
Modelling the Functions of Polo-Like Kinases in Mice and Their Applications as Cancer Targets with a Special Focus on Ovarian Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
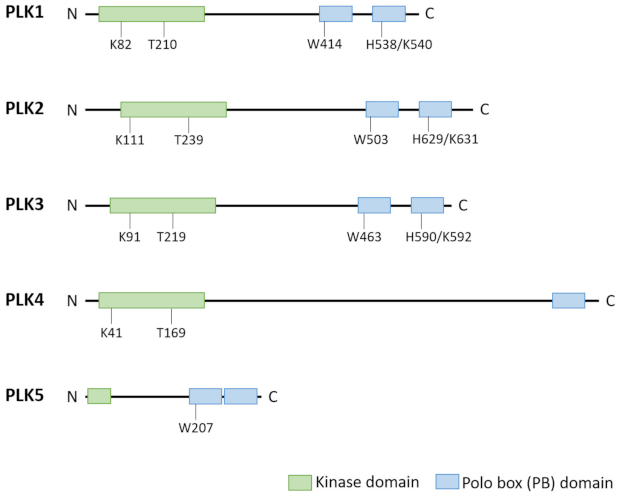
1. Polo-Like Kinases and Their Physiological Functions
2. PLKs and Tumor Development
2.1. PLK1 Is a Dual Game Player in Tumorigenesis
2.1.1. Oncogenic Potential of PLK1
2.1.2. Tumor Suppressor Potential of PLK1
2.2. PLK2 and Tumorigenesis
2.3. PLK3 and Tumorigenesis
2.4. PLK4 and Tumorigenesis
2.5. PLK5 and Tumorigenesis
3. Targeting PLKs in Cancer
4. Ovarian Cancer and PLKs
4.1. PLK1 in Ovarian Cancer
4.2. PLK2 in Ovarian Cancer
4.3. PLK3 in Ovarian Cancer
4.4. PLK4 in Ovarian Cancer
4.5. PLK 5 in Ovarian Cancer
5. Targeting PLKs in Ovarian Cancer
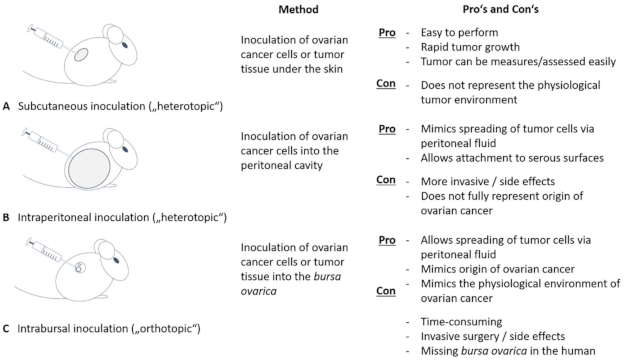
6. Mouse Models in Ovarian Cancer Research
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sunkel, C.E.; Glover, D.M. Polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell Sci. 1988, 89, 25–38. [Google Scholar] [CrossRef]
- Hamanaka, R.; Maloid, S.; Smith, M.R.; O’Connell, C.D.; Longo, D.L.; Ferris, D.K. Cloning and characterization of human and murine homologues of the Drosophila polo serine-threonine kinase. Cell Growth Differ. 1994, 5, 249–257. [Google Scholar] [PubMed]
- Barr, F.A.; Silljé, H.H.W.; Nigg, E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Lowery, D.M.; Lim, D.; Yaffe, M.B. Structure and function of Polo-like kinases. Oncogene 2005, 24, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Van de Weerdt, B.C.M.; Medema, R.H. Polo-like kinases: A team in control of the division. Cell Cycle 2006, 5, 853–864. [Google Scholar] [CrossRef]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [Google Scholar] [CrossRef]
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton 2018, 75, 481–494. [Google Scholar] [CrossRef]
- Slevin, L.K.; Nye, J.; Pinkerton, D.C.; Buster, D.W.; Rogers, G.C.; Slep, K.C. The structure of the plk4 cryptic polo box reveals two tandem polo boxes required for centriole duplication. Structure 2012, 20, 1905–1917. [Google Scholar] [CrossRef]
- Maniswami, R.R.; Prashanth, S.; Karanth, A.V.; Koushik, S.; Govindaraj, H.; Mullangi, R.; Rajagopal, S.; Jegatheesan, S.K. PLK4: A link between centriole biogenesis and cancer. Expert Opin. Ther. Targets 2018, 22, 59–73. [Google Scholar] [CrossRef]
- Garvey, D.R.; Chhabra, G.; Ndiaye, M.A.; Ahmad, N. Role of Polo-Like Kinase 4 (PLK4) in Epithelial Cancers and Recent Progress in its Small Molecule Targeting for Cancer Management. Mol. Cancer Ther. 2021, 20, 632–640. [Google Scholar] [CrossRef]
- Lee, K.S.; Grenfell, T.Z.; Yarm, F.R.; Erikson, R.L. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc. Natl. Acad. Sci. USA 1998, 95, 9301–9306. [Google Scholar] [CrossRef]
- Jang, Y.-J.; Lin, C.-Y.; Ma, S.; Erikson, R.L. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 1984–1989. [Google Scholar] [CrossRef]
