ser 87)/BCL-xL (Ser 62) Phosphorylation and BAX/BAK ER–Mitochondrial Oligomerization Associated with Apigenin-Induced ER–Mitochondrial Dysfunction of NPC Cells
3.2. Apigenin-Induced ER–Mitochondria Metabolic Dysfunction Involved in Dysregulation of the Anti-Apoptotic Function of BCL-xL/BCL-2
3.3. Induction of Unscheduled P-CDK1 (Thr 161)–Cyclin B1 Complexes by Apigenin Confers Dysregulation of the ER/Mitochondria Bioenergetic and Metabolic Control of BCL-2/BCL-xL
3.4. Induction of Lipid Raft-Associated ASM-Mediated Ceramide Generation by Apigenin-Induced Oxidative Stress Impedes the Lipid Raft Membrane-Associated p85α−GTP−Rac1- Akt Signaling, Leading to the Sustained Formation of p-CDK1 (Thr 161)–Cyclin B1 Complexes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Acid sphingomyelinase | ASM |
| B cell lymphoma 2 | BCL-2 |
| Bcl-2-antagonist of cell death | BAD |
| Bcl-2 antagonist killer 1 | BAK |
| Bcl-2-associated x protein | BAX |
| BCL-2/B-cell lymphoma-extra large | BCL-xL |
| Cyclin dependent kinase 1 | CDK1 |
| Cytochrome c | Cyt c |
| Endoplasmic reticulum | ER |
| Phosphatase and tensin homolog deleted from chromosome 10 | PTEN |
| Phosphatidylinositol 3-kinase | PI3K |
| Phosphatidylinositol-4,5-bisphosphate | PIP2 |
| Phosphatidylinositol-3,4,5-trisphosphate | PIP3 |
| Poly (ADP-ribose) polymerase | PARP |
| Protein kinase B | Akt |
| Ras-related C3 botulinum toxin substrate 1 | Rac1 |
| Short hairpin RNA | shRNA |
| Small interfering RNA | siRNA |
| Nasopharyngeal carcinoma | NPC |
References
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef]
- Szymanski, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziolkowski, W.; Duszynski, J.; Pinton, P.; Dobrzyn, A.; Wieckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Popgeorgiev, N.; Jabbour, L.; Gillet, G. Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Front. Cell Dev. Biol. 2018, 6, 13. [Google Scholar] [CrossRef]
- Gross, A.; Katz, S.G. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Tanaka, S.; Takayama, S.; Schibler, M.J.; Fenton, W.; Reed, J.C. Investigation of the subcellular distribution of the bcl-2 oncoprotein: Residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993, 53, 4701–4714. [Google Scholar]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Westphal, D.; Kluck, R.M.; Dewson, G. Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014, 21, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Terrano, D.T.; Upreti, M.; Chambers, T.C. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol. Cell. Biol. 2010, 30, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Deng, X.; May, W.S. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef]
- Veas-Perez de Tudela, M.; Delgado-Esteban, M.; Maestre, C.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Vecino, R.; Bolanos, J.P.; Almeida, A. Regulation of Bcl-xL-ATP Synthase Interaction by Mitochondrial Cyclin B1-Cyclin-Dependent Kinase-1 Determines Neuronal Survival. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 9287–9301. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Hermeking, H.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 1999, 401, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Lehtinen, M.; Donovan, N.; Bonni, A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol. Cell 2002, 9, 1005–1016. [Google Scholar] [CrossRef]
- Debatin, K.M.; Poncet, D.; Kroemer, G. Chemotherapy: Targeting the mitochondrial cell death pathway. Oncogene 2002, 21, 8786–8803. [Google Scholar] [CrossRef]
- Kim, H.; Tu, H.C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef]
- Lindqvist, A.; van Zon, W.; Karlsson Rosenthal, C.; Wolthuis, R.M. Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS Biol. 2007, 5, e123. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Wang, S.; Jiang, N.; Li, J.J. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 2019, 443, 56–66. [Google Scholar] [CrossRef]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Smits, V.A.; Klompmaker, R.; Vallenius, T.; Rijksen, G.; Makela, T.P.; Medema, R.H. p21 inhibits Thr161 phosphorylation of Cdc2 to enforce the G2 DNA damage checkpoint. J. Biol. Chem. 2000, 275, 30638–30643. [Google Scholar] [CrossRef]
