
Anti-Tumor Effects of MAPK-Dependent Tumor-Selective Oncolytic Vaccinia Virus Armed with CD/UPRT against Pancreatic Ductal Adenocarcinoma in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Lines
2.2. Virus Preparation
2.3. Viral Infection
2.4. Cytotoxicity Assay
2.5. In Vivo Experiments
2.6. Ex Vivo Infection and Immunohistochemical Analysis
2.7. Statistical Analysis
3. Results
3.1. Tumor Specificity of VGF−/O1−VV Is Dependent on Cellular MAPK/ERK1/2 Activity
3.2. Tumor Specificity of VGF-/O1-VV Enhances Its Therapeutic Index
3.3. In Vitro Viral Oncolytic Effect of CD/UPRT-Armed MDRVV and 5-FC Combination Therapy
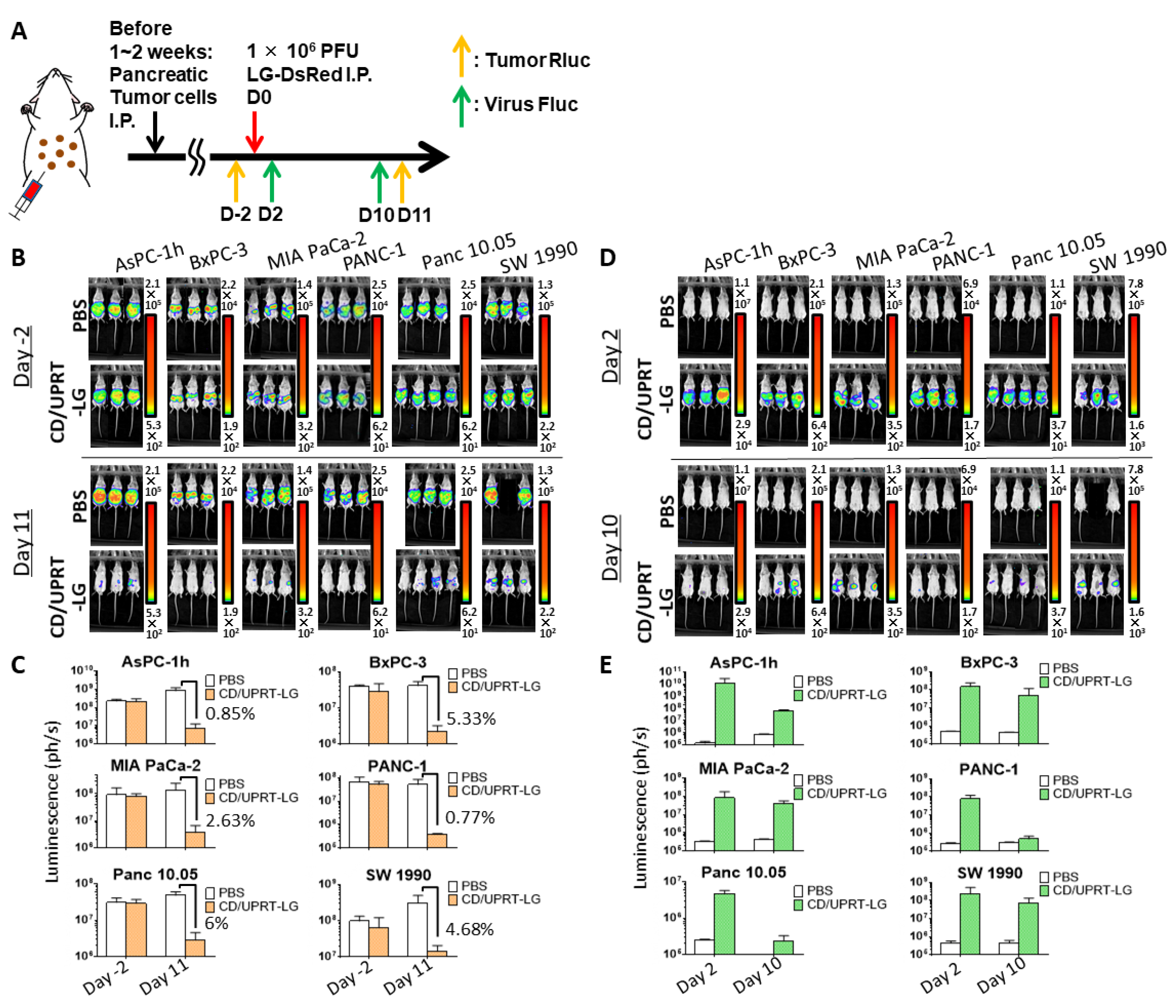
3.4. In Vivo Oncolytic Activity Against Pancreatic Cancers
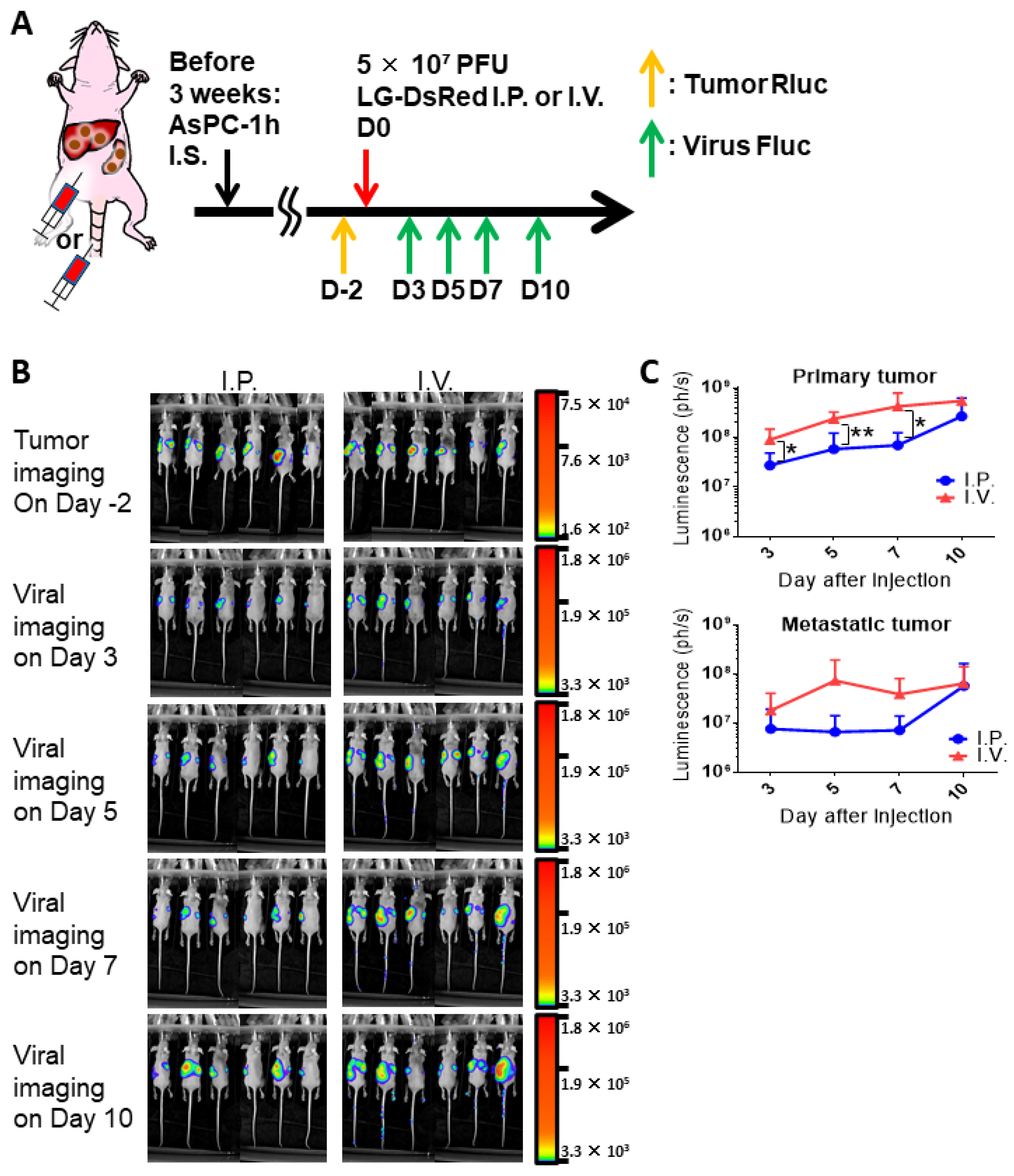
3.5. Combinational Therapy of Armed MDRVV and 5-FC in Clinically Relevant Mouse Tumor Model
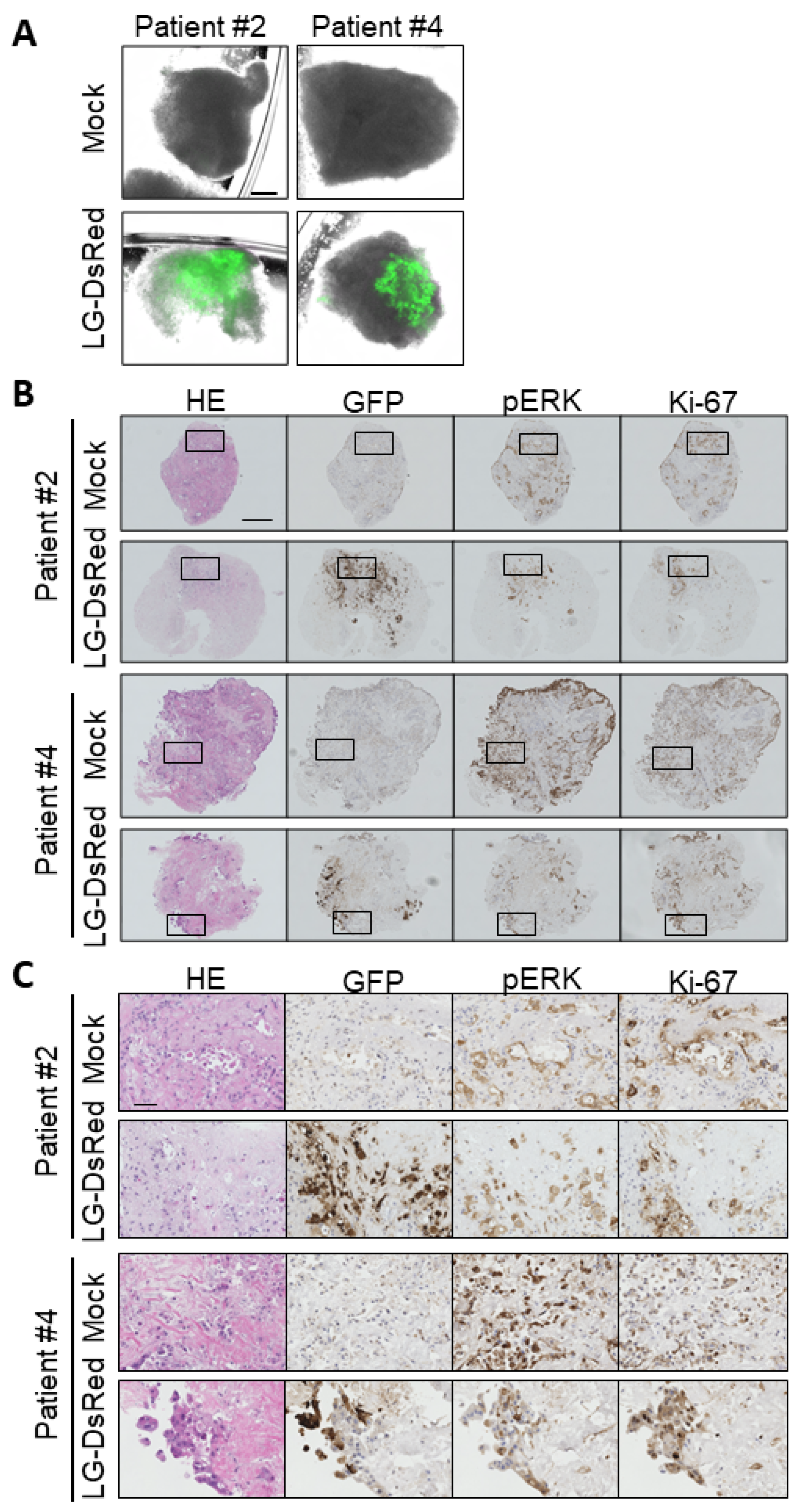
3.6. Ex Vivo Infection of Patient-Derived Tumor Tissue
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, M. Replicating Poxviruses for Human Cancer Therapy. J. Microbiol. 2015, 53, 209–218. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia Virus-mediated Cancer Immunotherapy: Cancer Vaccines and Oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic Armed Oncolytic and Immunologic Therapy for Cancer With JX-594, a Targeted Poxvirus Expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic Cancer Therapy with a Tumor-Selective Vaccinia Virus Mutant Lacking Thymidine Kinase and Vaccinia Growth Factor Genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar] [PubMed]
