
A Potential Role for HUWE1 in Modulating Cisplatin Sensitivity

 , , ,
, , ,  and
and 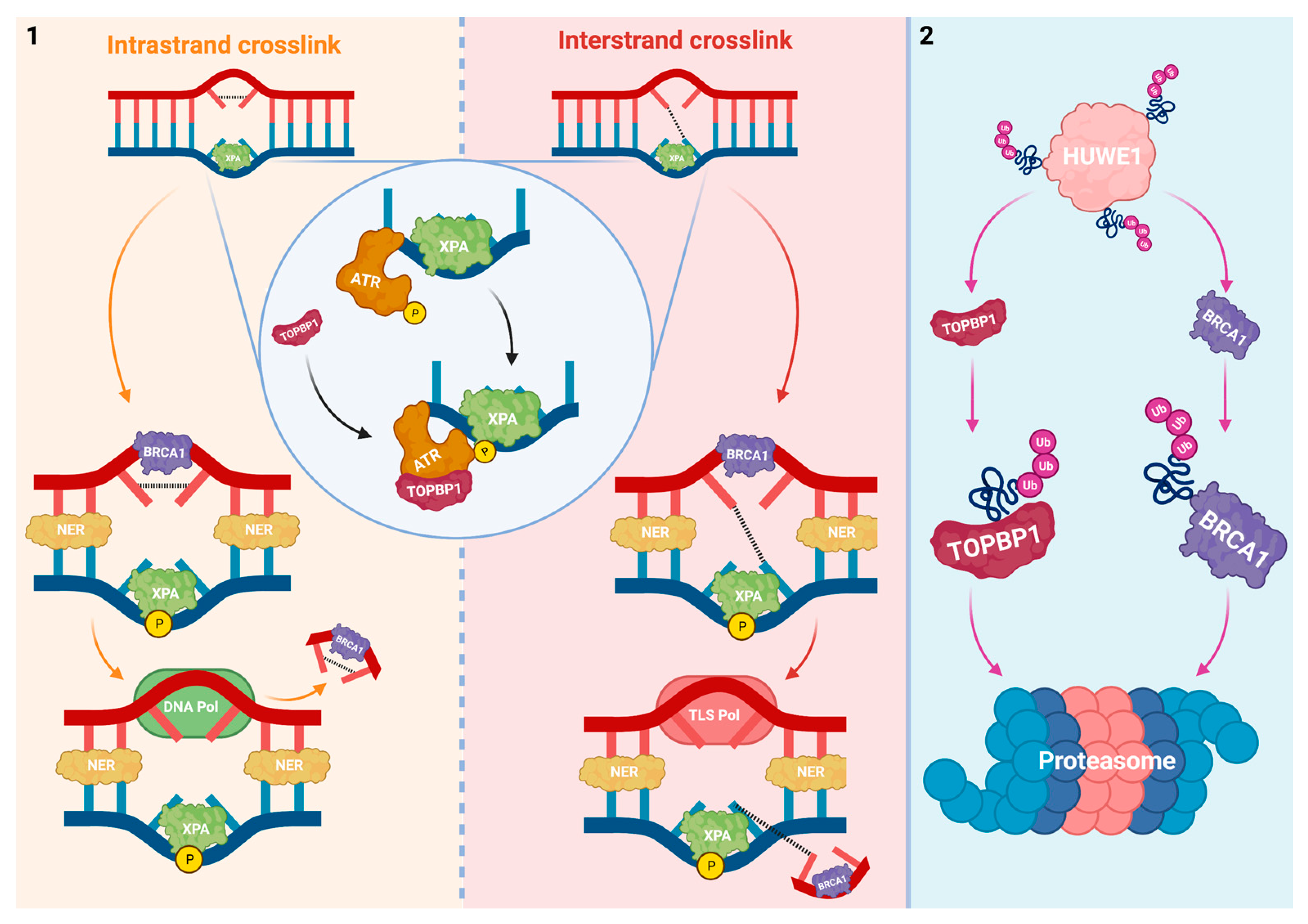{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. HUWE1 Interferes with DNA Damage Repair and Tolerance
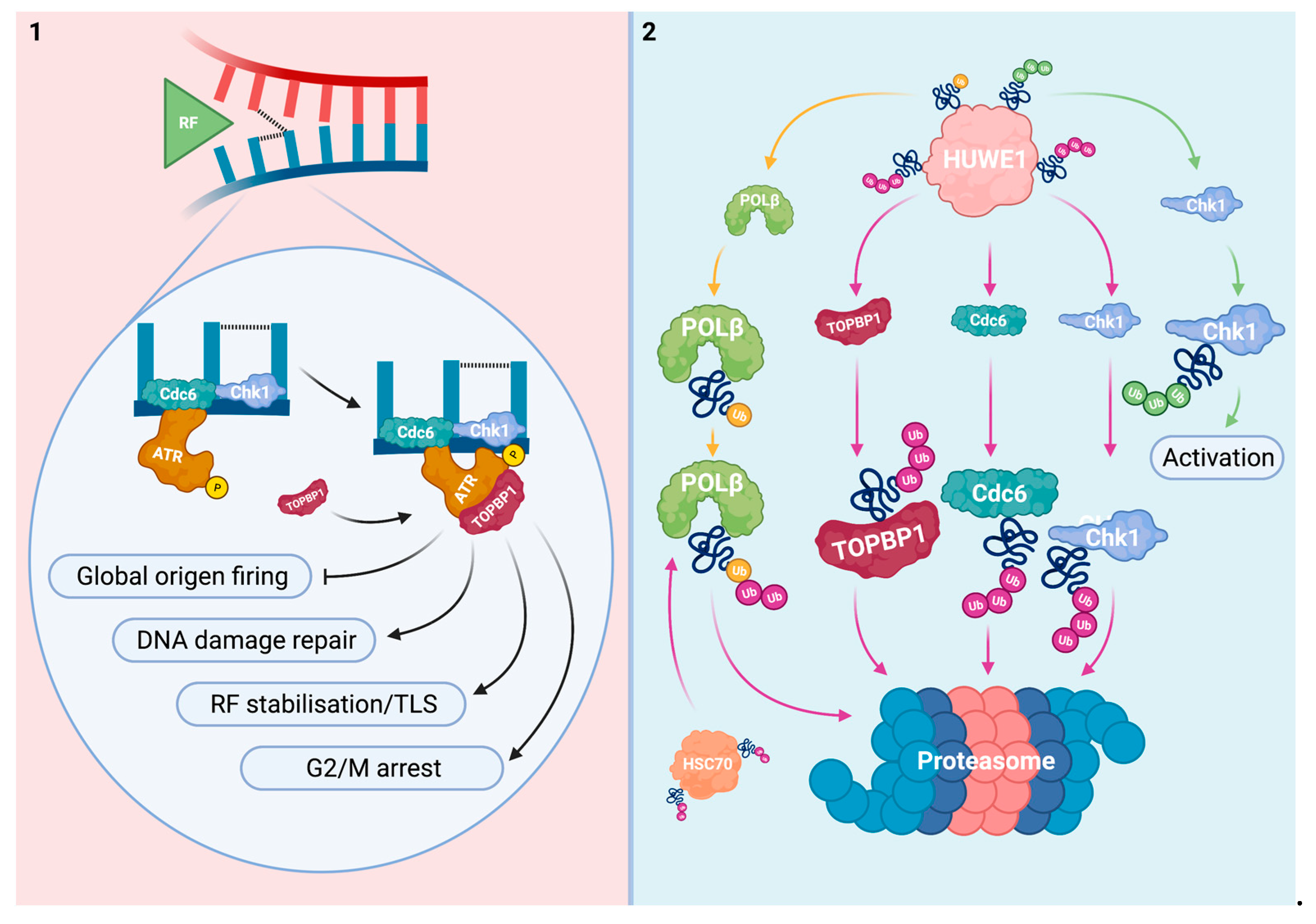
2.1. Interplay between HUWE1 and Mechanisms That Prevent Replication Fork Collision with Platinum-DNA Adducts
2.2. Interplay between HUWE1 and Pathways That Are Activated upon Replication Fork Collision with Platinum-DNA Adducts
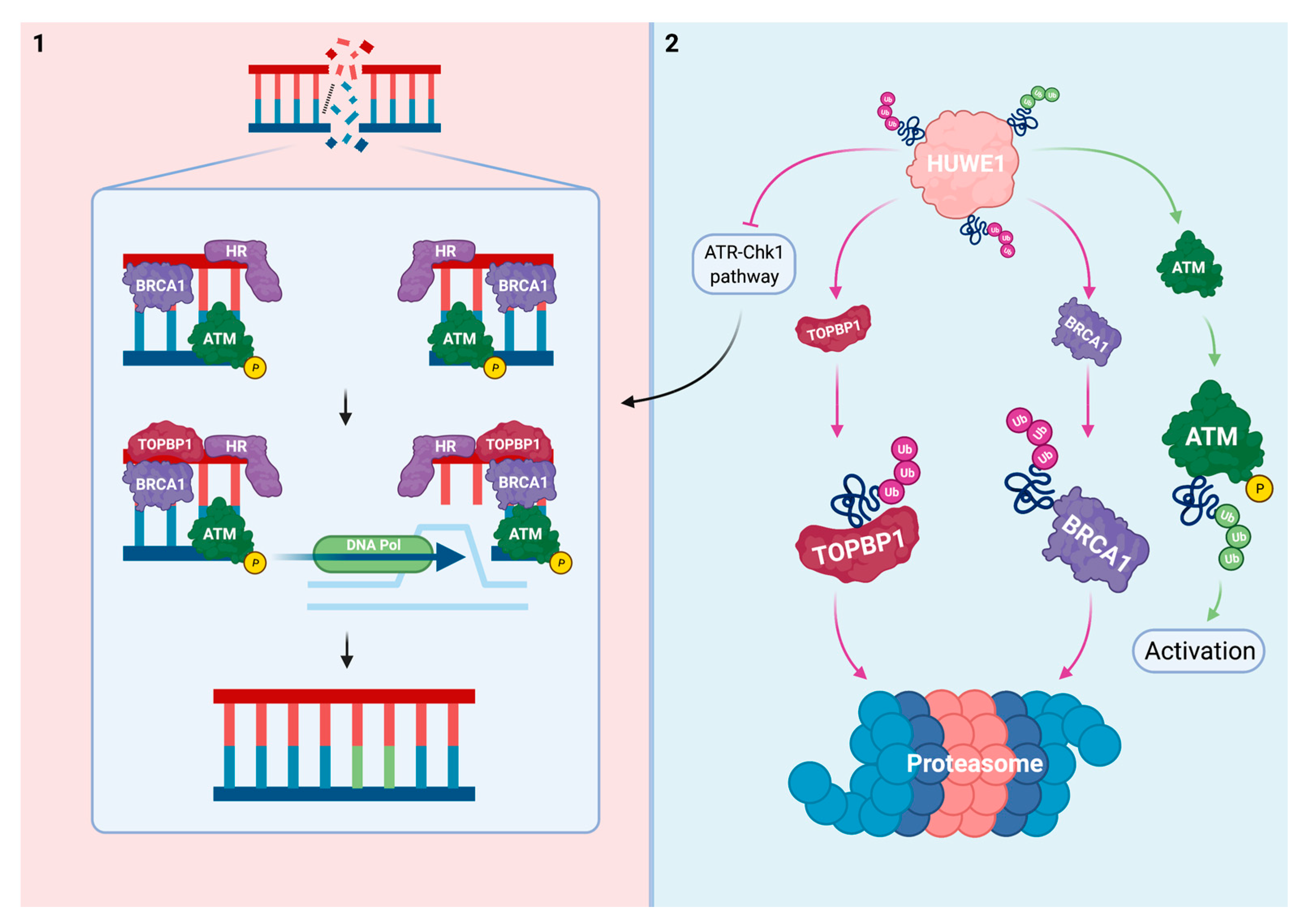
2.3. Interplay between HUWE1 and Mechanisms Induced by DNA Damage due to Replication Fork Collision with Platinum-DNA Adducts
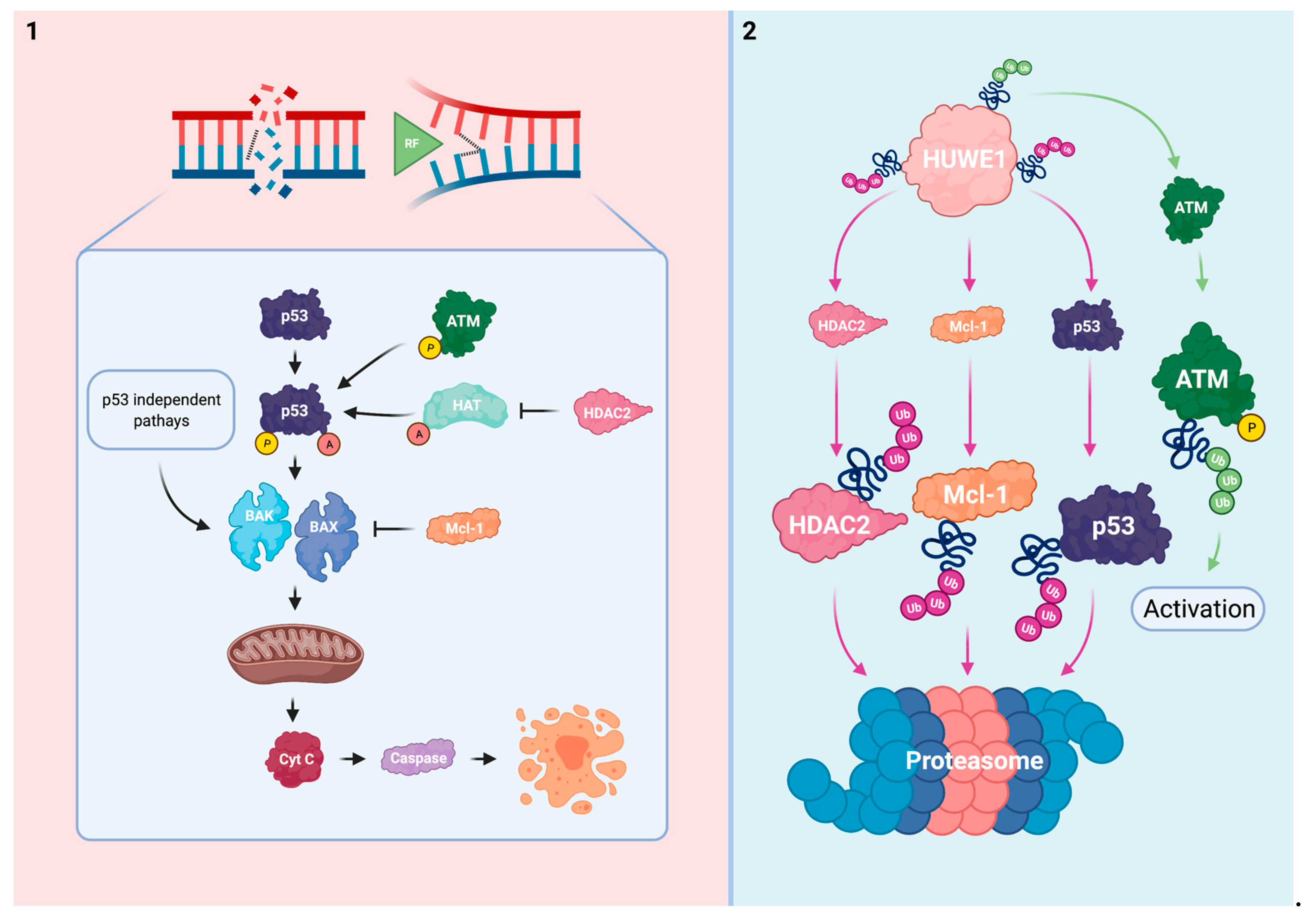
3. HUWE1 Modulates the Intrinsic Apoptotic Pathway
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Higby, D.J.; Wallace, H.J.; Albert, D.J.; Holland, J.F. Diaminodichloroplatinum: A phase I study showing responses in testicular and other tumors. Cancer 1974, 33, 1219–1225. [Google Scholar] [CrossRef]
- Gómez-Ruiz, S.; Maksimović-Ivanić, D.; Mijatović, S.; Kaluđerović, G.N. On the Discovery, Biological Effects, and Use of Cisplatin and Metallocenes in Anticancer Chemotherapy. Bioinorg. Chem. Appl. 2012, 2012, 1–14. [Google Scholar] [CrossRef]
- Witjes, J.A.; Babjuk, M.; Bellmunt, J.; Bruins, H.M.; De Reijke, T.M.; De Santis, M.; Gillessen, S.; James, N.; Maclennan, S.; Palou, J.; et al. EAU-ESMO Consensus Statements on the Management of Advanced and Variant Bladder Cancer—An International Collaborative Multistakeholder Effort. Eur. Urol. 2020, 77, 223–250. [Google Scholar] [CrossRef]
- Gilligan, T.; Lin, D.W.; Aggarwal, R.; Chism, D.; Cost, N.; Derweesh, I.H.; Emamekhoo, H.; Feldman, D.R.; Geynisman, D.M.; Hancock, S.L.; et al. Testicular Cancer, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 1529–1554. [Google Scholar] [CrossRef]
- Postmus, P.E.; Kerr, K.M.; Oudkerk, M.; Senan, S.; Waller, D.A.; Vansteenkiste, J.; Escriu, C.; Peters, S. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv1–iv21. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Characterization of the adducts produced in DNA by cis-diamminedichloroplatinum(II) and cis-dichloro(ethylenediamine)platinum(II). Biochemistry 1983, 22, 3927–3933. [Google Scholar] [CrossRef]
- Plooy, A.C.; Fichtinger-Schepman, A.M.J.; Schutte, H.H.; Van Dijk, M.; Lohman, P.H. The quantitative detection of various Pt-DNA-adducts in Chinese hamster ovary cells treated with cisplatin: Application of immunochemical techniques. Carcinogenesis 1985, 6, 561–566. [Google Scholar] [CrossRef]
- Donaldson, K.L.; Goolsby, G.L.; Wahl, A.F. Cytotoxicity of the anticancer agents cisplatin and taxol during cell proliferation and the cell cycle. Int. J. Cancer 1994, 57, 847–855. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Reevaluation of interaction of cis-dichloro(ethylenediamine)platinum(II) with DNA. Biochemistry 1986, 25, 3912–3915. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular Responses to Cisplatin-Induced DNA Damage. J. Nucleic Acids 2010, 2010, 1–16. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef]
- Yang, H.-T.; Shah, R.H.; Tegay, D.; Onel, K. Precision oncology: Lessons learned and challenges for the future. Cancer Manag. Res. 2019, 11, 7525–7536. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Meng, L.-H. CRISPR-cas9: A powerful tool towards precision medicine in cancer treatment. Acta Pharmacol. Sin. 2019, 41, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nat. Cell Biol. 2014, 509, 487–491. [Google Scholar] [CrossRef]