- Li, W.; Fan, J.; Banerjee, D.; Bertino, J.R. Overexpression of p21(waf1) decreases G2-M arrest and apoptosis induced by paclitaxel in human sarcoma cells lacking both p53 and functional Rb protein. Mol. Pharmacol. 1999, 55, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Charrier-Savournin, F.B.; Chateau, M.T.; Gire, V.; Sedivy, J.; Piette, J.; Dulic, V. p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Mol. Biol. Cell 2004, 15, 3965–3976. [Google Scholar] [CrossRef]
- Katayama, K.; Fujita, N.; Tsuruo, T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol. Cell. Biol. 2005, 25, 5725–5737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fujita, N.; Tsuruo, T. Caspase-mediated cleavage of p21Waf1/Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene 1999, 18, 1131–1318. [Google Scholar] [CrossRef]
- Jin, Y.H.; Yoo, K.J.; Lee, Y.H.; Lee, S.K. Caspase 3-mediated cleavage of p21WAF1/CIP1 associated with the cyclin A-cyclin-dependent kinase 2 complex is a prerequisite for apoptosis in SK-HEP-1 cells. J. Biol. Chem. 2000, 275, 30256–30263. [Google Scholar] [CrossRef]
- Li, Y.; Dowbenko, D.; Lasky, L.A. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.C.; Wang, S.G.; Lin, M.L.; Chen, S.S. Citrate-Induced p85alpha(-)PTEN Complex Formation Causes G2/M Phase Arrest in Human Pharyngeal Squamous Carcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 2105. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef]
- Brazil, D.P.; Yang, Z.Z.; Hemmings, B.A. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem. Sci. 2004, 29, 233–242. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Hu, Z.Y.; Zhu, X.F.; Zhong, Z.D.; Sun, J.; Wang, J.; Yang, D.; Zeng, Y.X. ApoG2, a novel inhibitor of antiapoptotic Bcl-2 family proteins, induces apoptosis and suppresses tumor growth in nasopharyngeal carcinoma xenografts. Int. J. Cancer 2008, 123, 2418–2429. [Google Scholar] [CrossRef]
- Wang, J.; Chang, L.; Lai, X.; Li, X.; Wang, Z.; Huang, Z.; Huang, J.; Zhang, G. Tetrandrine enhances radiosensitivity through the CDC25C/CDK1/cyclin B1 pathway in nasopharyngeal carcinoma cells. Cell Cycle 2018, 17, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.L.; Wang, S.G.; Chan, W.L.; Lee, C.H.; Wu, T.S.; Lin, M.L.; Chen, S.S. Impairment of Membrane Lipid Homeostasis by Bichalcone Analog TSWU-BR4 Attenuates Function of GRP78 in Regulation of the Oxidative Balance and Invasion of Cancer Cells. Cells 2020, 9, 371. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.L.; Lu, Y.C.; Chung, J.G.; Li, Y.C.; Wang, S.G.N.; Wu, C.Y.; Su, H.L.; Chen, S.S. Aloe-emodin induces apoptosis of human nasopharyngeal carcinoma cells via caspase-8-mediated activation of the mitochondrial death pathway. Cancer Lett. 2010, 291, 46–58. [Google Scholar] [CrossRef]
- Wu, C.W.; Wang, S.G.; Lee, C.H.; Chan, W.L.; Lin, M.L.; Chen, S.S. Enforced C-Src Activation Causes Compartmental Dysregulation of PI3K and PTEN Molecules in Lipid Rafts of Tongue Squamous Carcinoma Cells by Attenuating Rac1-Akt-GLUT-1-Mediated Sphingolipid Synthesis. Int. J. Mol. Sci. 2020, 21, 5812. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, X.H. Apigenin induces both intrinsic and extrinsic pathways of apoptosis in human colon carcinoma HCT-116 cells. Oncol. Rep. 2017, 37, 1132–1140. [Google Scholar] [CrossRef]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef]
- Pihan, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Opferman, J.T.; Pozzan, T.; Korsmeyer, S.J.; Scorrano, L. Regulation of endoplasmic reticulum Ca2+ dynamics by proapoptotic BCL-2 family members. Biochem. Pharmacol. 2003, 66, 1335–1340. [Google Scholar] [CrossRef]
- Lin, M.L.; Chen, S.S.; Huang, R.Y.; Lu, Y.C.; Liao, Y.R.; Reddy, M.V.; Lee, C.C.; Wu, T.S. Suppression of PI3K/Akt signaling by synthetic bichalcone analog TSWU-CD4 induces ER stress- and Bax/Bak-mediated apoptosis of cancer cells. Apoptosis Int. J. Program. Cell Death 2014, 19, 1637–1653. [Google Scholar] [CrossRef]