- Twardzik, D.R.; Brown, J.P.; Ranchalis, J.E.; Todaro, G.J.; Moss, B. Vaccinia Virus-infected Cells Release a Novel Polypeptide Functionally Related to Transforming and Epidermal Growth Factors. Proc. Natl. Acad. Sci. USA 1985, 82, 5300–5304. [Google Scholar] [CrossRef]
- Brown, J.P.; Twardzik, D.R.; Marquardt, H.; Todaro, G.J. Vaccinia Virus Encodes a Polypeptide Homologous to Epidermal Growth Factor and Transforming Growth Factor. Nature 1985, 313, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.M.; Chakrabarti, S.; Cooper, J.A.; Twardzik, D.R.; Moss, B. Deletion of the Vaccinia Virus Growth Factor Gene Reduces Virus Virulence. J. Virol. 1988, 62, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.A.; Silva, P.N.G.; Pereira, A.C.T.C.; De Sousa, L.P.; Ferreira, P.C.P.; Gazzinelli, R.T.; Kroon, E.G.; Ropert, C.; Bonjardim, C.A. The Vaccinia Virus-stimulated Mitogen-activated Protein Kinase (MAPK) Pathway is Required for Virus Multiplication. Biochem. J. 2004, 381, 437–446. [Google Scholar] [CrossRef]
- Buller, R.M.; Chakrabarti, S.; Moss, B.; Fredrickson, T. Cell Proliferative Response to Vaccinia Virus is Mediated by VGF. Virology 1988, 164, 182–192. [Google Scholar] [CrossRef]
- Tzahar, E.; Moyer, J.D.; Waterman, H.; Barbacci, E.G.; Bao, J.; Levkowitz, G.; Shelly, M.; Strano, S.; Pinkas-Kramarski, R.; Pierce, J.H.; et al. Pathogenic Poxviruses Reveal Viral Strategies to Exploit the ErbB Signaling Network. EMBO J. 1998, 17, 5948–5963. [Google Scholar] [CrossRef] [PubMed]