- Kumar, G.; Ritch, E.; Oo, H.; Wang, C.; Tortora, D.; Thaper, D.; Moskalev, I.; Wyatt, A.; Black, P.; Daugaard, M. Genome-wide CRISPR screen reveals SLFN11 as a potent mediator of cisplatin sensitivity in muscle-invasive bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 905. [Google Scholar] [CrossRef]
- Cassidy, K.B.; Bang, S.; Kurokawa, M.; Gerber, S.A. Direct regulation of Chk1 protein stability by E3 ubiquitin ligase HUWE1. FEBS J. 2020, 287, 1985–1999. [Google Scholar] [CrossRef]
- Adhikary, S.; Marinoni, F.; Hock, A.; Hulleman, E.; Popov, N.; Beier, R.; Bernard, S.; Quarto, M.; Capra, M.; Goettig, S.; et al. The Ubiquitin Ligase HectH9 Regulates Transcriptional Activation by Myc and Is Essential for Tumor Cell Proliferation. Cell 2005, 123, 409–421. [Google Scholar] [CrossRef]
- Parsons, J.; Tait, P.S.; Finch, D.; Dianova, I.; Edelmann, M.J.; Khoronenkova, S.V.; Kessler, B.M.; Sharma, R.A.; McKenna, W.G.; Dianov, G.L. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009, 28, 3207–3215. [Google Scholar] [CrossRef] [PubMed]
- Choe, K.N.; Nicolae, C.M.; Constantin, D.; Kawasawa, Y.I.; Delgado-Diaz, M.R.; De, S.; Freire, R.; Smits, V.A.; Moldovan, G. HUWE 1 interacts with PCNA to alleviate replication stress. EMBO Rep. 2016, 17, 874–886. [Google Scholar] [CrossRef]
- Markkanen, E.; Van Loon, B.; Ferrari, E.; Parsons, J.; Dianov, G.L.; Hubscher, U. Regulation of oxidative DNA damage repair by DNA polymerase and MutYH by cross-talk of phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2012, 109, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.A.; Swatek, K.N.; Hospenthal, M.K.; Komander, D. Ubiquitin Linkage-Specific Affimers Reveal Insights into K6-Linked Ubiquitin Signaling. Mol. Cell 2017, 68, 233–246.e5. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Du, D.; Deng, Y.; Zhou, Y.; Sun, L.; Yuan, S. The structure and regulation of the E3 ubiquitin ligase HUWE1 and its biological functions in cancer. Investig. New Drugs 2020, 38, 515–524. [Google Scholar] [CrossRef]
- Kao, S.-H.; Wu, H.-T.; Wu, K.-J. Ubiquitination by HUWE1 in tumorigenesis and beyond. J. Biomed. Sci. 2018, 25, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-Only E3 Ubiquitin Ligase, Catalyzes the Polyubiquitination of Mcl-1 and Regulates Apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar] [CrossRef]
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule Is a Critical Mediator of the ARF Tumor Suppressor. Cell 2005, 121, 1071–1083. [Google Scholar] [CrossRef]
- Wang, X.; Lu, G.; Li, L.; Yi, J.; Yan, K.; Wang, Y.; Zhu, B.; Kuang, J.; Lin, M.; Zhang, S.; et al. HUWE1 interacts with BRCA1 and promotes its degradation in the ubiquitin–proteasome pathway. Biochem. Biophys. Res. Commun. 2014, 444, 549–554. [Google Scholar] [CrossRef]
- Yang, D.; Cheng, D.; Tu, Q.; Yang, H.; Sun, B.; Yan, L.; Dai, H.; Luo, J.; Mao, B.; Cao, Y.; et al. HUWE1 controls the development of non-small cell lung cancer through down-regulation of p53. Theranostics 2018, 8, 3517–3529. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.T.; Vaisman, A.; Chaney, S.G.; Sancar, A. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and bis-acetoammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999, 59, 3968–3971. [Google Scholar] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 2016, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Park, J.-M.; Leem, S.-H.; Kang, T.-H. Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene 2012, 33, 19–25. [Google Scholar] [CrossRef]