- Eskes, R.; Desagher, S.; Antonsson, B.; Martinou, J.C. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 2000, 20, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Ruffolo, S.C.; Breckenridge, D.G.; Nguyen, M.; Goping, I.S.; Gross, A.; Korsmeyer, S.J.; Li, H.; Yuan, J.; Shore, G.C. BID-dependent and BID-independent pathways for BAX insertion into mitochondria. Cell Death Differ. 2000, 7, 1101–1108. [Google Scholar] [CrossRef]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar] [PubMed]
- Ellis, K.O.; Butterfield, J.L.; Wessels, F.L.; Carpenter, J.F. A comparison of skeletal, cardiac, and smooth muscle actions of dantrolene sodium—A skeletal muscle relaxant. Arch. Int. Pharmacodyn. Ther. 1976, 224, 118–132. [Google Scholar]
- Hainaut, K.; Desmedt, J.E. Effect of dantrolene sodium on calcium movements in single muscle fibres. Nature 1974, 252, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Elustondo, P.A.; Nichols, M.; Negoda, A.; Thirumaran, A.; Zakharian, E.; Robertson, G.S.; Pavlov, E.V. Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov. 2016, 2, 16070. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Martini, M.; De Santis, M.C.; Hirsch, E. How PI3K-derived lipids control cell division. Front. Cell Dev. Biol. 2015, 3, 61. [Google Scholar] [CrossRef] [PubMed]
- Kreis, N.N.; Sanhaji, M.; Rieger, M.A.; Louwen, F.; Yuan, J. p21Waf1/Cip1 deficiency causes multiple mitotic defects in tumor cells. Oncogene 2014, 33, 5716–5728. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [PubMed]
- Drevot, P.; Langlet, C.; Guo, X.J.; Bernard, A.M.; Colard, O.; Chauvin, J.P.; Lasserre, R.; He, H.T. TCR signal initiation machinery is pre-assembled and activated in a subset of membrane rafts. EMBO J. 2002, 21, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.W.; Walkiewicz, K.W.; Besong, T.M.; Guo, H.; Hawke, D.H.; Arold, S.T.; Mills, G.B. Regulation of the PI3K pathway through a p85alpha monomer-homodimer equilibrium. eLife 2015, 4, e06866. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Singh, D.; Phillips, G.; Kilkus, J.; Dawson, G. Ceramide regulation of the tumor suppressor phosphatase PTEN in rafts isolated from neurotumor cell lines. J. Neurosci. Res. 2005, 81, 541–550. [Google Scholar] [CrossRef]
- Kreis, P.; Leondaritis, G.; Lieberam, I.; Eickholt, B.J. Subcellular targeting and dynamic regulation of PTEN: Implications for neuronal cells and neurological disorders. Front. Mol. Neurosci. 2014, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gulbins, E.; Zhang, Y. Oxidative stress triggers Ca-dependent lysosome trafficking and activation of acid sphingomyelinase. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2012, 30, 815–826. [Google Scholar] [CrossRef]
- Oninla, V.O.; Breiden, B.; Babalola, J.O.; Sandhoff, K. Acid sphingomyelinase activity is regulated by membrane lipids and facilitates cholesterol transfer by NPC2. J. Lipid Res. 2014, 55, 2606–2619. [Google Scholar] [CrossRef]
- Guo, Q.; Lu, T.; Chen, Y.; Su, Y.; Zheng, Y.; Chen, Z.; Chen, C.; Lin, S.; Pan, J.; Yuan, X. Genetic variations in the PI3K-PTEN-AKT-mTOR pathway are associated with distant metastasis in nasopharyngeal carcinoma patients treated with intensity-modulated radiation therapy. Sci. Rep. 2016, 6, 37576. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Zhu, G.; Liao, S.; Yi, W.; Luo, G.; He, J.; Pei, Z.; Li, G.; Zhou, Y. Dysregulation of the PI3K/Akt signaling pathway affects cell cycle and apoptosis of side population cells in nasopharyngeal carcinoma. Oncol. Lett. 2015, 10, 182–188. [Google Scholar] [CrossRef]
- Tulalamba, W.; Janvilisri, T. Nasopharyngeal carcinoma signaling pathway: An update on molecular biomarkers. Int. J. Cell Biol. 2012, 2012, 594681. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Rieusset, J. Mitochondria-Associated Membranes Response to Nutrient Availability and Role in Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza-Alaguero, I.; Canto, C.; Zorzano, A. Metabolic implications of organelle-mitochondria communication. EMBO Rep. 2019, 20, e47928. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.L.S.; Kwok-Wai, L.; Soo-Beng, A.K.; Mohana-Kumaran, N. Single Agent and Synergistic Activity of Maritoclax with ABT-263 in Nasopharyngeal Carcinoma (NPC) Cell Lines. Trop. Life Sci. Res. 2020, 31, 1–13. [Google Scholar] [PubMed]