- Schweneker, M.; Lukassen, S.; Späth, M.; Wolferstätter, M.; Babel, E.; Brinkmann, K.; Wielert, U.; Chaplin, P.; Suter, M.; Hausmann, J. The Vaccinia Virus O1 Protein is Required for Sustained Activation of Extracellular Signal-regulated Kinase 1/2 and Promotes Viral Virulence. J. Virol. 2012, 86, 2323–2336. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic Cancer Biology and Genetics from an Evolutionary Perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes with Outcomes of Patients with Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the Undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Knudsen, E.S.; O’Reilly, E.M.; Brody, J.R.; Witkiewicz, A.K. Genetic Diversity of Pancreatic Ductal Adenocarcinoma and Opportunities for Precision Medicine. Gastroenterology 2016, 150, 48–63. [Google Scholar] [CrossRef]
- Li, S.; Balmain, A.; Counter, C.M. A Model for RAS Mutation Patterns in Cancers: Finding the Sweet Spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF Receptor is Required for KRAS-induced Pancreatic Tumorigenesis. Cancer Cell. 2012, 22, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. Pancreatic Cancer: EGFR Inhibition is Effective against KRAS-wild-type Disease. Nat. Rev. Clin. Oncol. 2017, 14, 524–525. [Google Scholar] [CrossRef]
- Yuan, T.L.; Amzallag, A.; Bagni, R.; Yi, M.; Afghani, S.; Burgan, W.; Fer, N.; Strathern, L.A.; Powell, K.; Smith, B.; et al. Differential Effector Engagement by Oncogenic KRAS. Cell Rep. 2018, 22, 1889–1902. [Google Scholar] [CrossRef]
- Fountzilas, C.; Chhatrala, R.; Khushalani, N.; Tan, W.; LeVea, C.; Hutson, A.; Tucker, C.; Ma, W.W.; Warren, G.; Boland, P.; et al. A Phase II Trial of Erlotinib Monotherapy in Advanced Pancreatic Cancer as a First- Or Second-line Agent. Cancer Chemother. Pharmacol. 