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008, 22, 1478–1489. [Google Scholar] [CrossRef]
- Lindsey-Boltz, L.A.; Kemp, M.G.; Reardon, J.T.; DeRocco, V.; Iyer, R.R.; Modrich, P.; Sancar, A. Coupling of Human DNA Excision Repair and the DNA Damage Checkpoint in a Defined in Vitro System. J. Biol. Chem. 2014, 289, 5074–5082. [Google Scholar] [CrossRef]
- Herold, S.; Hock, A.; Herkert, B.; Berns, K.; Mullenders, J.; Beijersbergen, R.; Bernards, R.; Eilers, M. Miz1 and HectH9 regulate the stability of the checkpoint protein, TopBP1. EMBO J. 2008, 27, 2851–2861. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-P.; Yan, X.-B.; Liu, L.-G.; Tian, C.; Han, K.; Zhang, H.; Min, D.-L. TopBP1 promotes malignant progression and correlates with poor prognosis in osteosarcoma. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4022–4031. [Google Scholar] [PubMed]
- Wang, L.-R.; He, L.-J.; Wang, Y.; Li, Y.-Y.; Lou, Y.; Zhang, G.-B.; Li, Y.; Chen, J. Correlation between BRCA1 and TopBP1 protein expression and clinical outcome of non-small cell lung cancer treated with platinum-based chemotherapy. Cancer Chemother. Pharmacol. 2015, 76, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Lin, G.E.; Liu, K.; Song, Y.; Lin, F.-T.; Lin, W.-C. Targeting TopBP1 at a convergent point of multiple oncogenic pathways for cancer therapy. Nat. Commun. 2014, 5, 1–15. [Google Scholar] [CrossRef]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Taron, M.; Barnadas, A.; Scagliotti, G.; Sarries, C.; Roig, B. Nucleotide Excision Repair Pathways Involved in Cisplatin Resistance in Non-Small-Cell Lung Cancer. Cancer Control. 2003, 10, 297–305. [Google Scholar] [CrossRef]
- Metzger, R.; Leichman, C.G.; Danenberg, K.D.; Danenberg, P.V.; Lenz, H.J.; Hayashi, K.; Groshen, S.; Salonga, D.; Cohen, H.; Laine, L.; et al. ERCC1 mRNA levels complement thymidylate synthase mRNA levels in predicting response and survival for gastric cancer patients receiving combination cisplatin and fluorouracil chemotherapy. J. Clin. Oncol. 1998, 16, 309–316. [Google Scholar] [CrossRef]
- Li, Q.; Yu, J.J.; Mu, C.; Yunmbam, M.K.; Slavsky, D.; Cross, C.L.; Bostick-Bruton, F.; Reed, E. Association between the level of ERCC-1 expression and the repair of cisplatin-induced DNA damage in human ovarian cancer cells. Anticancer Res. 2000, 20, 645–652. [Google Scholar]
- Köberle, B.; Grimaldi, K.A.; Sunters, A.; Hartley, J.A.; Kelland, L.R.; Masters, J.R. DNA Repair capacity and cisplatin sensitivity of human testis tumour cells. Int. J. Cancer 1997, 70, 551–555. [Google Scholar] [CrossRef]
- Mendoza, J.; Martínez, J.; Hernandez, C.; Pérez-Montiel, D.; Castro, C.; Fabián-Morales, E.; Santibañez, M.; González-Barrios, R.; Diaz-Chavez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumours. Br. J. Cancer 2013, 109, 68–75. [Google Scholar] [CrossRef]
- Usanova, S.; Piée-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Köberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cierna, Z.; Miskovska, V.; Roska, J.; Jurkovicova, D.; Pulzova, L.B.; Sestakova, Z.; Hurbanova, L.; Machalekova, K.; Chovanec, M.; Rejlekova, K.; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Barakat, K.H.; Jordheim, L.P.; Pérez-Pineiro, R.; Wishart, D.S.; Dumontet, C.; Tuszynski, J.A. Virtual Screening and Biological Evaluation of Inhibitors Targeting the XPA-ERCC1 Interaction. PLoS ONE 2012, 7, e51329. [Google Scholar] [CrossRef] [PubMed]
- Pulzová, L.B.; Ward, T.A.; Chovanec, M. XPA: DNA Repair Protein of Significant Clinical Importance. Int. J. Mol. Sci. 2020, 21, 2182. [Google Scholar] [CrossRef] [PubMed]