- Elia, A.E.H.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.C.; Smerdon, S.J.; Yaffe, M.B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Archambault, V.; Lépine, G.; Kachaner, D. Understanding the Polo Kinase machine. Oncogene 2015, 34, 4799–4807. [Google Scholar] [CrossRef]
- De Cárcer, G.; Manning, G.; Malumbres, M. From Plk1 to Plk5: Functional evolution of polo-like kinases. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef]
- Schmit, T.L.; Ahmad, N. Regulation of mitosis via mitotic kinases: New opportunities for cancer management. Mol. Cancer Ther. 2007, 6, 1920–1931. [Google Scholar] [CrossRef]
- Winkles, J.A.; Alberts, G.F. Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene 2005, 24, 260–266. [Google Scholar] [CrossRef]
- Archambault, V.; Glover, D.M. Polo-like kinases: Conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 2009, 10, 265–275. [Google Scholar] [CrossRef]
- Lu, L.-Y.; Wood, J.L.; Minter-Dykhouse, K.; Ye, L.; Saunders, T.L.; Yu, X.; Chen, J. Polo-like kinase 1 is essential for early embryonic development and tumor suppression. Mol. Cell. Biol. 2008, 28, 6870–6876. [Google Scholar] [CrossRef]
- Wachowicz, P.; Fernández-Miranda, G.; Marugán, C.; Escobar, B.; de Cárcer, G. Genetic depletion of Polo-like kinase 1 leads to embryonic lethality due to mitotic aberrancies. Bioessays 2016, 38 (Suppl. 1), S96–S106. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, A.R.; Sharma, G.; Chakraborty, C.; Kim, J. PLK-1: Angel or devil for cell cycle progression. Biochim. Biophys. Acta 2016, 1865, 190–203. [Google Scholar] [CrossRef]
- Combes, G.; Alharbi, I.; Braga, L.G.; Elowe, S. Playing polo during mitosis: PLK1 takes the lead. Oncogene 2017, 36, 4819–4827. [Google Scholar] [CrossRef]
- Seki, A.; Coppinger, J.A.; Jang, C.-Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef]
- Eckerdt, F.; Yuan, J.; Strebhardt, K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276. [Google Scholar] [CrossRef]
- Spänkuch, B.; Steinhauser, I.; Wartlick, H.; Kurunci-Csacsko, E.; Strebhardt, K.I.; Langer, K. Downregulation of Plk1 expression by receptor-mediated uptake of antisense oligonucleotide-loaded nanoparticles. Neoplasia 2008, 10, 223–234. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, B.T.; Strebhardt, K. Polo-like kinase 1: Target and regulator of transcriptional control. Cell Cycle 2006, 5, 2881–2885. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Fan, H.-Y.; Lian, L.; Li, S.-W.; Chen, D.-Y.; Schatten, H.; Sun, Q.-Y. Polo-like kinase-1 is a pivotal regulator of microtubule assembly during mouse oocyte meiotic maturation, fertilization, and early embryonic mitosis. Biol. Reprod 2002, 67, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Solc, P.; Kitajima, T.S.; Yoshida, S.; Brzakova, A.; Kaido, M.; Baran, V.; Mayer, A.; Samalova, P.; Motlik, J.; Ellenberg, J. Multiple requirements of PLK1 during mouse oocyte maturation. PLoS ONE 2015, 10, e0116783. [Google Scholar] [CrossRef]