2017, 80, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Bodoky, G.; Timcheva, C.; Spigel, D.R.; La Stella, P.J.; Ciuleanu, T.E.; Pover, G.; Tebbutt, N.C. A Phase II Open-label Randomized Study to Assess the Efficacy and Safety of Selumetinib (AZD6244 [ARRY-142886]) Versus Capecitabine in Patients with Advanced or Metastatic Pancreatic Cancer Who Have Failed First-line Gemcitabine Therapy. Investig. New Drugs 2012, 30, 1216–1223. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Malek, S. Targeting the MAPK Pathway in RAS Mutant Cancers. Cold Spring Harb. Perspect. Med. 2018, 8, a031492. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Hanker, A.B.; Garrett, J.T. Genomic Alterations of ERBB Receptors in Cancer: Clinical Implications. Oncotarget 2017, 8, 114371–114392. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Gopinath, P.; Ghosh, S.S. Implication of Functional Activity for Determining Therapeutic Efficacy of Suicide Genes In Vitro. Biotechnol. Lett. 2008, 30, 1913–1921. [Google Scholar] [CrossRef]
- Chalikonda, S.; Kivlen, M.H.; O’Malley, M.E.; Eric Dong, X.D.; McCart, J.A.; Gorry, M.C.; Yin, X.Y.; Brown, C.K.; Zeh, H.J., 3rd; Guo, Z.S.; et al. Oncolytic virotherapy for ovarian carcinomatosis using a replication-selective vaccinia virus armed with a yeast cytosine deaminase gene. Cancer Gene Ther. 2008, 15, 115–125. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.J.; et al. Effect of Vocimagene Amiretrorepvec in Combination with Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients with Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1939–1946. [Google Scholar] [CrossRef]