- Morishima, K.-I.; Sakamoto, S.; Kobayashi, J.; Izumi, H.; Suda, T.; Matsumoto, Y.; Tauchi, H.; Ide, H.; Komatsu, K.; Matsuura, S. TopBP1 associates with NBS1 and is involved in homologous recombination repair. Biochem. Biophys. Res. Commun. 2007, 362, 872–879. [Google Scholar] [CrossRef]
- Fujikawa, Y.; Kawanishi, M.; Kuraoka, I.; Yagi, T. Frequencies of mutagenic translesion DNA synthesis over cisplatin-guanine intra-strand crosslinks in lacZ plasmids propagated in human cells. Mutat. Res. Toxicol. Environ. Mutagen. 2014, 770, 23–28. [Google Scholar] [CrossRef]
- Lambert, S.; Mizuno, K.; Blaisonneau, J.; Martineau, S.; Chanet, R.; Fréon, K.; Murray, J.M.; Carr, A.M.; Baldacci, G. Homologous Recombination Restarts Blocked Replication Forks at the Expense of Genome Rearrangements by Template Exchange. Mol. Cell 2010, 39, 346–359. [Google Scholar] [CrossRef]
- Choi, J.-H.; Lindsey-Boltz, L.A.; Kemp, M.; Mason, A.C.; Wold, M.S.; Sancar, A. Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 13660–13665. [Google Scholar] [CrossRef] [PubMed]
- Borlado, L.R.; Méndez, J. CDC6: From DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis 2007, 29, 237–243. [Google Scholar] [CrossRef]
- Duursma, A.; Agami, R. p53-Dependent Regulation of Cdc6 Protein Stability Controls Cellular Proliferation. Mol. Cell. Biol. 2005, 25, 6937–6947. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sugimoto, N.; Iwahori, S.; Yugawa, T.; Narisawa-Saito, M.; Kiyono, T.; Fujita, M. CDC6 interaction with ATR regulates activation of a replication checkpoint in higher eukaryotic cells. J. Cell Sci. 2010, 123, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Kow, E.; Nevis, K.R.; Lu, C.K.; Luce, K.S.; Zhong, Q.; Cook, J.G. Cdc6 Stability Is Regulated by the Huwe1 Ubiquitin Ligase after DNA Damage. Mol. Biol. Cell 2007, 18, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Lee, H.O.; Bunker, B.D.; Dorn, E.S.; Rogers, G.C.; Duronio, R.J.; Cook, J.G. Cdt1 and Cdc6 Are Destabilized by Rereplication-induced DNA Damage. J. Biol. Chem. 2008, 283, 25356–25363. [Google Scholar] [CrossRef]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef]
- Leung-Pineda, V.; Huh, J.; Piwnica-Worms, H. DDB1 Targets Chk1 to the Cul4 E3 Ligase Complex in Normal Cycling Cells and in Cells Experiencing Replication Stress. Cancer Res. 2009, 69, 2630–2637. [Google Scholar] [CrossRef]
- Xu, Y.; Anderson, D.E.; Ye, Y. The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation. Cell Discov. 2016, 2, 16040. [Google Scholar] [CrossRef] [PubMed]
- Knobel, P.A.; Marti, T. Translesion DNA synthesis in the context of cancer research. Cancer Cell Int. 2011, 11, 39. [Google Scholar] [CrossRef]
- Singhal, R.K.; Wilson, S.H. Short gap-filling synthesis by DNA polymerase beta is processive. J. Biol. Chem. 1993, 268, 15906–15911. [Google Scholar] [CrossRef]
- Koren, A. The role of the DNA damage checkpoint in regulation of translesion DNA synthesis. Mutagenesis 2007, 22, 155–160. [Google Scholar] [CrossRef][Green Version]
- Hoffmann, J.S.; Pillaire, M.J.; Maga, G.; Podust, V.; Hubscher, U.; Villani, G. DNA polymerase beta bypasses in vitro a single d(GpG)-cisplatin adduct placed on codon 13 of the HRAS gene. Proc. Natl. Acad. Sci. USA 1995, 92, 5356–5360. [Google Scholar] [CrossRef]