- Cunningham, C.E.; MacAuley, M.J.; Vizeacoumar, F.S.; Abuhussein, O.; Freywald, A.; Vizeacoumar, F.J. The CINs of Polo-Like Kinase 1 in Cancer. Cancers 2020, 12, 2953. [Google Scholar] [CrossRef]
- Lane, H.A.; Nigg, E.A. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J. Cell Biol. 1996, 135, 1701–1713. [Google Scholar] [CrossRef]
- De Luca, M.; Lavia, P.; Guarguaglini, G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle 2006, 5, 296–303. [Google Scholar] [CrossRef]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Shinya, N.; Iwamatsu, A.; Nishida, E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 2001, 410, 215–220. [Google Scholar] [CrossRef]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 2002, 3, 341–348. [Google Scholar] [CrossRef]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef]
- Sumara, I.; Giménez-Abián, J.F.; Gerlich, D.; Hirota, T.; Kraft, C.; de La Torre, C.; Ellenberg, J.; Peters, J.-M. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr. Biol. 2004, 14, 1712–1722. [Google Scholar] [CrossRef]
- Van Vugt, M.A.T.M.; Brás, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef]
- Li, H.; Liu, X.S.; Yang, X.; Wang, Y.; Wang, Y.; Turner, J.R.; Liu, X. Phosphorylation of CLIP-170 by Plk1 and CK2 promotes timely formation of kinetochore-microtubule attachments. EMBO J. 2010, 29, 2953–2965. [Google Scholar] [CrossRef]
- Liu, D.; Davydenko, O.; Lampson, M.A. Polo-like kinase-1 regulates kinetochore-microtubule dynamics and spindle checkpoint silencing. J. Cell Biol. 2012, 198, 491–499. [Google Scholar] [CrossRef]
- Sumara, I.; Vorlaufer, E.; Stukenberg, P.T.; Kelm, O.; Redemann, N.; Nigg, E.A.; Peters, J.-M. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell 2002, 9, 515–525. [Google Scholar] [CrossRef]
- Kang, Y.H.; Park, J.-E.; Yu, L.-R.; Soung, N.-K.; Yun, S.-M.; Bang, J.K.; Seong, Y.-S.; Yu, H.; Garfield, S.; Veenstra, T.D.; et al. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol. Cell 2006, 24, 409–422. [Google Scholar] [CrossRef]
- Brennan, I.M.; Peters, U.; Kapoor, T.M.; Straight, A.F. Polo-like kinase controls vertebrate spindle elongation and cytokinesis. PLoS ONE 2007, 2, e409. [Google Scholar] [CrossRef]
- Petronczki, M.; Glotzer, M.; Kraut, N.; Peters, J.-M. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev. Cell 2007, 12, 713–725. [Google Scholar] [CrossRef]
- Wolfe, B.A.; Takaki, T.; Petronczki, M.; Glotzer, M. Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol. 2009, 7, e1000110. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, G.; Chakraborty, C.; Sharma, A.R.; Kim, J. Regulatory functional territory of PLK-1 and their substrates beyond mitosis. Oncotarget 2017, 8, 37942–37962. [Google Scholar] [CrossRef]
- Raab, C.A.; Raab, M.; Becker, S.; Strebhardt, K. Non-mitotic functions of polo-like kinases in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188467. [Google Scholar] [CrossRef]