- Zaoui, K.; Bossow, S.; Grossardt, C.; Leber, M.F.; Springfeld, C.; Plinkert, P.K.; Kalle, C.; Ungerechts, G. Chemovirotherapy for head and neck squamous cell carcinoma with EGFR-targeted and CD/UPRT-armed oncolytic measles virus. Cancer Gene Ther. 2012, 19, 181–191. [Google Scholar] [CrossRef]
- Foloppe, J.; Kempf, J.; Futin, N.; Kintz, J.; Cordier, P.; Pichon, C.; Findeli, A.; Vorburger, F.; Quemeneur, E.; Erbs, P. The Enhanced Tumor Specificity of TG6002, an Armed Oncolytic Vaccinia Virus Deleted in Two Genes Involved in Nucleotide Metabolism. Mol. Ther. Oncolytics 2019, 14, 1–14. [Google Scholar] [CrossRef]
- Franck, C.; Müller, C.; Rosania, R.; Croner, R.S.; Pech, M.; Venerito, M. Advanced Pancreatic Ductal Adenocarcinoma: Moving Forward. Cancers 2020, 12, 1995. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, S.; Ikeshita, S.; Miyatake, Y.; Kasahara, M. Pancreatic Cancer Cells Express CD44 Variant 9 and Multidrug Resistance Protein 1 During Mitosis. Exp. Mol. Pathol. 2015, 98, 41–46. [Google Scholar] [CrossRef]
- Hasegawa, K.; Nakamura, T.; Harvey, M.; Ikeda, Y.; Oberg, A.; Figini, M.; Canevari, S.; Hartmann, L.C.; Peng, K.W. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin. Cancer Res. 2006, 12, 6170–6178. [Google Scholar] [CrossRef] [PubMed]
- Hikichi, M.; Kidokoro, M.; Haraguchi, T.; Iba, H.; Shida, H.; Tahara, H.; Nakamura, T. MicroRNA Regulation of Glycoprotein B5R in Oncolytic Vaccinia Virus Reduces Viral Pathogenicity without Impairing Its Antitumor Efficacy. Mol. Ther. 2011, 19, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T. National Center for Biotechnology Information. “PubChem Patent Summary for US-9809803-B2”. Available online: https://pubchem.ncbi.nlm.nih.gov/patent/US-9809803-B2 (accessed on 27 January 2021).
- Nakamura, T.; Nakatake, M.; Kurosaki, H.; Horita, K. Method for Producing Vaccinia Virus Expressing Foreign Gene. U.S. Patent 16/317,198, 5 December 2019. [Google Scholar]
- Horita, K.; Kurosaki, H.; Nakatake, M.; Kuwano, N.; Oishi, T.; Itamochi, H.; Sato, S.; Kono, H.; Ito, M.; Hasegawa, K.; et al. lncRNA UCA1-Mediated Cdc42 Signaling Promotes Oncolytic Vaccinia Virus Cell-to-Cell Spread in Ovarian Cancer. Mol. Ther. Oncolytics 2019, 13, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.M.; Oke, P.G.; Coupar, B.E. A Synthetic Vaccinia Virus Promoter with Enhanced Early and Late Activity. J. Virol. Methods 1997, 66, 135–138. [Google Scholar] [CrossRef]
- Kenner, J.; Cameron, F.; Empig, C.; Jobes, D.V.; Gurwith, M. LC16m8: An Attenuated Smallpox Vaccine. Vaccine 2006, 24, 7009–7022. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, K.; Yoshizawa, H.; Morita, M.; Suzuki, K. Properties of Attenuated Mutant of Vaccinia Virus, LC16m8, Derived from Lister Strain; Elsevier Science Publishing Co. Inc.: New York, NY, USA, 1985; pp. 421–428. [Google Scholar]
- Recommendations of the Advisory Committee on Immunization Practices (ACIP): Use of vaccines and immune globulins for persons with altered immunocompetence. MMWR Recomm Rep. 1993, 42, 1–18.
- Lun, X.Q.; Jang, J.H.; Tang, N.; Deng, H.; Head, R.; Bell, J.C.; Stojdl, D.F.; Nutt, C.L.; Senger, D.L.; Forsyth, P.A.; et al. Efficacy of Systemically Administered Oncolytic Vaccinia Virotherapy for Malignant Gliomas is Enhanced by Combination Therapy with Rapamycin or Cyclophosphamide. Clin. Cancer Res. 2009, 15, 2777–2788. [Google Scholar] [CrossRef]
- Ikeda, K.; Ichikawa, T.; Wakimoto, H.; Silver, J.S.; Deisboeck, T.S.; Finkelstein, D.; Harsh, G.R., 4th; Louis, D.N.; Bartus, R.T.; Hochberg, F.H.; et al. Oncolytic Virus Therapy of Multiple Tumors in The Brain Requires Suppression of Innate and Elicited Antiviral Responses. Nat. Med. 1999, 5, 881–887. [Google Scholar] [CrossRef]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Moz, S.; Basso, D.; Bozzato, D.; Galozzi, P.; Navaglia, F.; Negm, O.H.; Arrigoni, G.; Zambon, C.F.; Padoan, A.; Tighe, P.; et al. SMAD4 Loss Enables EGF, TGFβ1 and S100A8/A9 Induced Activation of Critical Pathways to Invasion in Human Pancreatic Adenocarcinoma Cells. Oncotarget 2016, 7, 69927–69944. [Google Scholar] [CrossRef]
- Yasutome, M.; Gunn, J.; Korc, M. Restoration of Smad4 in BxPC3 Pancreatic Cancer Cells Attenuates Proliferation Without Altering Angiogenesis. Clin. Exp. Metastasis 2005, 22, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Nakatake, M.; Kurosaki, H.; Kuwano, N.; Horita, K.; Ito, M.; Kono, H.; Okamura, T.; Hasegawa, K.; Yasutomi, Y.; Nakamura, T. Partial Deletion of Glycoprotein B5R Enhances Vaccinia Virus Neutralization Escape while Preserving Oncolytic Function. Mol. Ther. Oncolytics 2019, 14, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Niu, M.; Yuan, X.; Wu, K.; Liu, A. CD44 as a Tumor Biomarker and Therapeutic Target. Exp. Hematol. Oncol. 2020, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, S.; Tsugawa, H.; Matsuzaki, J.; Hirata, K.; Mori, H.; Saya, H.; Kanai, T.; Suzuki, H. Inhibiting xCT Improves 5-Fluorouracil Resistance of Gastric Cancer Induced by CD44 Variant 9 Expression. Anticancer Res. 2018, 38, 6163–6170. [Google Scholar] [CrossRef]
- Nakao, S.; Arai, Y.; Tasaki, M.; Yamashita, M.; Murakami, R.; Kawase, T.; Amino, N.; Nakatake, M.; Kurosaki, H.; Mori, M.; et al. Intratumoral Expression of IL-7 and IL-12 using an Oncolytic Virus Increases Systemic Sensitivity to Immune Checkpoint Blockade. Sci. Transl. Med. 2020, 12, eaax7992. [Google Scholar] [CrossRef]
- Nakatake, M.; Kuwano, N.; Kaitsurumaru, E.; Kurosaki, H.; Nakamura, T. Fusogenic Oncolytic Vaccinia Virus Enhances Systemic Antitumor Immune Response by Modulating the Tumor Microenvironment. Mol. Ther. 2020. [Google Scholar] [CrossRef]
- Ferguson, M.S.; Dunmall, L.S.C.; Gangeswaran, R.; Marelli, G.; Tysome, J.R.; Burns, E.; Whitehead, M.A.; Aksoy, E.; Alusi, G.; Hiley, C.; et al. Transient Inhibition of PI3Kdelta Enhances the Therapeutic Effect of Intravenous Delivery of Oncolytic Vaccinia Virus. Mol. Ther. 2020, 28, 1263–1275. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurosaki, H.; Nakatake, M.; Sakamoto, T.; Kuwano, N.; Yamane, M.; Ishii, K.; Fujiwara, Y.; Nakamura, T. Anti-Tumor Effects of MAPK-Dependent Tumor-Selective Oncolytic Vaccinia Virus Armed with CD/UPRT against Pancreatic Ductal Adenocarcinoma in Mice. Cells 2021, 10, 985. https://doi.org/10.3390/cells10050985
Kurosaki H, Nakatake M, Sakamoto T, Kuwano N, Yamane M, Ishii K, Fujiwara Y, Nakamura T. Anti-Tumor Effects of MAPK-Dependent Tumor-Selective Oncolytic Vaccinia Virus Armed with CD/UPRT against Pancreatic Ductal Adenocarcinoma in Mice. Cells. 2021; 10(5):985. https://doi.org/10.3390/cells10050985
Chicago/Turabian StyleKurosaki, Hajime, Motomu Nakatake, Teruhisa Sakamoto, Nozomi Kuwano, Masato Yamane, Kenta Ishii, Yoshiyuki Fujiwara, and Takafumi Nakamura. 2021. "Anti-Tumor Effects of MAPK-Dependent Tumor-Selective Oncolytic Vaccinia Virus Armed with CD/UPRT against Pancreatic Ductal Adenocarcinoma in Mice" Cells 10, no. 5: 985. https://doi.org/10.3390/cells10050985
APA StyleKurosaki, H., Nakatake, M., Sakamoto, T., Kuwano, N., Yamane, M., Ishii, K., Fujiwara, Y., & Nakamura, T. (2021). Anti-Tumor Effects of MAPK-Dependent Tumor-Selective Oncolytic Vaccinia Virus Armed with CD/UPRT against Pancreatic Ductal Adenocarcinoma in Mice. Cells, 10(5), 985. https://doi.org/10.3390/cells10050985