
An Arrhythmic Mutation E7K Facilitates TRPM4 Channel Activation via Enhanced PIP2 Interaction

 , , , , and
, , , , and 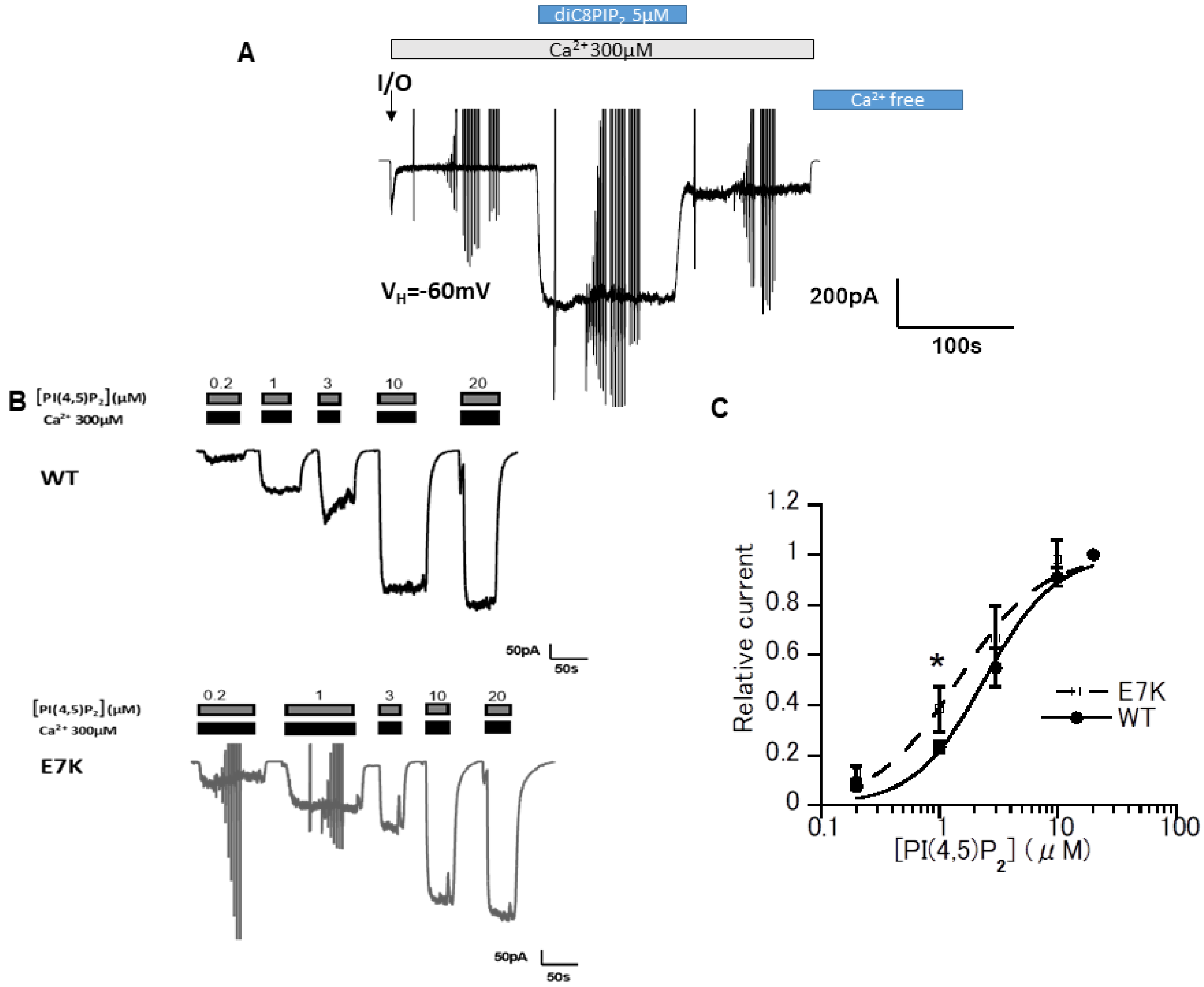{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Gene Transfection
2.2. Solution
2.3. Electrophysiology
2.4. FRET Measurement
2.5. Statistical Evaluation
3. Results
3.1. DiC8-PIP2 More Potently Reactivates E7K Than WT-TRPM4 Channels
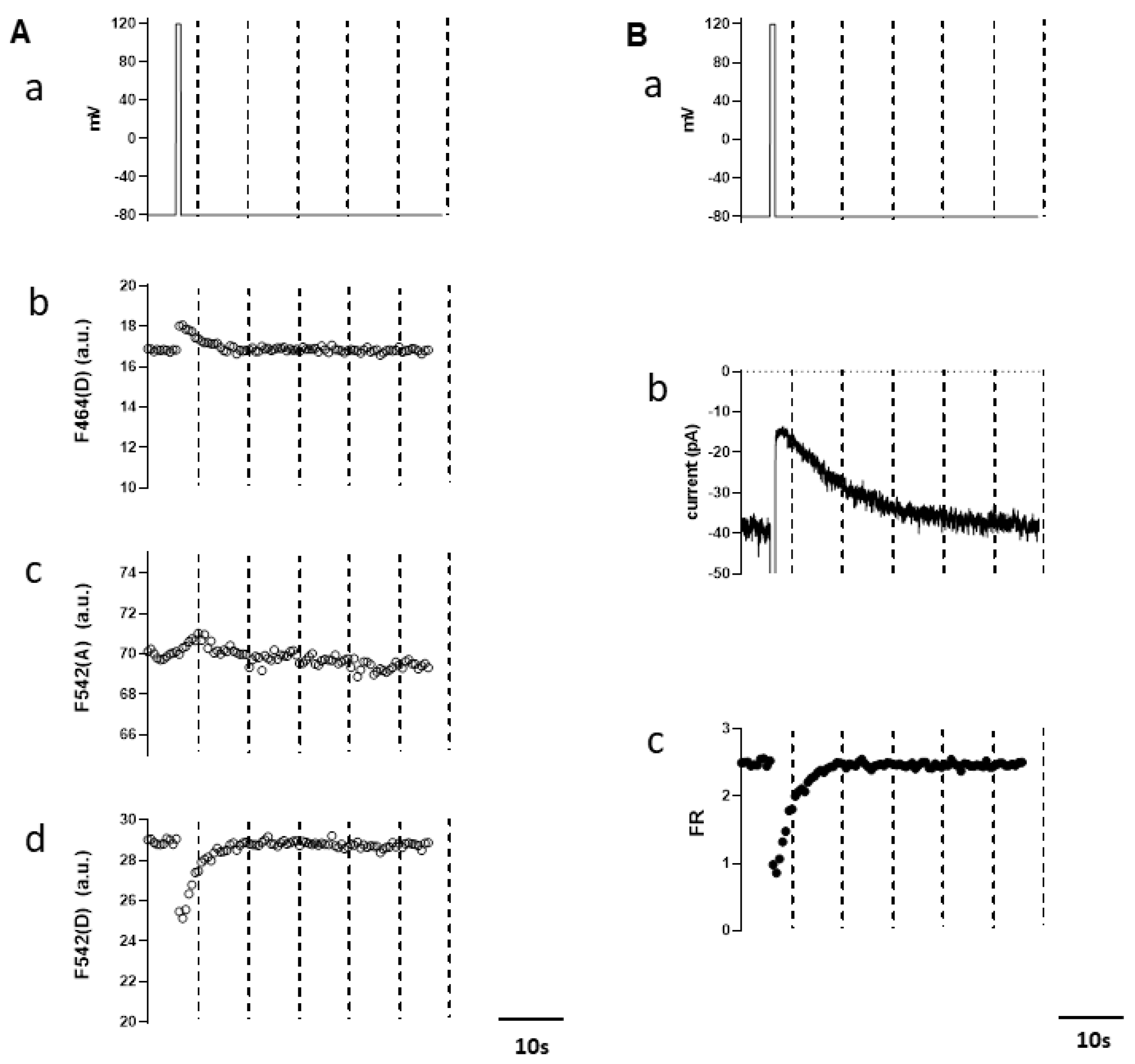
3.2. Simultaneous Measurements of Endogenous PI(4,5)P2 and TRPM4 Channel Activity
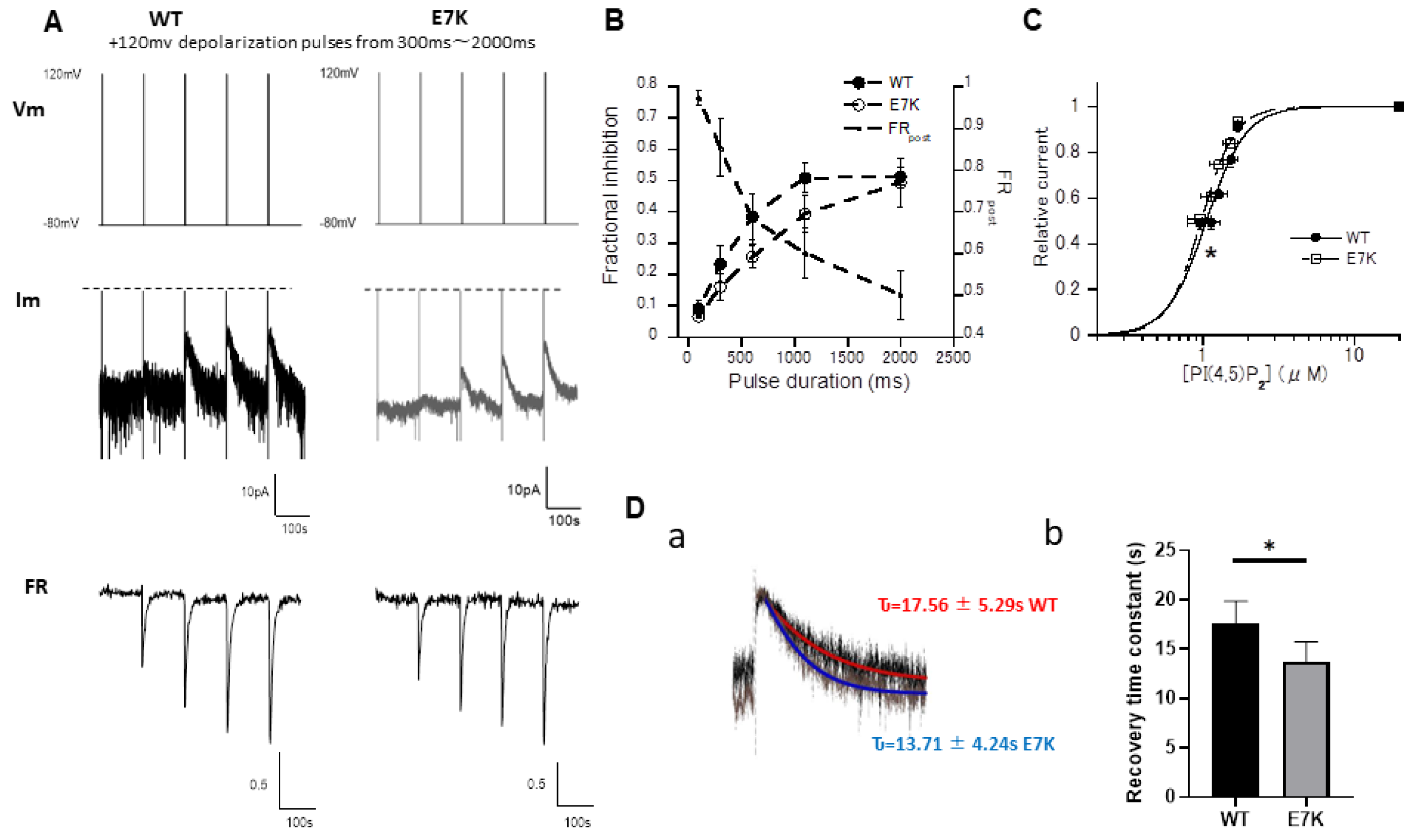
3.3. Differential PIP2 Sensitivities of WT and E7K-Mutant TRPM4 Channels
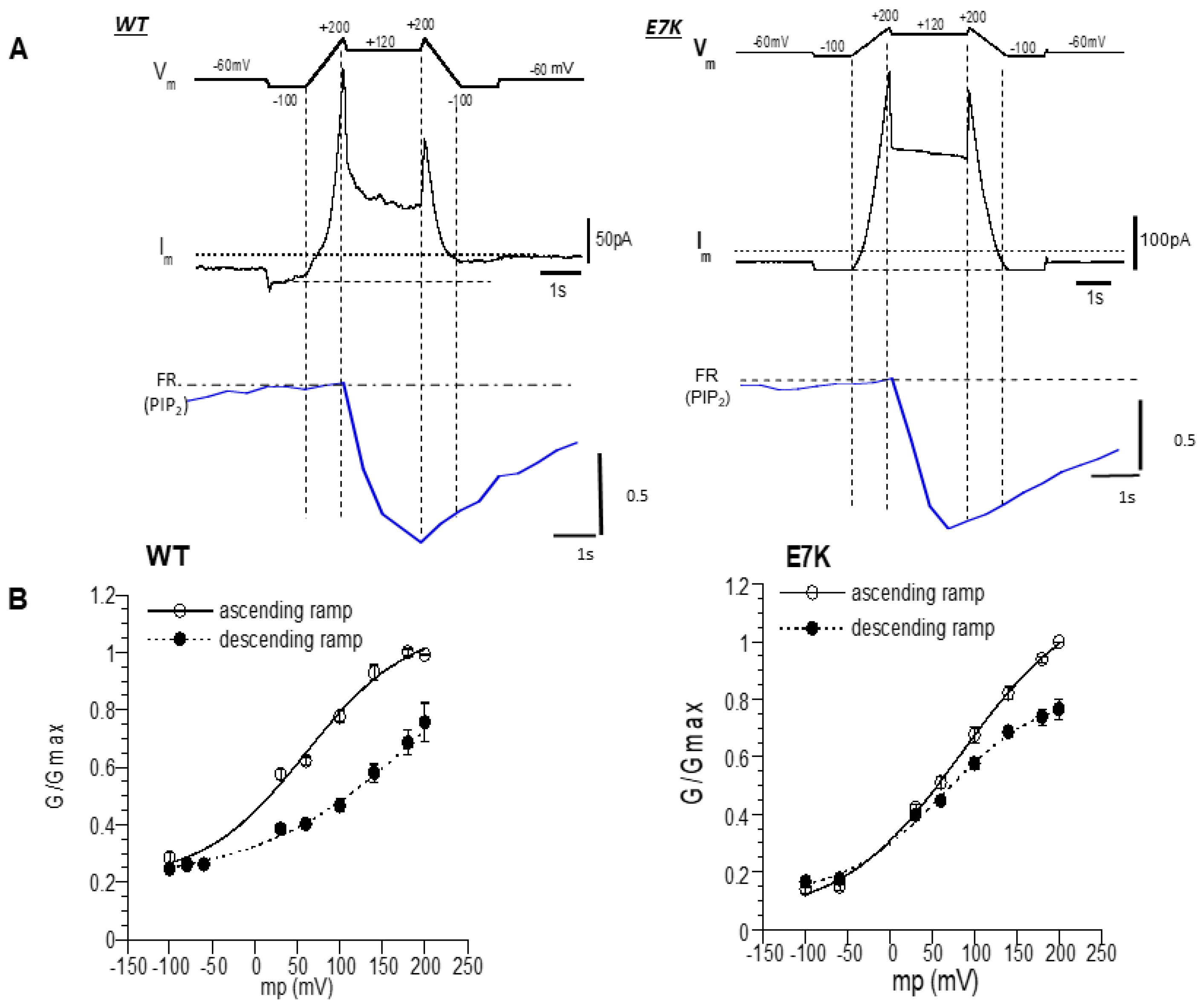
3.4. PIP2 Depletion Only Modestly Affects the Voltage-Dependent Activation of E7K Mutant Because of Its Higher PIP2 Affinity
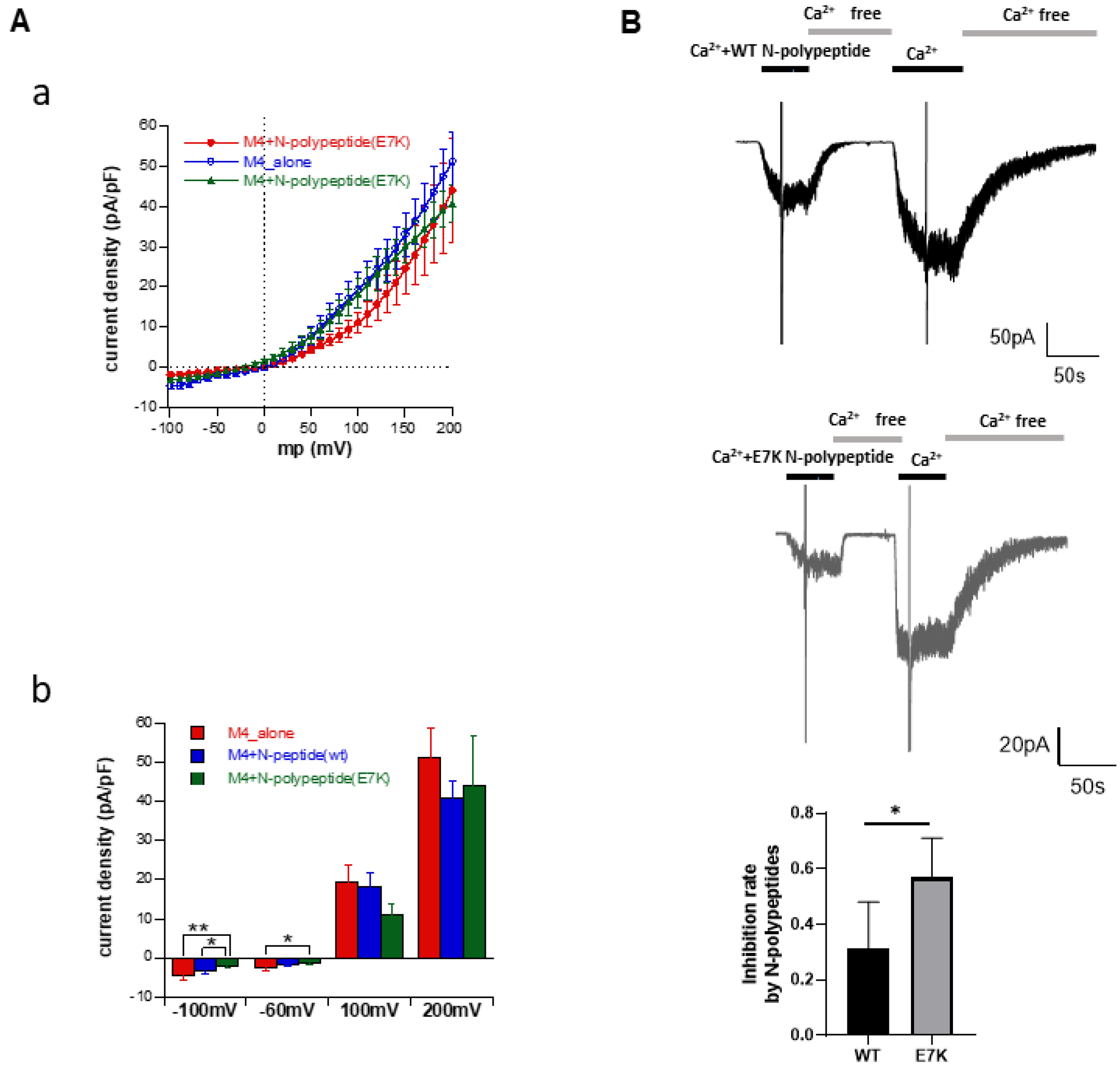
3.5. N-Terminal Polypeptides Inhibit TRPM4 Channel Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, C.X.; Brown, A.; Lau, D.H.; Chugh, S.S.; Albert, C.M.; Kalman, J.M.; Sanders, P. Epidemiology of sudden cardiac death: Global and regional perspectives. Heart Lung Circ. 2019, 28, 6–14. [Google Scholar] [CrossRef]