- Liu, X.S.; Li, H.; Song, B.; Liu, X. Polo-like kinase 1 phosphorylation of G2 and S-phase-expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep. 2010, 11, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Strebhardt, K. Plk1: Unexpected roles in DNA replication. Cell Res. 2013, 23, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Matthess, Y.; Raab, M.; Knecht, R.; Becker, S.; Strebhardt, K. Sequential Cdk1 and Plk1 phosphorylation of caspase-8 triggers apoptotic cell death during mitosis. Mol. Oncol. 2014, 8, 596–608. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, X. Kinases Involved in Both Autophagy and Mitosis. Int. J. Mol. Sci. 2017, 18, 1884. [Google Scholar] [CrossRef]
- Ruf, S.; Heberle, A.M.; Langelaar-Makkinje, M.; Gelino, S.; Wilkinson, D.; Gerbeth, C.; Schwarz, J.J.; Holzwarth, B.; Warscheid, B.; Meisinger, C.; et al. PLK1 (polo like kinase 1) inhibits MTOR complex 1 and promotes autophagy. Autophagy 2017, 13, 486–505. [Google Scholar] [CrossRef]
- Tao, Y.-F.; Li, Z.-H.; Du, W.-W.; Xu, L.-X.; Ren, J.-L.; Li, X.-L.; Fang, F.; Xie, Y.; Li, M.; Qian, G.-H.; et al. Inhibiting PLK1 induces autophagy of acute myeloid leukemia cells via mammalian target of rapamycin pathway dephosphorylation. Oncol. Rep. 2017, 37, 1419–1429. [Google Scholar] [CrossRef]
- Simmons, D.L.; Neel, B.G.; Stevens, R.; Evett, G.; Erikson, R.L. Identification of an early-growth-response gene encoding a novel putative protein kinase. Mol. Cell. Biol. 1992, 12, 4164–4169. [Google Scholar] [CrossRef]
- Liby, K.; Wu, H.; Ouyang, B.; Wu, S.; Chen, J.; Dai, W. Identification of the human homologue of the early-growth response gene Snk, encoding a serum-inducible kinase. DNA Seq. 2001, 11, 527–533. [Google Scholar] [CrossRef]
- Ma, S.; Charron, J.; Erikson, R.L. Role of Plk2 (Snk) in mouse development and cell proliferation. Mol. Cell. Biol. 2003, 23, 6936–6943. [Google Scholar] [CrossRef]
- Villegas, E.; Kabotyanski, E.B.; Shore, A.N.; Creighton, C.J.; Westbrook, T.F.; Rosen, J.M. Plk2 regulates mitotic spindle orientation and mammary gland development. Development 2014, 141, 1562–1571. [Google Scholar] [CrossRef]
- Kauselmann, G.; Weiler, M.; Wulff, P.; Jessberger, S.; Konietzko, U.; Scafidi, J.; Staubli, U.; Bereiter-Hahn, J.; Strebhardt, K.; Kuhl, D. The polo-like protein kinases Fnk and Snk associate with a Ca(2+)- and integrin-binding protein and are regulated dynamically with synaptic plasticity. EMBO J. 1999, 18, 5528–5539. [Google Scholar] [CrossRef]
- Pak, D.T.S.; Sheng, M. Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science 2003, 302, 1368–1373. [Google Scholar] [CrossRef]
- Oueslati, A.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E3945–E3954. [Google Scholar] [CrossRef] [PubMed]
- Walkup, W.G.; Sweredoski, M.J.; Graham, R.L.; Hess, S.; Kennedy, M.B. Phosphorylation of synaptic GTPase-activating protein (synGAP) by polo-like kinase (Plk2) alters the ratio of its GAP activity toward HRas, Rap1 and Rap2 GTPases. Biochem. Biophys. Res. Commun. 2018, 503, 1599–1604. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, D.P.; Feliu-Mojer, M.; Gaiottino, J.; Pak, D.T.S.; Sheng, M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron 2008, 58, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Warnke, S.; Kemmler, S.; Hames, R.S.; Tsai, H.-L.; Hoffmann-Rohrer, U.; Fry, A.M.; Hoffmann, I. Polo-like kinase-2 is required for centriole duplication in mammalian cells. Curr. Biol. 2004, 14, 1200–1207. [Google Scholar] [CrossRef]