- Sakurikar, N.; Eichhorn, J.M.; Chambers, T.C. Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J. Biol. Chem. 2012, 287, 39193–39204. [Google Scholar] [CrossRef] [PubMed]
- Chagpar, R.B.; Links, P.H.; Pastor, M.C.; Furber, L.A.; Hawrysh, A.D.; Chamberlain, M.D.; Anderson, D.H. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 5471–5476. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Rieusset, J. Metabolic signaling functions of ER-mitochondria contact sites: Role in metabolic diseases. J. Mol. Endocrinol. 2017, 58, R87–R106. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Min, K.T. The Interface Between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cells 2018, 41, 1000–1007. [Google Scholar] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Hill, M.M.; Feng, J.; Hemmings, B.A. Identification of a plasma membrane Raft-associated PKB Ser473 kinase activity that is distinct from ILK and PDK1. Curr. Biol. 2002, 12, 1251–1255. [Google Scholar] [CrossRef]
- Grassme, H.; Jendrossek, V.; Bock, J.; Riehle, A.; Gulbins, E. Ceramide-rich membrane rafts mediate CD40 clustering. J. Immunol. 2002, 168, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar] [CrossRef] [PubMed]
- Megha; london, E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): Implications for lipid raft structure and function. J. Biol. Chem. 2004, 279, 9997–10004. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.P.; Barr, P.; Yee, V.C.; Distelhorst, C.W. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim. Biophys. Acta 2009, 1793, 971–978. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, X.; Eno, C.O.; Zhao, G.; Li, C.; White, C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J. Biol. Chem. 2013, 288, 19870–19881. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Decrock, E.; Arbel, N.; van Vliet, A.R.; La Rovere, R.M.; De Smedt, H.; Parys, J.B.; Agostinis, P.; Leybaert, L.; Shoshan-Barmatz, V.; et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 2015, 290, 9150–9161. [Google Scholar] [CrossRef]
- Li, C.; Fox, C.J.; Master, S.R.; Bindokas, V.P.; Chodosh, L.A.; Thompson, C.B. Bcl-X(L) affects Ca(2+) homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 9830–9835. [Google Scholar] [CrossRef] [PubMed]
- Distelhorst, C.W.; Bootman, M.D. Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: Role in Ca(2+) signaling and disease. Cell Calcium 2011, 50, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.W.; De Smedt, H.; et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012, 19, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Scorrano, L.; Opferman, J.T.; Bassik, M.C.; Nishino, M.; Pozzan, T.; Korsmeyer, S.J. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Avanzato, D.; Pupo, E.; Ducano, N.; Isella, C.; Bertalot, G.; Luise, C.; Pece, S.; Bruna, A.; Rueda, O.M.; Caldas, C.; et al. High USP6NL Levels in Breast Cancer Sustain Chronic AKT Phosphorylation and GLUT1 Stability Fueling Aerobic Glycolysis. Cancer Res. 2018, 78, 3432–3444. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.-G.; Tien, N.; Chang, Y.-C.; Lin, M.-L.; Chen, S.-S. Oxidative Stress-Induced Unscheduled CDK1–Cyclin B1 Activity Impairs ER–Mitochondria-Mediated Bioenergetic Metabolism. Cells 2021, 10, 1280. https://doi.org/10.3390/cells10061280
Chang J-G, Tien N, Chang Y-C, Lin M-L, Chen S-S. Oxidative Stress-Induced Unscheduled CDK1–Cyclin B1 Activity Impairs ER–Mitochondria-Mediated Bioenergetic Metabolism. Cells. 2021; 10(6):1280. https://doi.org/10.3390/cells10061280
Chicago/Turabian StyleChang, Jan-Gowth, Ni Tien, Yi-Chih Chang, Meng-Liang Lin, and Shih-Shun Chen. 2021. "Oxidative Stress-Induced Unscheduled CDK1–Cyclin B1 Activity Impairs ER–Mitochondria-Mediated Bioenergetic Metabolism" Cells 10, no. 6: 1280. https://doi.org/10.3390/cells10061280
APA StyleChang, J.-G., Tien, N., Chang, Y.-C., Lin, M.-L., & Chen, S.-S. (2021). Oxidative Stress-Induced Unscheduled CDK1–Cyclin B1 Activity Impairs ER–Mitochondria-Mediated Bioenergetic Metabolism. Cells, 10(6), 1280. https://doi.org/10.3390/cells10061280