- Fang, Q.; Inanç, B.; Schamus, S.; Wang, X.-H.; Wei, L.; Brown, A.R.; Svilar, D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nat. Commun. 2014, 5, 5513. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.; Gu, Y.; Zhang, Z.; Zheng, G.; He, Z. TCRP1 contributes to cisplatin resistance by preventing Pol β degradation in lung cancer cells. Mol. Cell. Biochem. 2015, 398, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.K.; Srivastava, D.K.; Zmudzka, B.Z.; Wilson, S.H. Strategic down-regulation of DNA polymerase β by antisense RNA sensitizes mammalian cells to specific DNA damaging agents. Nucleic Acids Res. 1995, 23, 3810–3815. [Google Scholar] [CrossRef]
- Wang, M.; Li, E.; Lin, L.; Kumar, A.K.; Pan, F.; He, L.; Zhang, J.; Hu, Z.; Guo, Z. Enhanced Activity of Variant DNA Polymerase β (D160G) Contributes to Cisplatin Therapy by Impeding the Efficiency of NER. Mol. Cancer Res. 2019, 17, 2077–2088. [Google Scholar] [CrossRef] [PubMed]
- Kothandapani, A.; Sawant, A.; Dangeti, V.S.M.N.; Sobol, R.W.; Patrick, S.M. Epistatic role of base excision repair and mismatch repair pathways in mediating cisplatin cytotoxicity. Nucleic Acids Res. 2013, 41, 7332–7343. [Google Scholar] [CrossRef]
- Ummat, A.; Rechkoblit, O.; Jain, R.; Choudhury, J.R.; Johnson, R.E.; Silverstein, T.D.; Buku, A.; Lone, S.; Prakash, L.; Prakash, S.; et al. Structural basis for cisplatin DNA damage tolerance by human polymerase η during cancer chemotherapy. Nat. Struct. Mol. Biol. 2012, 19, 628–632. [Google Scholar] [CrossRef]
- Chen, S.; Chen, X.; Xie, G.; He, Y.; Yan, D.; Zheng, D.; Li, S.; Fu, X.; Li, Y.; Pang, X.; et al. Cdc6 contributes to cisplatin-resistance by activation of ATR-Chk1 pathway in bladder cancer cells. Oncotarget 2016, 7, 40362–40376. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Chen, S.; Zhang, Y.; Xie, W.; Hu, Z.; Li, H.; Li, J.; Zhou, Z.; Tan, W. Norcantharidin inhibits the DDR of bladder cancer stem-like cells through cdc6 degradation. OncoTargets Ther. 2019, 12, 4403–4413. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Cheng, Y.-C.; Lin, T.-Y.; Shieh, S.-Y. Candidate tumor suppressor BTG3 maintains genomic stability by promoting Lys63-linked ubiquitination and activation of the checkpoint kinase CHK1. Proc. Natl. Acad. Sci. USA 2013, 110, 5993–5998. [Google Scholar] [CrossRef]
- Brandsma, I.; van Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 1–10. [Google Scholar] [CrossRef]
- Hao, Z.; Duncan, G.S.; Su, Y.-W.; Li, W.Y.; Silvester, J.; Hong, C.; You, H.; Brenner, D.; Gorrini, C.; Haight, J.; et al. The E3 ubiquitin ligase Mule acts through the ATM–p53 axis to maintain B lymphocyte homeostasis. J. Exp. Med. 2012, 209, 173–186. [Google Scholar] [CrossRef]
- Welcsh, P.L. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Ludwig, T.; Fisher, P.; Ganesan, S.; Efstratiadis, A. Tumorigenesis in mice carrying a truncating Brca1 mutation. Genes Dev. 2001, 15, 1188–1193. [Google Scholar] [CrossRef]
- Wu, W.; Sato, K.; Koike, A.; Nishikawa, H.; Koizumi, H.; Venkitaraman, A.R.; Ohta, T. HERC2 Is an E3 Ligase That Targets BRCA1 for Degradation. Cancer Res. 2010, 70, 6384–6392. [Google Scholar] [CrossRef]
- Liu, F.; Cao, L.; Zhang, T.; Chang, F.; Xu, Y.; Li, Q.; Deng, J.; Li, L.; Shao, G. CRL4BRBBP7 targets HUWE1 for ubiquitination and proteasomal degradation. Biochem. Biophys. Res. Commun. 2018, 501, 440–447. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Hu, X.-C.; Zhang, J.; Xu, B.-H.; Cai, L.; Ragaz, J.; Wang, Z.-H.; Wang, B.-Y.; Teng, Y.-E.; Tong, Z.-S.; Pan, Y.-Y.; et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 436–446. [Google Scholar] [CrossRef]
- Husain, A.; He, G.; Venkatraman, E.S.; Spriggs, D.R. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diamminedichloroplatinum(II). Cancer Res. 1998, 58, 1120–1123. [Google Scholar]
- Tassone, P.; Tagliaferri, P.; Perricelli, A.; Blotta, S.; Quaresima, B.; Martelli, M.L.; Goel, A.; Barbieri, V.; Di Costanzo, F.; Boland, C.R.; et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer 2003, 88, 1285–1291. [Google Scholar] [CrossRef]
- Promkan, M.; Liu, G.; Patmasiriwat, P.; Chakrabarty, S. BRCA1 modulates malignant cell behavior, the expression of survivin and chemosensitivity in human breast cancer cells. Int. J. Cancer 2009, 125, 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Kato, T.; Kiyotani, K.; Tarhan, Y.E.; Saloura, V.; Chung, S.; Ueda, K.; Nakamura, Y.; Park, J.-H. p53-independent p21 induction by MELK inhibition. Oncotarget 2017, 8, 57938–57947. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Perez, R.E.; Davaadelger, B.; Maki, C.G. Two 4N Cell-Cycle Arrests Contribute to Cisplatin-Resistance. PLoS ONE 2013, 8, e59848. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.-R.; Ford, J.M. BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat. Genet. 2002, 32, 180–184. [Google Scholar] [CrossRef]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef]
- Clements, K.E.; Schleicher, E.M.; Thakar, T.; Hale, A.; Dhoonmoon, A.; Tolman, N.J.; Sharma, A.; Liang, X.; Kawasawa, Y.I.; Nicolae, C.M.; et al. Identification of regulators of poly-ADP-ribose polymerase inhibitor response through complementary CRISPR knockout and activation screens. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Lee, E.K.; Matulonis, U.A. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers 2020, 12, 2054. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef]
- Henkels, K.M.; Turchi, J.J. Cisplatin-induced apoptosis proceeds by caspase-3-dependent and -independent pathways in cisplatin-resistant and -sensitive human ovarian cancer cell lines. Cancer Res. 1999, 59, 3077–3083. [Google Scholar]
- Han, J.Y.; Chung, Y.J.; Kim, J.S.; Rhyu, M.G.; Kim, H.K.; Lee, K.S.; Park, S.W. The relationship between cisplatin—induced apoptosis and p53, bcl-2 and bax expression in human lung cancer cells. Korean J. Intern. Med. 1999, 14, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. New insights into p53 activation. Cell Res. 2010, 20, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Lehman, T.A.; Modali, R.; Boukamp, P.; Stanek, J.; Bennett, W.P.; Welsh, J.A.; Metcalf, R.A.; Stampfer, M.R.; Fusenig, N.; Rogan, E.M.; et al. p53 Mutations in human immortalized epithelial cell lines. Carcinogenesis 1993, 14, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Leu, J.J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D. Mitochondrial p53 activates BAK and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, K.; O’Neill, K.L.; Pang, X.; Luo, X. Bax/Bak activation in the absence of Bid, Bim, Puma, and p53. Cell Death Dis. 2016, 7, e2266. [Google Scholar] [CrossRef]
- Qi, C.-F.; Kim, Y.-S.; Xiang, S.; Abdullaev, Z.; Torrey, T.A.; Janz, S.; Kovalchuk, A.L.; Sun, J.; Chen, D.; Cho, W.C.; et al. Characterization of ARF-BP1/HUWE1 Interactions with CTCF, MYC, ARF and p53 in MYC-Driven B Cell Neoplasms. Int. J. Mol. Sci. 2012, 13, 6204–6219. [Google Scholar] [CrossRef]
- Ma, W.; Zhao, P.; Zang, L.; Zhang, K.; Liao, H.; Hu, Z. Tumour suppressive function of HUWE1 in thyroid cancer. J. Biosci. 2016, 41, 395–405. [Google Scholar] [CrossRef]
- Zhang, J.; Kan, S.; Huang, B.; Hao, Z.; Mak, T.W.; Zhong, Q. Mule determines the apoptotic response to HDAC inhibitors by targeted ubiquitination and destruction of HDAC2. Genes Dev. 2011, 25, 2610–2618. [Google Scholar] [CrossRef]
- Germain, M.; Milburn, J.; Duronio, V. MCL-1 Inhibits BAX in the Absence of MCL-1/BAX Interaction. J. Biol. Chem. 2008, 283, 6384–6392. [Google Scholar] [CrossRef] [PubMed]
- Warr, M.R.; Acoca, S.; Liu, Z.; Germain, M.; Watson, M.; Blanchette, M.; Wing, S.S.; Shore, G.C. BH3-ligand regulates access of MCL-1 to its E3 ligase. FEBS Lett. 2005, 579, 5603–5608. [Google Scholar] [CrossRef] [PubMed]
- Myant, K.B.; Cammareri, P.; Hodder, M.C.; Wills, J.; Von Kriegsheim, A.; Győrffy, B.; Rashid, M.; Polo, S.; Maspero, E.; Vaughan, L.; et al. HUWE 1 is a critical colonic tumour suppressor gene that prevents MYC signalling, DNA damage accumulation and tumour initiation. EMBO Mol. Med. 2016, 9, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Pervin, S.; Tran, A.; Tran, L.; Urman, R.; Braga, M.; Chaudhuri, G.; Singh, R. Reduced association of anti-apoptotic protein Mcl-1 with E3 ligase Mule increases the stability of Mcl-1 in breast cancer cells. Br. J. Cancer 2011, 105, 428–437. [Google Scholar] [CrossRef]
- He, G.-Q.; Xu, W.-M.; Liao, H.-J.; Jiang, C.; Li, C.-Q.; Zhang, W. Silencing Huwe1 reduces apoptosis of cortical neurons exposed to oxygen-glucose deprivation and reperfusion. Neural Regen. Res. 2019, 14, 1977–1985. [Google Scholar] [CrossRef]
- Liao, M.; Zhao, J.; Wang, T.; Duan, J.; Zhang, Y.; Deng, X. Role of bile salt in regulating Mcl-1 phosphorylation and chemoresistance in hepatocellular carcinoma cells. Mol. Cancer 2011, 10, 44. [Google Scholar] [CrossRef]
- Stewart, D.P.; Koss, B.; Bathina, M.; Perciavalle, R.M.; Bisanz, K.; Opferman, J.T. Ubiquitin-Independent Degradation of Antiapoptotic MCL-1. Mol. Cell. Biol. 2010, 30, 3099–3110. [Google Scholar] [CrossRef]
- Ding, Q.; He, X.; Hsu, J.-M.; Xia, W.; Chen, C.-T.; Li, L.-Y.; Lee, D.-F.; Liu, J.-C.; Zhong, Q.; Wang, X.; et al. Degradation of Mcl-1 by β-TrCP Mediates Glycogen Synthase Kinase 3-Induced Tumor Suppression and Chemosensitization. Mol. Cell. Biol. 2006, 27, 4006–4017. [Google Scholar] [CrossRef]
- Senichkin, V.V.; Kopeina, G.S.; Prokhorova, E.A.; Zamaraev, A.V.; Lavrik, I.N.; Zhivotovsky, B. Modulation of Mcl-1 transcription by serum deprivation sensitizes cancer cells to cisplatin. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 557–566. [Google Scholar] [CrossRef]
- Yu, X.; Lijun, L.; Xia, Z.; Xie, L.; Ma, X.; Liang, Q.; Liu, L.; Wang, J.; Zhou, X.; Yang, Y.; et al. Targeting MCL-1 sensitizes human esophageal squamous cell carcinoma cells to cisplatin-induced apoptosis. BMC Cancer 2017, 17, 1–13. [Google Scholar] [CrossRef]
- Fletcher, S. MCL-1 inhibitors—Where are we now (2019)? Expert Opin. Ther. Pat. 2019, 29, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Perego, P.; Giarola, M.; Righetti, S.C.; Supino, R.; Caserini, C.; Delia, D.; Pierotti, M.A.; Miyashita, T.; Reed, J.C.; Zunino, F. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996, 56, 556–562. [Google Scholar]
- Di Pietro, A.; Koster, R.; Eck, W.B.-V.; Dam, W.A.; Mulder, N.H.; Gietema, J.A.; De Vries, E.G.; De Jong, S. Pro- and anti-apoptotic effects of p53 in cisplatin-treated human testicular cancer are cell context-dependent. Cell Cycle 2012, 11, 4552–4562. [Google Scholar] [CrossRef]
- Itahana, Y.; Ke, H.; Zhang, Y. p53 Oligomerization Is Essential for Its C-terminal Lysine Acetylation. J. Biol. Chem. 2009, 284, 5158–5164. [Google Scholar] [CrossRef]
- Chee, J.L.; Saidin, S.; Lane, D.P.; Leong, S.M.; Noll, J.E.; Neilsen, P.M.; Phua, Y.T.; Gabra, H.; Lim, T.M. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 2013, 12, 278–288. [Google Scholar] [CrossRef]
- Grabbe, C.; Husnjak, K.; Dikic, I. Europe PMC Funders Group. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 2013, 12, 295–307. [Google Scholar] [CrossRef]
- Deben, C.; Wouters, A.; De Beeck, K.O.; Bossche, J.V.D.; Jacobs, J.; Zwaenepoel, K.; Peeters, M.; Van Meerbeeck, J.; Lardon, F.; Rolfo, C.; et al. The MDM2-inhibitor Nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in non-small cell lung cancer. Oncotarget 2015, 6, 22666–22679. [Google Scholar] [CrossRef]
- Zanjirband, M.; Edmondson, R.J.; Lunec, J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget 2016, 7, 40115–40134. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor Suppressive Functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef]
- Huang, R.; Langdon, S.P.; Tse, M.; Mullen, P.; Um, I.H.; Faratian, D.; Harrison, D.J. The role of HDAC2 in chromatin remodelling and response to chemotherapy in ovarian cancer. Oncotarget 2015, 7, 4695–4711. [Google Scholar] [CrossRef]
- Alzoubi, S.; Brody, L.; Rahman, S.; Mahul-Mellier, A.-L.; Mercado, N.; Ito, K.; El-Bahrawy, M.; Silver, A.; Boobis, A.; Bell, J.D.; et al. Synergy between histone deacetylase inhibitors and DNA-damaging agents is mediated by histone deacetylase 2 in colorectal cancer. Oncotarget 2016, 7, 44505–44521. [Google Scholar] [CrossRef]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schäfer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2016, 36, 1804–1815. [Google Scholar] [CrossRef]
- Ji, P.; Yeh, V.; Ramirez, T.; Murata-Hori, M.; Lodish, H.F. Histone deacetylase 2 is required for chromatin condensation and subsequent enucleation of cultured mouse fetal erythroblasts. Haematologica 2010, 95, 2013–2021. [Google Scholar] [CrossRef]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin Compaction Protects Genomic DNA from Radiation Damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Bandolik, J.J.; Hamacher, A.; Schrenk, C.; Weishaupt, R.; Kassack, M.U. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3052. [Google Scholar] [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar]
- Heinen, C.; Ács, K.; Hoogstraten, D.; Dantuma, N.P. C-terminal UBA domains protect ubiquitin receptors by preventing initiation of protein degradation. Nat. Commun. 2011, 2, 191. [Google Scholar] [CrossRef]
- Sander, B.; Xu, W.; Eilers, M.; Popov, N.; Lorenz, S. A conformational switch regulates the ubiquitin ligase HUWE1. eLife 2017, 6, e21036. [Google Scholar] [CrossRef]
- Cui, L.; Zhou, F.; Chen, C.; Wang, C.C. Overexpression of CCDC69 activates p14ARF/MDM2/p53 pathway and confers cisplatin sensitivity. J. Ovarian Res. 2019, 12, 1–8. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wenmaekers, S.; Viergever, B.J.; Kumar, G.; Kranenburg, O.; Black, P.C.; Daugaard, M.; Meijer, R.P. A Potential Role for HUWE1 in Modulating Cisplatin Sensitivity. Cells 2021, 10, 1262. https://doi.org/10.3390/cells10051262
Wenmaekers S, Viergever BJ, Kumar G, Kranenburg O, Black PC, Daugaard M, Meijer RP. A Potential Role for HUWE1 in Modulating Cisplatin Sensitivity. Cells. 2021; 10(5):1262. https://doi.org/10.3390/cells10051262
Chicago/Turabian StyleWenmaekers, Stijn, Bastiaan J. Viergever, Gunjan Kumar, Onno Kranenburg, Peter C. Black, Mads Daugaard, and Richard P. Meijer. 2021. "A Potential Role for HUWE1 in Modulating Cisplatin Sensitivity" Cells 10, no. 5: 1262. https://doi.org/10.3390/cells10051262
APA StyleWenmaekers, S., Viergever, B. J., Kumar, G., Kranenburg, O., Black, P. C., Daugaard, M., & Meijer, R. P. (2021). A Potential Role for HUWE1 in Modulating Cisplatin Sensitivity. Cells, 10(5), 1262. https://doi.org/10.3390/cells10051262