- Hof, T.; Chaigne, S.; Récalde, A.; Sallé, L.; Brette, F.; Guinamard, R. Transient receptor potential channels in cardiac health and disease. Nat. Rev. Cardiol. 2019, 16, 344–360. [Google Scholar] [CrossRef]
- Inoue, R.; Jensen, L.J.; Shi, J.; Morita, H.; Nishida, M.; Honda, A.; Ito, Y. Transient receptor potential channels in cardiovascular function and disease. Circ. Res. 2006, 99, 119–131. [Google Scholar] [CrossRef]
- Launay, P.; Fleig, A.; Perraud, A.-L.; Scharenberg, A.M.; Penner, R.; Kinet, J.-P. Trpm4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef]
- Simard, C.; Sallé, L.; Rouet, R.; Guinamard, R. Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. Br. J. Pharmacol. 2012, 165, 2354–2364. [Google Scholar] [CrossRef]
- Kruse, M.; Schulze-Bahr, E.; Corfield, V.; Beckmann, A.; Stallmeyer, B.; Kurtbay, G.; Ohmert, I.; Schulze-Bahr, E.; Brink, P.; Pongs, O. Impaired endocytosis of the ion channel trpm4 is associated with human progressive familial heart block type i. J. Clin. Investig. 2009, 119, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, S.; Duff, H.J. Genetic determinants of hereditary bradyarrhythmias: A contemporary review of a diverse group of disorders. Can. J. Cardiol. 2017, 33, 758–767. [Google Scholar] [CrossRef]
- Rosenhouse-Dantsker, A.; Mehta, D.; Levitan, I. Regulation of ion channels by membrane lipids. Compr. Physiol. 2011, 2, 31–68. [Google Scholar]
- Hilgemann, D.W.; Feng, S.; Nasuhoglu, C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001, 2001, re19. [Google Scholar] [CrossRef]
- Barki-Harrington, L.; Luttrell, L.M.; Rockman, H.A. Dual inhibition of β-adrenergic and angiotensin ii receptors by a single antagonist: A functional role for receptor–receptor interaction in vivo. Circulation 2003, 108, 1611–1618. [Google Scholar] [CrossRef]
- Tappia, P.S.; Liu, S.-Y.; Shatadal, S.; Takeda, N.; Dhalla, N.S.; Panagia, V. Changes in sarcolemmal plc isoenzymes in postinfarct congestive heart failure: Partial correction by imidapril. Am. J. Physiol. Heart Circ. Physiol. 1999, 277, H40–H49. [Google Scholar] [CrossRef]
- Nilius, B.; Mahieu, F.; Prenen, J.; Janssens, A.; Owsianik, G.; Vennekens, R.; Voets, T. The Ca2+-activated cation channel trpm4 is regulated by phosphatidylinositol 4, 5-biphosphate. EMBO J. 2006, 25, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Okawa, H.; Wang, Y.; Liman, E.R. Phosphatidylinositol 4, 5-bisphosphate rescues trpm4 channels from desensitization. J. Biol. Chem. 2005, 280, 39185–39192. [Google Scholar] [CrossRef]
- Murata, Y.; Iwasaki, H.; Sasaki, M.; Inaba, K.; Okamura, Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 2005, 435, 1239–1243. [Google Scholar] [CrossRef]
- van der Wal, J.; Habets, R.; Várnai, P.; Balla, T.; Jalink, K. Monitoring agonist-induced phospholipase c activation in live cells by fluorescence resonance energy transfer. J. Biol. Chem. 2001, 276, 15337–15344. [Google Scholar] [CrossRef] [PubMed]
- Itsuki, K.; Imai, Y.; Hase, H.; Okamura, Y.; Inoue, R.; Mori, M.X. PLC-mediated PI(4, 5) P2 hydrolysis regulates activation and inactivation of trpc6/7 channels. J. Gen. Physiol. 2014, 143, 183–201. [Google Scholar] [CrossRef]
- Inoue, R.; Okada, T.; Onoue, H.; Hara, Y.; Shimizu, S.; Naitoh, S.; Ito, Y.; Mori, Y. The transient receptor potential protein homologue trp6 is the essential component of vascular α1-adrenoceptor–activated Ca2+-permeable cation channel. Circ. Res. 2001, 88, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.G.; Liang, H.; Mori, M.X.; Yue, D.T. Fret two-hybrid mapping reveals function and location of L-type Ca2+ channel cam preassociation. Neuron 2003, 39, 97–107. [Google Scholar] [CrossRef]
- Hu, Y.; Duan, Y.; Takeuchi, A.; Hai-Kurahara, L.; Ichikawa, J.; Hiraishi, K.; Numata, T.; Ohara, H.; Iribe, G.; Nakaya, M. Uncovering the arrhythmogenic potential of trpm4 activation in atrial-derived HL-1 cells using novel recording and numerical approaches. Cardiovasc. Res. 2017, 113, 1243–1255. [Google Scholar] [CrossRef]
- Falkenburger, B.H.; Jensen, J.B.; Hille, B. Kinetics of PIP2 metabolism and kcnq2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol. 2010, 135, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Itsuki, K.; Okamura, Y.; Inoue, R.; Mori, M.X. A self-limiting regulation of vasoconstrictor-activated trpc3/c6/c7 channels coupled to PI(4, 5) P2-diacylglycerol signalling. J. Physiol. 2012, 590, 1101–1119. [Google Scholar] [CrossRef]
- Ishikawa, Y. The 95th annual meeting of the physiological society of Japan. J. Physiol. Sci. 2018, 68, 1–210. [Google Scholar] [CrossRef]
- Bousova, K.; Jirku, M.; Bumba, L.; Bednarova, L.; Sulc, M.; Franek, M.; Vyklicky, L.; Vondrasek, J.; Teisinger, J. PIP2 and PIP3 interact with n-terminus region of trpm4 channel. Biophys. Chem. 2015, 205, 24–32. [Google Scholar] [CrossRef]
- Holendova, B.; Grycova, L.; Jirku, M.; Teisinger, J. Ptdins (4, 5) P2 interacts with cam binding domains on trpm3 n-terminus. Channels 2012, 6, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human trpm4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232. [Google Scholar] [CrossRef]
- Zheng, W.; Cai, R.; Hofmann, L.; Nesin, V.; Hu, Q.; Long, W.; Fatehi, M.; Liu, X.; Hussein, S.; Kong, T. Direct binding between pre-s1 and trp-like domains in trpp channels mediates gating and functional regulation by PIP2. Cell Rep. 2018, 22, 1560–1573. [Google Scholar] [CrossRef]
- Hille, B. Ionic channels in excitable membranes. Current problems and biophysical approaches. Biophys. J. 1978, 22, 283–294. [Google Scholar] [CrossRef]
- Thompson, M.J.; Baenziger, J.E. Ion channels as lipid sensors: From structures to mechanisms. Nat. Chem. Biol. 2020, 16, 1331–1342. [Google Scholar] [CrossRef]
- Duan, J.; Li, Z.; Li, J.; Santa-Cruz, A.; Sanchez-Martinez, S.; Zhang, J.; Clapham, D.E. Structure of full-length human trpm4. Proc. Natl. Acad. Sci. USA 2018, 115, 2377–2382. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Demion, M.; Magaud, C.; Potreau, D.; Bois, P. Functional expression of the trpm4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension 2006, 48, 587–594. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Li, Q.; Kurahara, L.-H.; Shioi, N.; Hiraishi, K.; Fujita, T.; Zhu, X.; Inoue, R. An Arrhythmic Mutation E7K Facilitates TRPM4 Channel Activation via Enhanced PIP2 Interaction. Cells 2021, 10, 983. https://doi.org/10.3390/cells10050983
Hu Y, Li Q, Kurahara L-H, Shioi N, Hiraishi K, Fujita T, Zhu X, Inoue R. An Arrhythmic Mutation E7K Facilitates TRPM4 Channel Activation via Enhanced PIP2 Interaction. Cells. 2021; 10(5):983. https://doi.org/10.3390/cells10050983
Chicago/Turabian StyleHu, Yaopeng, Qin Li, Lin-Hai Kurahara, Narumi Shioi, Keizo Hiraishi, Takayuki Fujita, Xin Zhu, and Ryuji Inoue. 2021. "An Arrhythmic Mutation E7K Facilitates TRPM4 Channel Activation via Enhanced PIP2 Interaction" Cells 10, no. 5: 983. https://doi.org/10.3390/cells10050983
APA StyleHu, Y., Li, Q., Kurahara, L.-H., Shioi, N., Hiraishi, K., Fujita, T., Zhu, X., & Inoue, R. (2021). An Arrhythmic Mutation E7K Facilitates TRPM4 Channel Activation via Enhanced PIP2 Interaction. Cells, 10(5), 983. https://doi.org/10.3390/cells10050983