- Cizmecioglu, O.; Warnke, S.; Arnold, M.; Duensing, S.; Hoffmann, I. Plk2 regulated centriole duplication is dependent on its localization to the centrioles and a functional polo-box domain. Cell Cycle 2008, 7, 3548–3555. [Google Scholar] [CrossRef]
- Matthew, E.M.; Yen, T.J.; Dicker, D.T.; Dorsey, J.F.; Yang, W.; Navaraj, A.; El-Deiry, W.S. Replication stress, defective S-phase checkpoint and increased death in Plk2-deficient human cancer cells. Cell Cycle 2007, 6, 2571–2578. [Google Scholar] [CrossRef]
- Burns, T.F.; Fei, P.; Scata, K.A.; Dicker, D.T.; El-Deiry, W.S. Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol. Cell. Biol. 2003, 23, 5556–5571. [Google Scholar] [CrossRef]
- Yang, Y.; Bai, J.; Shen, R.; Brown, S.A.N.; Komissarova, E.; Huang, Y.; Jiang, N.; Alberts, G.F.; Costa, M.; Lu, L.; et al. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res. 2008, 68, 4077–4085. [Google Scholar] [CrossRef]
- Myer, D.L.; Robbins, S.B.; Yin, M.; Boivin, G.P.; Liu, Y.; Greis, K.D.; Bahassi, E.M.; Stambrook, P.J. Absence of polo-like kinase 3 in mice stabilizes Cdc25A after DNA damage but is not sufficient to produce tumors. Mutat. Res. 2011, 714, 1–10. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, S.; Chen, J.; Fukasawa, K.; Naik, U.; Traganos, F.; Darzynkiewicz, Z.; Jhanwar-Uniyal, M.; Dai, W. Cell cycle arrest and apoptosis induced by human Polo-like kinase 3 is mediated through perturbation of microtubule integrity. Mol. Cell. Biol. 2002, 22, 3450–3459. [Google Scholar] [CrossRef]
- Holtrich, U.; Wolf, G.; Yuan, J.; Bereiter-Hahn, J.; Karn, T.; Weiler, M.; Kauselmann, G.; Rehli, M.; Andreesen, R.; Kaufmann, M.; et al. Adhesion induced expression of the serine/threonine kinase Fnk in human macrophages. Oncogene 2000, 19, 4832–4839. [Google Scholar] [CrossRef]
- Aquino Perez, C.; Palek, M.; Stolarova, L.; von Morgen, P.; Macurek, L. Phosphorylation of PLK3 Is Controlled by Protein Phosphatase 6. Cells 2020, 9, 1506. [Google Scholar] [CrossRef]
- Helmke, C.; Becker, S.; Strebhardt, K. The role of Plk3 in oncogenesis. Oncogene 2016, 35, 135–147. [Google Scholar] [CrossRef]
- Rödel, F.; Martin, D.; Helmke, C.; Balermpas, P.; Fokas, E.; Wieland, U.; Rave-Fränk, M.; Kitz, J.; Matthess, Y.; Raab, M.; et al. Polo-like kinase 3 and phosphoT273 caspase-8 are associated with improved local tumor control and survival in patients with anal carcinoma treated with concomitant chemoradiotherapy. Oncotarget 2016, 7, 53339–53349. [Google Scholar] [CrossRef]
- Zimmerman, W.C.; Erikson, R.L. Polo-like kinase 3 is required for entry into S phase. Proc. Natl. Acad. Sci. USA 2007, 104, 1847–1852. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Hennigan, R.F.; Myer, D.L.; Stambrook, P.J. Cdc25C phosphorylation on serine 191 by Plk3 promotes its nuclear translocation. Oncogene 2004, 23, 2658–2663. [Google Scholar] [CrossRef]
- Xie, S.; Wu, H.; Wang, Q.; Cogswell, J.P.; Husain, I.; Conn, C.; Stambrook, P.; Jhanwar-Uniyal, M.; Dai, W. Plk3 functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway. J. Biol. Chem. 2001, 276, 43305–43312. [Google Scholar] [CrossRef]
- Xie, S.; Wu, H.; Wang, Q.; Kunicki, J.; Thomas, R.O.; Hollingsworth, R.E.; Cogswell, J.; Dai, W. Genotoxic stress-induced activation of Plk3 is partly mediated by Chk2. Cell Cycle 2002, 1, 424–429. [Google Scholar] [CrossRef][Green Version]
- Bahassi, E.M.; Conn, C.W.; Myer, D.L.; Hennigan, R.F.; McGowan, C.H.; Sanchez, Y.; Stambrook, P.J. Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene 2002, 21, 6633–6640. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Myer, D.L.; McKenney, R.J.; Hennigan, R.F.; Stambrook, P.J. Priming phosphorylation of Chk2 by polo-like kinase 3 (Plk3) mediates its full activation by ATM and a downstream checkpoint in response to DNA damage. Mutat. Res. 2006, 596, 166–176. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Q.; Jiang, Y.; Zhang, Y.; Vega-Saenzdemiera, E.; Osman, I.; Dai, W. Roles of Polo-like kinase 3 in suppressing tumor angiogenesis. Exp. Hematol. Oncol. 2012, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Karn, T.; Holtrich, U.; Wolf, G.; Hock, B.; Strebhardt, K.; Rubsamenwaigmann, H. Human SAK related to the PLK/polo family of cell cycle kinases shows high mRNA expression in testis. Oncol. Rep. 1997, 4, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.W.; Kozarova, A.; Cheung, P.; Macmillan, J.C.; Swallow, C.J.; Cross, J.C.; Dennis, J.W. Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr. Biol. 2001, 11, 441–446. [Google Scholar] [CrossRef]
- Habedanck, R.; Stierhof, Y.-D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Habedanck, R.; Stierhof, Y.-D.; Nigg, E.A. Plk4-induced centriole biogenesis in human cells. Dev. Cell 2007, 13, 190–202. [Google Scholar] [CrossRef]
- Sillibourne, J.E.; Bornens, M. Polo-like kinase 4: The odd one out of the family. Cell Div. 2010, 5, 25. [Google Scholar] [CrossRef]
- Park, J.-E.; Zhang, L.; Bang, J.K.; Andresson, T.; DiMaio, F.; Lee, K.S. Phase separation of Polo-like kinase 4 by autoactivation and clustering drives centriole biogenesis. Nat. Commun. 2019, 10, 4959. [Google Scholar] [CrossRef]
- Breslow, D.K.; Holland, A.J. Mechanism and Regulation of Centriole and Cilium Biogenesis. Annu. Rev. Biochem. 2019, 88, 691–724. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, C.; Liang, H.; Han, L. Polo-Like Kinase 4′s Critical Role in Cancer Development and Strategies for Plk4-Targeted Therapy. Front. Oncol. 2021, 11, 587554. [Google Scholar] [CrossRef]
- Rosario, C.O.; Ko, M.A.; Haffani, Y.Z.; Gladdy, R.A.; Paderova, J.; Pollett, A.; Squire, J.A.; Dennis, J.W.; Swallow, C.J. Plk4 is required for cytokinesis and maintenance of chromosomal stability. Proc. Natl. Acad. Sci. USA 2010, 107, 6888–6893. [Google Scholar] [CrossRef]
- Press, M.F.; Xie, B.; Davenport, S.; Zhou, Y.; Guzman, R.; Nolan, G.P.; O’Brien, N.; Palazzolo, M.; Mak, T.W.; Brugge, J.S.; et al. Role for polo-like kinase 4 in mediation of cytokinesis. Proc. Natl. Acad. Sci. USA 2019, 116, 11309–11318. [Google Scholar] [CrossRef]
- Bonni, S.; Ganuelas, M.L.; Petrinac, S.; Hudson, J.W. Human Plk4 phosphorylates Cdc25C. Cell Cycle 2008, 7, 545–547. [Google Scholar] [CrossRef]
- Montenegro Gouveia, S.; Zitouni, S.; Kong, D.; Duarte, P.; Ferreira Gomes, B.; Sousa, A.L.; Tranfield, E.M.; Hyman, A.; Loncarek, J.; Bettencourt-Dias, M. PLK4 is a microtubule-associated protein that self-assembles promoting de novo MTOC formation. J. Cell Sci. 2018, 132. [Google Scholar] [CrossRef]
- Byrne, D.P.; Clarke, C.J.; Brownridge, P.J.; Kalyuzhnyy, A.; Perkins, S.; Campbell, A.; Mason, D.; Jones, A.R.; Eyers, P.A.; Eyers, C.E. Use of the Polo-like kinase 4 (PLK4) inhibitor centrinone to investigate intracellular signalling networks using SILAC-based phosphoproteomics. Biochem. J. 2020, 477, 2451–2475. [Google Scholar] [CrossRef]
- Andrysik, Z.; Bernstein, W.Z.; Deng, L.; Myer, D.L.; Li, Y.-Q.; Tischfield, J.A.; Stambrook, P.J.; Bahassi, E.M. The novel mouse Polo-like kinase 5 responds to DNA damage and localizes in the nucleolus. Nucleic Acids Res. 2010, 38, 2931–2943. [Google Scholar] [CrossRef]
- De Cárcer, G.; Escobar, B.; Higuero, A.M.; García, L.; Ansón, A.; Pérez, G.; Mollejo, M.; Manning, G.; Meléndez, B.; Abad-Rodríguez, J.; et al. Plk5, a polo box domain-only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol. Cell. Biol. 2011, 31, 1225–1239. [Google Scholar] [CrossRef]
- Holtrich, U.; Wolf, G.; Bräuninger, A.; Karn, T.; Böhme, B.; Rübsamen-Waigmann, H.; Strebhardt, K. Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 1736–1740. [Google Scholar] [CrossRef]
- Raab, M.; Kappel, S.; Krämer, A.; Sanhaji, M.; Matthess, Y.; Kurunci-Csacsko, E.; Calzada-Wack, J.; Rathkolb, B.; Rozman, J.; Adler, T.; et al. Toxicity modelling of Plk1-targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat. Commun. 2011, 2, 395. [Google Scholar] [CrossRef]
- Raab, M.; Sanhaji, M.; Matthess, Y.; Hörlin, A.; Lorenz, I.; Dötsch, C.; Habbe, N.; Waidmann, O.; Kurunci-Csacsko, E.; Firestein, R.; et al. PLK1 has tumor-suppressive potential in APC-truncated colon cancer cells. Nat. Commun. 2018, 9, 1106. [Google Scholar] [CrossRef]
- De Cárcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montañés, P.; Sanclemente, M.; Escobar, B.; de Martino, A.; et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 2018, 9, 3012. [Google Scholar] [CrossRef]
- Smith, M.R.; Wilson, M.L.; Hamanaka, R.; Chase, D.; Kung, H.; Longo, D.L.; Ferris, D.K. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem. Biophys. Res. Commun. 1997, 234, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Elez, R.; Doermer, A.; Holtrich, U.; Ackermann, H.; Stutte, H.J.; