
Proteoglycan 4 Modulates Osteogenic Smooth Muscle Cell Differentiation during Vascular Remodeling and Intimal Calcification

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Highlights:
- Proteoglycan 4 (PRG4) induction by smooth muscle cells (SMCs) appears as an early reaction to vascular intimal remodeling, preceding, and, likely facilitating, the later formation of macro-calcifications.
- Osteogenic and inflammatory growth factors, lipids, high calcium and particularly high phosphate conditions induce PRG4 expression regulated by SMAD3 and SOX9 transcription factors, which accompanies the osteogenic phenotypic switch of SMCs.
- As a feedback loop, PRG4-enriched extracellular matrix leads to the recovery of typical SMC markers and cellular quiescence under calcifying conditions.
- The association among PRG4, SMC phenotypic modulation and atherosclerotic plaque calcification warrants further translational investigations to explore PRG4 as a clinical marker of plaque phenotype.
1. Introduction
2. Materials and Methods
2.1. Human Material
2.2. Animal Studies
2.2.1. Mouse Carotid Ligation Model
2.2.2. Rat Carotid Artery Balloon Injury
2.2.3. Mouse Atherosclerotic Calcification Model
2.3. In Vitro Assays
2.3.1. Cytokine Stimulations
2.3.2. Transfection and PRG4 Silencing
2.3.3. Migration Assay
2.3.4. Proliferation Assay
2.3.5. Calcification Assay
2.3.6. OxLDL Assay
2.4. Recombinant Human PRG4
2.5. RNA Extraction and Gene Expression Analyses by Quantitative PCR (qPCR)
2.6. Histological Analyses
2.6.1. Antibodies
2.6.2. Immunohistochemistry (IHC)
2.6.3. Semi-Quantitative IHC Scoring
2.6.4. Immunofluorescence (IFL)
2.7. Computed Tomography (CT) Angiography Image Analysis
2.8. Bioinformatic and Statistical Analyses
3. Results
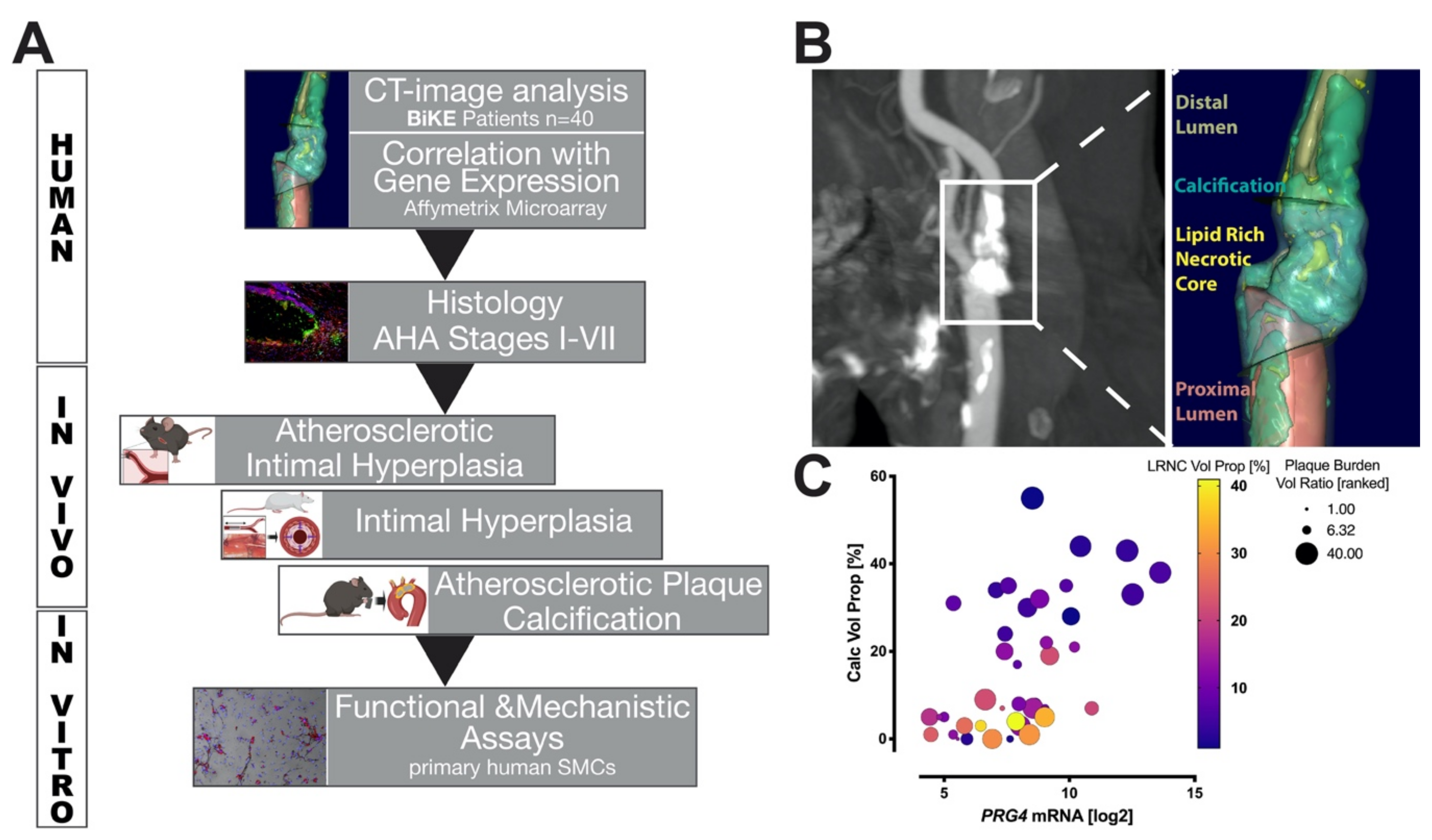
3.1. Study Design and Correlation of PRG4 with Human Plaque Composition
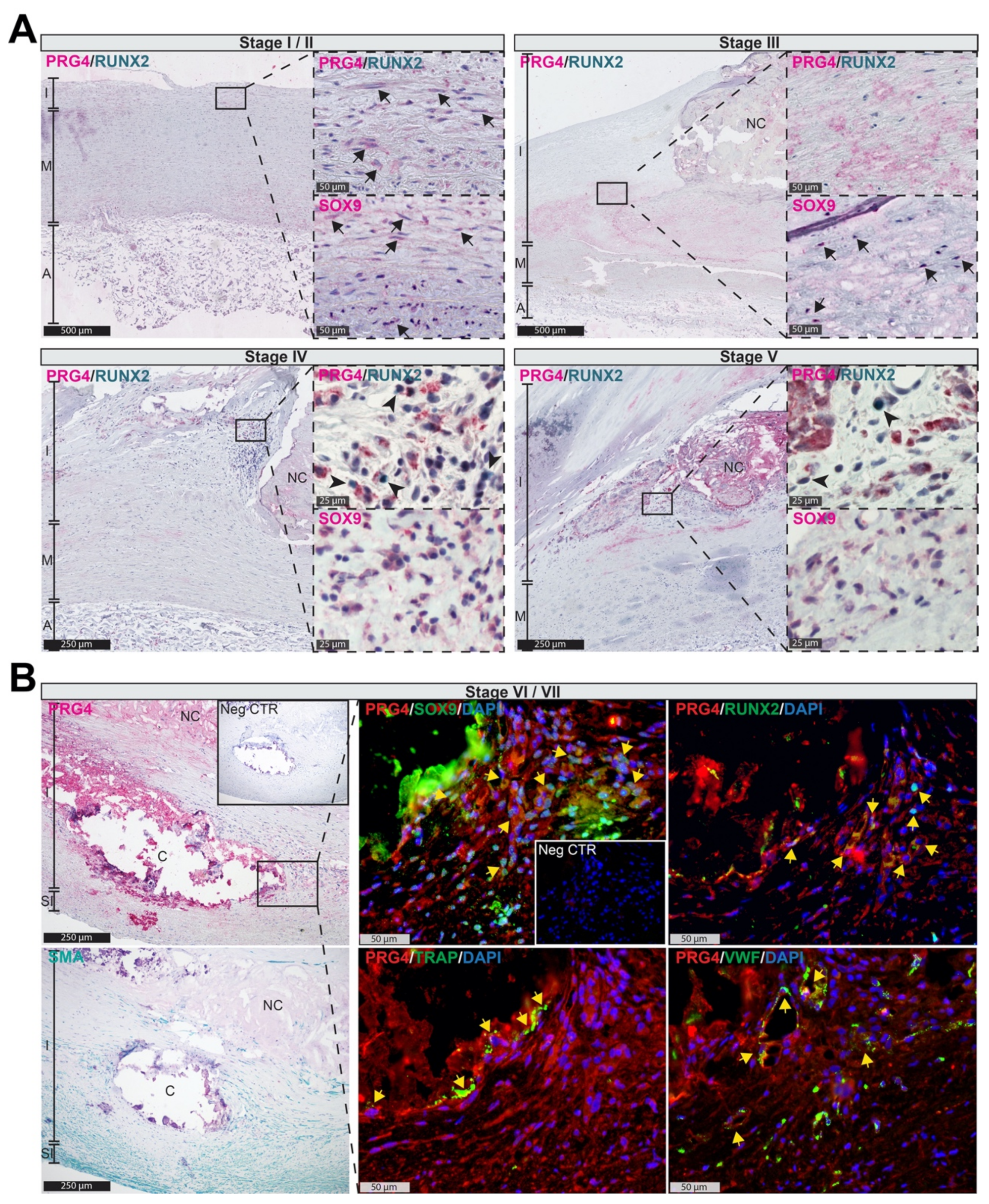
3.2. PRG4 Is Detectable in Human Adaptive Intimal Thickening and Intimal Xanthomas
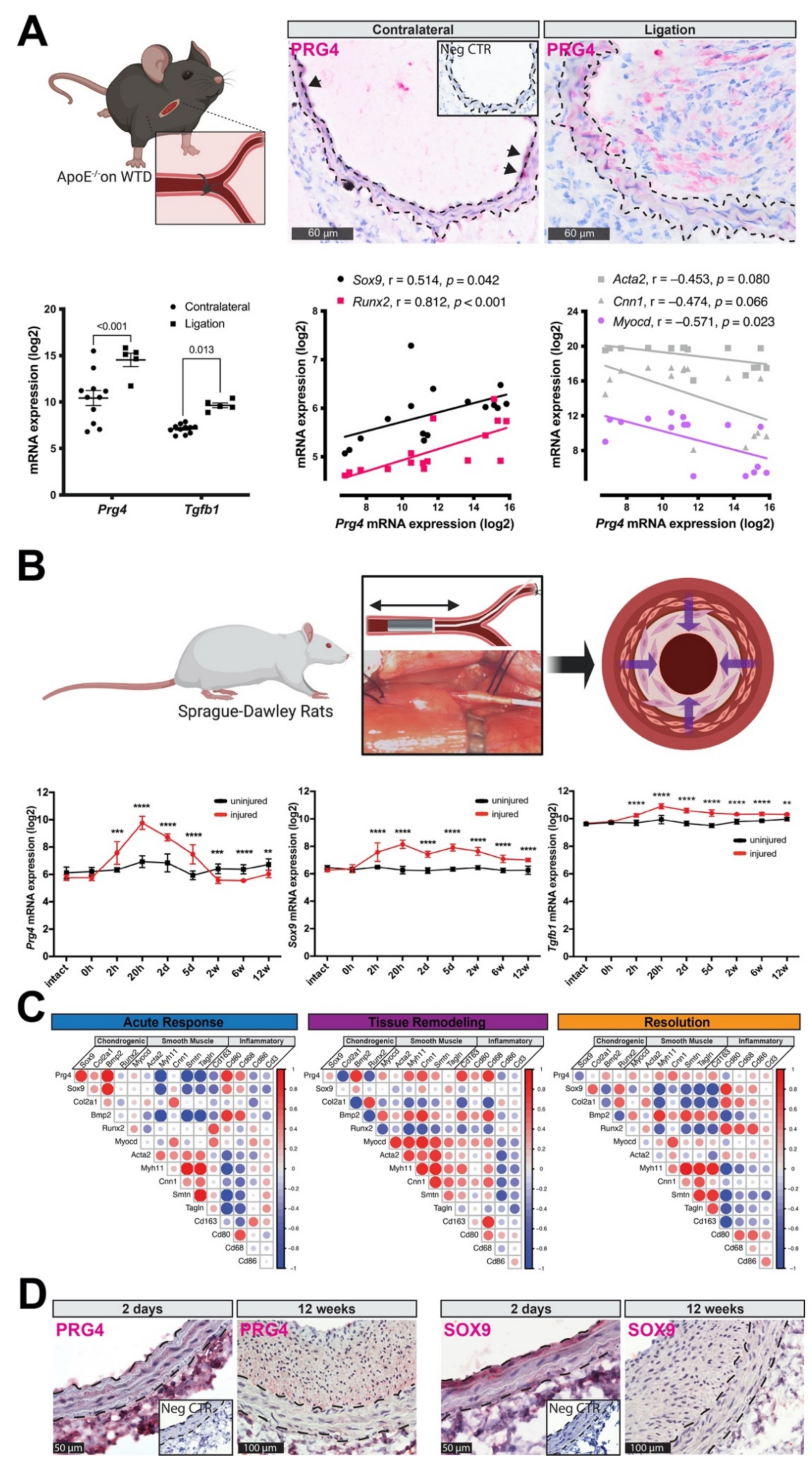
3.3. PRG4 Is Expressed Early during Vascular Remodeling In Vivo
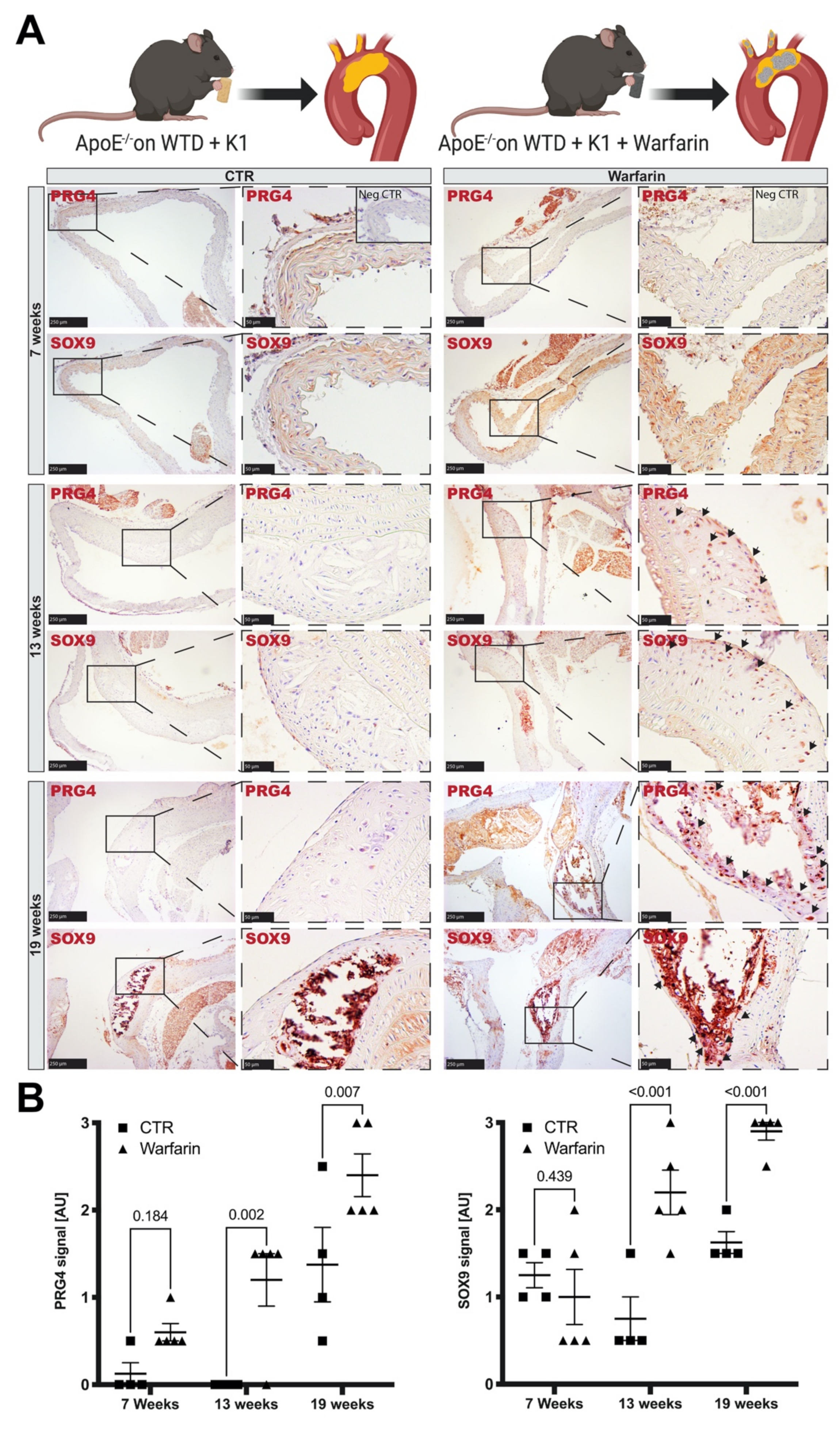
3.4. Accumulation of PRG4 Precedes Intimal Macro-Calcification In Vivo
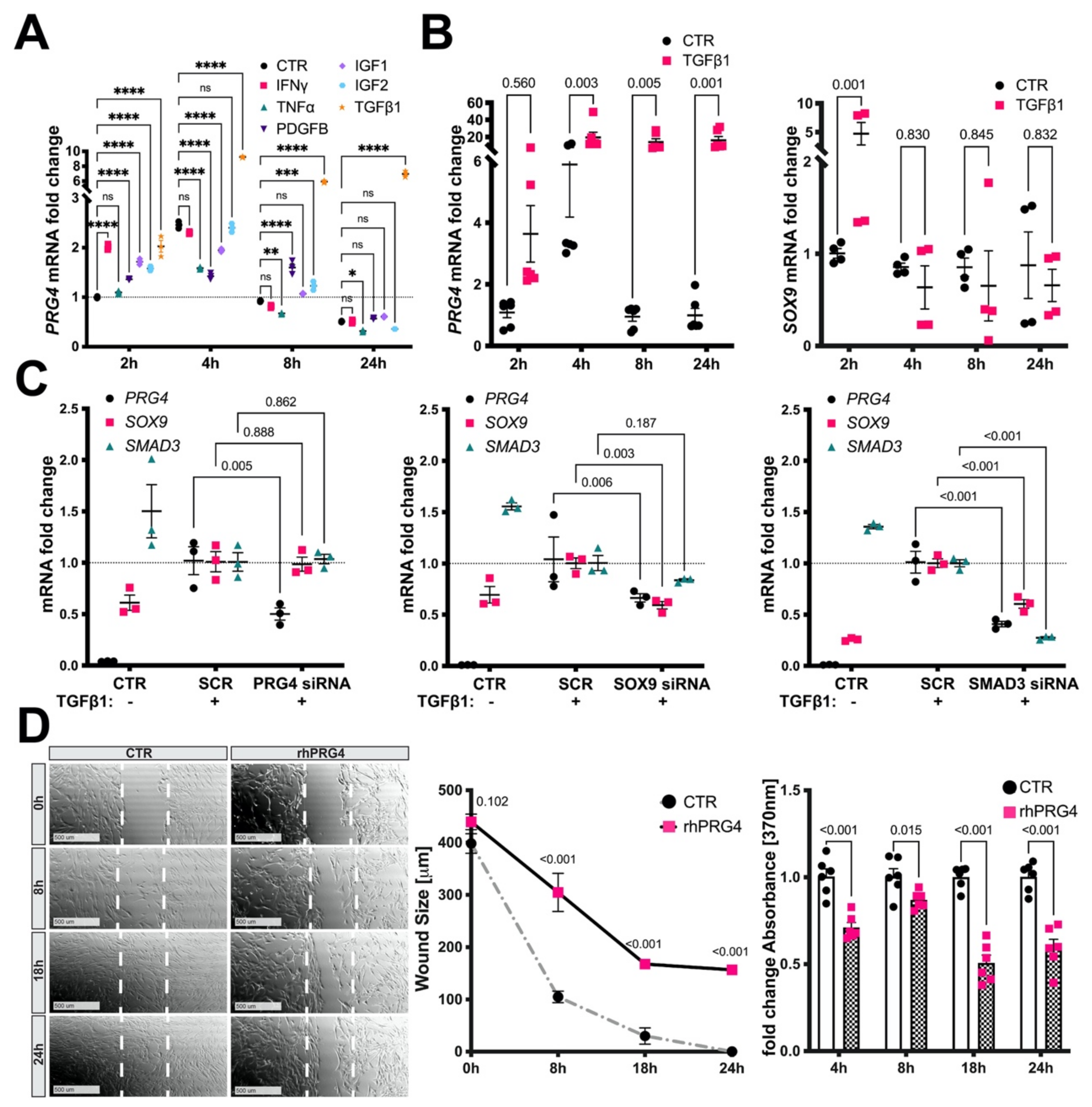
3.5. TGFb1, SMAD3, and SOX9 Control PRG4 Induction in SMCs
3.6. Exogenous PRG4 Inhibits SMC Migration and Proliferation In Vitro
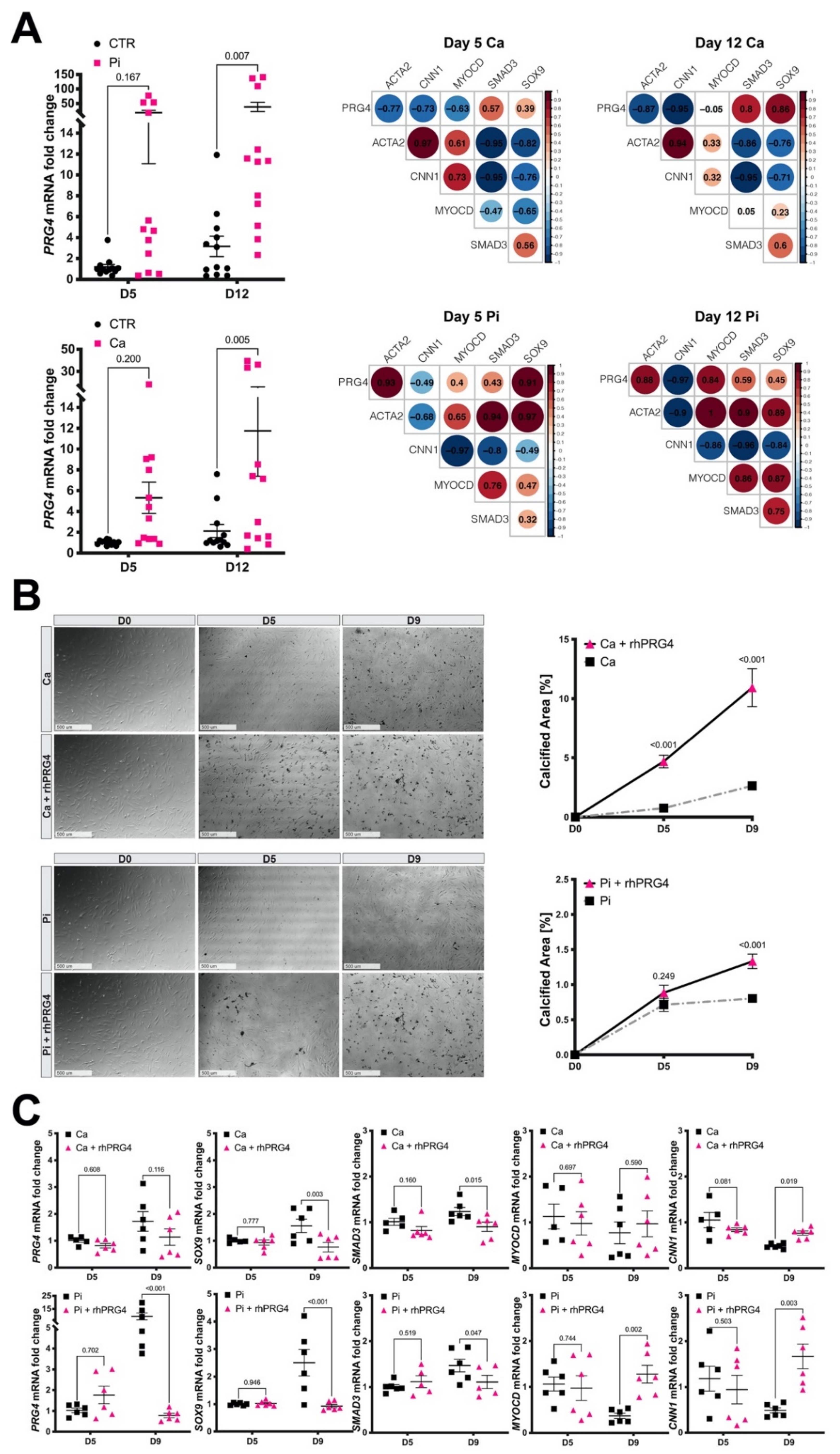
3.7. Calcifying SMCs Upregulate PRG4 Expression
3.8. Exogenous PRG4 Elevates Calcification and Counteracts SMC Phenotypic Switch
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Non-Standard Abbreviations and Acronyms
| CALC | coronary artery calcification |
| CALC Vol Prop | calcification volume proportion |
| ECM | extracellular matrix |
| HAoSMCs | human aortic smooth muscle cells |
| HCtSMCs | human carotid smooth muscle cells |
| LRNC Vol Prop | lipid rich necrotic core volume proportion |
| rhPRG4 | recombinant human proteoglycan 4 |
| TMA | tissue microarray |
| WTD | western type diet |
References
- Finn, A.V.; Nakano, M.; Narula, J.; Kolodgie, F.D.; Virmani, R. Concept of vulnerable/unstable plaque. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1282–1292. [Google Scholar] [CrossRef]
- Motoyama, S.; Kondo, T.; Sarai, M.; Sugiura, A.; Harigaya, H.; Sato, T.; Inoue, K.; Okumura, M.; Ishii, J.; Anno, H.; et al. Multislice computed tomographic characteristics of coronary lesions in acute coronary syndromes. J. Am. Coll. Cardiol. 2007, 50, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Nahrendorf, M.; Figueiredo, J.L.; Swirski, F.K.; Shtatland, T.; Kohler, R.H.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 2007, 116, 2841–2850. [Google Scholar] [CrossRef] [PubMed]
- Karlof, E.; Seime, T.; Dias, N.; Lengquist, M.; Witasp, A.; Almqvist, H.; Kronqvist, M.; Gadin, J.R.; Odeberg, J.; Maegdefessel, L.; et al. Correlation of computed tomography with carotid plaque transcriptomes associates calcification with lesion-stabilization. Atherosclerosis 2019. [Google Scholar] [CrossRef] [PubMed]
- Nandalur, K.R.; Hardie, A.D.; Raghavan, P.; Schipper, M.J.; Baskurt, E.; Kramer, C.M. Composition of the stable carotid plaque: Insights from a multidetector computed tomography study of plaque volume. Stroke 2007, 38, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Perisic, L.; Aldi, S.; Sun, Y.; Folkersen, L.; Razuvaev, A.; Roy, J.; Lengquist, M.; Akesson, S.; Wheelock, C.E.; Maegdefessel, L.; et al. Gene expression signatures, pathways and networks in carotid atherosclerosis. J. Intern. Med. 2016, 279, 293–308. [Google Scholar] [CrossRef]
- Shaalan, W.E.; Cheng, H.; Gewertz, B.; McKinsey, J.F.; Schwartz, L.B.; Katz, D.; Cao, D.; Desai, T.; Glagov, S.; Bassiouny, H.S. Degree of carotid plaque calcification in relation to symptomatic outcome and plaque inflammation. J. Vasc. Surg. 2004, 40, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Pjanic, M.; Wang, T.; Nguyen, T.; Cohain, A.; Lee, J.D.; Perisic, L.; Hedin, U.; Kundu, R.K.; Majmudar, D.; et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun. 2016, 7, 12092. [Google Scholar] [CrossRef] [PubMed]
- Speer, M.Y.; Yang, H.Y.; Brabb, T.; Leaf, E.; Look, A.; Lin, W.L.; Frutkin, A.; Dichek, D.; Giachelli, C.M. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ. Res. 2009, 104, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Flannery, C.R.; Hughes, C.E.; Schumacher, B.L.; Tudor, D.; Aydelotte, M.B.; Kuettner, K.E.; Caterson, B. Articular cartilage superficial zone protein (SZP) is homologous to megakaryocyte stimulating factor precursor and Is a multifunctional proteoglycan with potential growth-promoting, cytoprotective, and lubricating properties in cartilage metabolism. Biochem. Biophys. Res. Commun. 1999, 254, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Al-Sharif, A.; Jamal, M.; Zhang, L.X.; Larson, K.; Schmidt, T.A.; Jay, G.D.; Elsaid, K.A. Lubricin/Proteoglycan 4 Binding to CD44 Receptor: A Mechanism of the Suppression of Proinflammatory Cytokine-Induced Synoviocyte Proliferation by Lubricin. Arthritis Rheumatol. 2015, 67, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Alquraini, A.; Garguilo, S.; D’Souza, G.; Zhang, L.X.; Schmidt, T.A.; Jay, G.D.; Elsaid, K.A. The interaction of lubricin/proteoglycan 4 (PRG4) with toll-like receptors 2 and 4: An anti-inflammatory role of PRG4 in synovial fluid. Arthritis Res. Ther. 2015, 17, 353. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.M.; Leonard, C.; Regmi, S.C.; De Rantere, D.; Tailor, P.; Ren, G.; Ishida, H.; Hsu, C.; Abubacker, S.; Pang, D.S.; et al. Lubricin/Proteoglycan 4 binds to and regulates the activity of Toll-Like Receptors In Vitro. Sci. Rep. 2016, 6, 18910. [Google Scholar] [CrossRef]
- Novince, C.M.; Michalski, M.N.; Koh, A.J.; Sinder, B.P.; Entezami, P.; Eber, M.R.; Pettway, G.J.; Rosol, T.J.; Wronski, T.J.; Kozloff, K.M.; et al. Proteoglycan 4: A dynamic regulator of skeletogenesis and parathyroid hormone skeletal anabolism. J. Bone Miner. Res. 2012, 27, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Xu, C.; Li, X.; Song, J.; Yu, B. Treatment with recombinant lubricin attenuates osteoarthritis by positive feedback loop between articular cartilage and subchondral bone in ovariectomized rats. Bone 2015, 74, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Artiach, G.; Carracedo, M.; Seime, T.; Plunde, O.; Laguna-Fernandez, A.; Matic, L.; Franco-Cereceda, A.; Bäck, M. Proteoglycan 4 is Increased in Human Calcified Aortic Valves and Enhances Valvular Interstitial Cell Calcification. Cells 2020, 9, 684. [Google Scholar] [CrossRef] [PubMed]
- Niikura, T.; Reddi, A.H. Differential regulation of lubricin/superficial zone protein by transforming growth factor beta/bone morphogenetic protein superfamily members in articular chondrocytes and synoviocytes. Arthritis Rheum. 2007, 56, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Naylor, A.R.; Rothwell, P.M.; Bell, P.R. Overview of the principal results and secondary analyses from the European and North American randomised trials of endarterectomy for symptomatic carotid stenosis. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2003, 26, 115–129. [Google Scholar] [CrossRef]
- Halliday, A.; Harrison, M.; Hayter, E.; Kong, X.; Mansfield, A.; Marro, J.; Pan, H.; Peto, R.; Potter, J.; Rahimi, K.; et al. 10-year stroke prevention after successful carotid endarterectomy for asymptomatic stenosis (ACST-1): A multicentre randomised trial. Lancet 2010, 376, 1074–1084. [Google Scholar] [CrossRef]
- Perisic, L.; Hedin, E.; Razuvaev, A.; Lengquist, M.; Osterholm, C.; Folkersen, L.; Gillgren, P.; Paulsson-Berne, G.; Ponten, F.; Odeberg, J.; et al. Profiling of atherosclerotic lesions by gene and tissue microarrays reveals PCSK6 as a novel protease in unstable carotid atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2432–2443. [Google Scholar] [CrossRef]
- Perisic Matic, L.; Rykaczewska, U.; Razuvaev, A.; Sabater-Lleal, M.; Lengquist, M.; Miller, C.L.; Ericsson, I.; Rohl, S.; Kronqvist, M.; Aldi, S.; et al. Phenotypic Modulation of Smooth Muscle Cells in Atherosclerosis Is Associated With Downregulation of LMOD1, SYNPO2, PDLIM7, PLN, and SYNM. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Natural history and histological classification of atherosclerotic lesions: An update. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef]
- van Dijk, R.A.; Virmani, R.; von der Thusen, J.H.; Schaapherder, A.F.; Lindeman, J.H. The natural history of aortic atherosclerosis: A systematic histopathological evaluation of the peri-renal region. Atherosclerosis 2010, 210, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Li, D.Y.; Chernogubova, E.; Sun, C.; Busch, A.; Eken, S.M.; Saliba-Gustafsson, P.; Winter, H.; Winski, G.; Raaz, U.; et al. Local Delivery of miR-21 Stabilizes Fibrous Caps in Vulnerable Atherosclerotic Lesions. Mol. Ther. 2018, 26, 1040–1055. [Google Scholar] [CrossRef] [PubMed]
- Razuvaev, A.; Henderson, B.; Girnita, L.; Larsson, O.; Axelson, M.; Hedin, U.; Roy, J. The cyclolignan picropodophyllin attenuates intimal hyperplasia after rat carotid balloon injury by blocking insulin-like growth factor-1 receptor signaling. J. Vasc. Surg. 2007, 46, 108–115. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Röhl, S.; Rykaczewska, U.; Seime, T.; Suur, B.E.; GonzalezDiez, M.; Gådin, J.R.; Gainullina, A.; Sergushichev, A.A.; Wirka, R.; Lengquist, M.; et al. Transcriptomic profiling of experimental arterial injury reveals new mechanisms and temporal dynamics in vascular healing response. J. Vasc. Surg. Vasc. Sci. 2020. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Joosen, I.A.; Laufer, E.M.; Chatrou, M.L.; Herfs, M.; Winkens, M.H.; Westenfeld, R.; Veulemans, V.; Krueger, T.; Shanahan, C.M.; et al. Vitamin K-antagonists accelerate atherosclerotic calcification and induce a vulnerable plaque phenotype. PLoS ONE 2012, 7, e43229. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. JASN 2004, 15, 2857–2867. [Google Scholar] [CrossRef]
- Willems, B.A.; Furmanik, M.; Caron, M.M.J.; Chatrou, M.L.L.; Kusters, D.H.M.; Welting, T.J.M.; Stock, M.; Rafael, M.S.; Viegas, C.S.B.; Simes, D.C.; et al. Ucma/GRP inhibits phosphate-induced vascular smooth muscle cell calcification via SMAD-dependent BMP signalling. Sci. Rep. 2018, 8, 4961. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef]
- Abubacker, S.; Dorosz, S.G.; Ponjevic, D.; Jay, G.D.; Matyas, J.R.; Schmidt, T.A. Full-Length Recombinant Human Proteoglycan 4 Interacts with Hyaluronan to Provide Cartilage Boundary Lubrication. Ann. Biomed. Eng. 2016, 44, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Waller, K.A.; Chin, K.E.; Jay, G.D.; Zhang, L.X.; Teeple, E.; McAllister, S.; Badger, G.J.; Schmidt, T.A.; Fleming, B.C. Intra-articular Recombinant Human Proteoglycan 4 Mitigates Cartilage Damage After Destabilization of the Medial Meniscus in the Yucatan Minipig. Am. J. Sports Med. 2017, 45, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Uceda, D.E.; Dey, A.K.; Abdelrahman, K.M.; Aksentijevich, M.; Rodante, J.A.; Elnabawi, Y.A.; Reddy, A.; Keel, A.; Erb-Alvarez, J.; et al. Treatment of Psoriasis with Biologic Therapy Is Associated With Improvement of Coronary Artery Plaque Lipid-Rich Necrotic Core: Results From a Prospective, Observational Study. Circ. Cardiovasc. Imaging 2020, 13, e011199. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, K.M.; Chen, M.Y.; Dey, A.K.; Virmani, R.; Finn, A.V.; Khamis, R.Y.; Choi, A.D.; Min, J.K.; Williams, M.C.; Buckler, A.J.; et al. Coronary Computed Tomography Angiography From Clinical Uses to Emerging Technologies: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1226–1243. [Google Scholar] [CrossRef]
- Van Assen, M.; Varga-Szemes, A.; Egorova, S.; Johnson, K.; St. Pierre, S.; Zaki, B.; Schoepf, U.J.; Buckler, A.J. Automated plaque analysis for the prognostication of major adverse cardiac events. Eur. Soc. Cardiol. 2019, 116, 8. [Google Scholar] [CrossRef]
- Zhu, G.; Li, Y.; Ding, V.; Jiang, B.; Ball, R.L.; Rodriguez, F.; Fleischmann, D.; Desai, M.; Saloner, D.; Gupta, A.; et al. Semiautomated Characterization of Carotid Artery Plaque Features From Computed Tomography Angiography to Predict Atherosclerotic Cardiovascular Disease Risk Score. J. Comput. Assist. Tomogr. 2019, 43, 452–459. [Google Scholar] [CrossRef]
- Sheahan, M.; Ma, X.; Paik, D.; Obuchowski, N.A.; St Pierre, S.; Newman, W.P., 3rd; Rae, G.; Perlman, E.S.; Rosol, M.; Keith, J.C., Jr.; et al. Atherosclerotic Plaque Tissue: Noninvasive Quantitative Assessment of Characteristics with Software-aided Measurements from Conventional CT Angiography. Radiology 2018, 286, 622–631. [Google Scholar] [CrossRef]
- Chrencik, M.T.; Khan, A.A.; Luther, L.; Anthony, L.; Yokemick, J.; Patel, J.; Sorkin, J.D.; Sikdar, S.; Lal, B.K. Quantitative assessment of carotid plaque morphology (geometry and tissue composition) using computed tomography angiography. J. Vasc. Surg. 2019, 70, 858–868. [Google Scholar] [CrossRef]
- Wickham, H. RStudio, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Wei, T.; Simko, V. R package “corrplot”: Visualization of a Correlation Matrix (Version 0.88). Available online: https://github.com/taiyun/corrplot (accessed on 21 May 2021).
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W., Jr.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, N.; Raggi, P. Calcification in atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; de Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef]
- Augstein, A.; Mierke, J.; Poitz, D.M.; Strasser, R.H. Sox9 is increased in arterial plaque and stenosis, associated with synthetic phenotype of vascular smooth muscle cells and causes alterations in extracellular matrix and calcification. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2526–2537. [Google Scholar] [CrossRef]
- Speer, M.Y.; Li, X.; Hiremath, P.G.; Giachelli, C.M. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J. Cell Biochem. 2010, 110, 935–947. [Google Scholar] [CrossRef]
- Deckers, M.M.; Van Beek, E.R.; Van Der Pluijm, G.; Wetterwald, A.; Van Der Wee-Pals, L.; Cecchini, M.G.; Papapoulos, S.E.; Lowik, C.W. Dissociation of angiogenesis and osteoclastogenesis during endochondral bone formation in neonatal mice. J. Bone Miner. Res. 2002, 17, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Daoudi, M.; Rosa, M.; Vinod, M.; Louvet, L.; Copin, C.; Fanchon, M.; Vanhoutte, J.; Derudas, B.; Belloy, L.; et al. Human Alternative Macrophages Populate Calcified Areas of Atherosclerotic Lesions and Display Impaired RANKL-Induced Osteoclastic Bone Resorption Activity. Circ. Res. 2017, 121, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R.; Astudillo, L.; Elmariah, S.; Purushothaman, K.R.; Purushothaman, M.; Lento, P.A.; Sharma, S.K.; Fuster, V.; Adams, D.H. Increased macrophage infiltration and neovascularization in congenital bicuspid aortic valve stenosis. J. Thorac. Cardiovasc. Surg. 2011, 142, 895–901. [Google Scholar] [CrossRef]
- Xu, Z.; Ji, G.; Shen, J.; Wang, X.; Zhou, J.; Li, L. SOX9 and myocardin counteract each other in regulating vascular smooth muscle cell differentiation. Biochem. Biophys. Res. Commun. 2012, 422, 285–290. [Google Scholar] [CrossRef]
- Majesky, M.W.; Schwartz, S.M.; Clowes, M.M.; Clowes, A.W. Heparin regulates smooth muscle S phase entry in the injured rat carotid artery. Circ. Res. 1987, 61, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Thyberg, J.; Blomgren, K.; Hedin, U.; Dryjski, M. Phenotypic modulation of smooth muscle cells during the formation of neointimal thickenings in the rat carotid artery after balloon injury: An electron-microscopic and stereological study. Cell Tissue Res. 1995, 281, 421–433. [Google Scholar] [CrossRef]
- Majd, S.E.; Kuijer, R.; Kowitsch, A.; Groth, T.; Schmidt, T.A.; Sharma, P.K. Both hyaluronan and collagen type II keep proteoglycan 4 (lubricin) at the cartilage surface in a condition that provides low friction during boundary lubrication. Langmuir 2014, 30, 14566–14572. [Google Scholar] [CrossRef]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Wong, W.; Reginato, A.M.; Sun, C.; Schmidt, T.A.; Elsaid, K.A. Recombinant human proteoglycan-4 reduces phagocytosis of urate crystals and downstream nuclear factor kappa B and inflammasome activation and production of cytokines and chemokines in human and murine macrophages. Arthritis Res. Ther. 2018, 20, 192. [Google Scholar] [CrossRef] [PubMed]
- Nahon, J.E.; Hoekstra, M.; Havik, S.R.; Van Santbrink, P.J.; Dallinga-Thie, G.M.; Kuivenhoven, J.A.; Geerling, J.J.; Van Eck, M. Proteoglycan 4 regulates macrophage function without altering atherosclerotic lesion formation in a murine bone marrow-specific deletion model. Atherosclerosis 2018, 274, 120–127. [Google Scholar] [CrossRef] [PubMed]
- DuRaine, G.D.; Chan, S.M.; Reddi, A.H. Effects of TGF-β1 on alternative splicing of Superficial Zone Protein in articular cartilage cultures. Osteoarthr. Cartil. 2011, 19, 103–110. [Google Scholar] [CrossRef]
- Chavez, R.D.; Coricor, G.; Perez, J.; Seo, H.S.; Serra, R. SOX9 protein is stabilized by TGF-β and regulates PAPSS2 mRNA expression in chondrocytes. Osteoarthr. Cartil. 2017, 25, 332–340. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Kalampogias, A.; Siasos, G.; Oikonomou, E.; Tsalamandris, S.; Mourouzis, K.; Tsigkou, V.; Vavuranakis, M.; Zografos, T.; Deftereos, S.; Stefanadis, C.; et al. Basic Mechanisms in Atherosclerosis: The Role of Calcium. Med. Chem. 2016, 12, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Jay, G.D.; Tantravahi, U.; Britt, D.E.; Barrach, H.J.; Cha, C.J. Homology of lubricin and superficial zone protein (SZP): Products of megakaryocyte stimulating factor (MSF) gene expression by human synovial fibroblasts and articular chondrocytes localized to chromosome 1q25. J. Orthop. Res. 2001, 19, 677–687. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wirka, R.C.; Kim, J.B.; Nguyen, T.; Kundu, R.; Zhao, Q.; Pedroza, A.; Nagao, M.; Iyer, D.; Fischbein, M.P.; et al. Smad3 Regulates Smooth Muscle Cell Fate and Governs Adverse Remodeling and Calcification of Atherosclerotic Plaque. bioRxiv 2020. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seime, T.; Akbulut, A.C.; Liljeqvist, M.L.; Siika, A.; Jin, H.; Winski, G.; van Gorp, R.H.; Karlöf, E.; Lengquist, M.; Buckler, A.J.; et al. Proteoglycan 4 Modulates Osteogenic Smooth Muscle Cell Differentiation during Vascular Remodeling and Intimal Calcification. Cells 2021, 10, 1276. https://doi.org/10.3390/cells10061276
Seime T, Akbulut AC, Liljeqvist ML, Siika A, Jin H, Winski G, van Gorp RH, Karlöf E, Lengquist M, Buckler AJ, et al. Proteoglycan 4 Modulates Osteogenic Smooth Muscle Cell Differentiation during Vascular Remodeling and Intimal Calcification. Cells. 2021; 10(6):1276. https://doi.org/10.3390/cells10061276
Chicago/Turabian StyleSeime, Till, Asim Cengiz Akbulut, Moritz Lindquist Liljeqvist, Antti Siika, Hong Jin, Greg Winski, Rick H. van Gorp, Eva Karlöf, Mariette Lengquist, Andrew J. Buckler, and et al. 2021. "Proteoglycan 4 Modulates Osteogenic Smooth Muscle Cell Differentiation during Vascular Remodeling and Intimal Calcification" Cells 10, no. 6: 1276. https://doi.org/10.3390/cells10061276
APA StyleSeime, T., Akbulut, A. C., Liljeqvist, M. L., Siika, A., Jin, H., Winski, G., van Gorp, R. H., Karlöf, E., Lengquist, M., Buckler, A. J., Kronqvist, M., Waring, O. J., Lindeman, J. H. N., Biessen, E. A. L., Maegdefessel, L., Razuvaev, A., Schurgers, L. J., Hedin, U., & Matic, L. (2021). Proteoglycan 4 Modulates Osteogenic Smooth Muscle Cell Differentiation during Vascular Remodeling and Intimal Calcification. Cells, 10(6), 1276. https://doi.org/10.3390/cells10061276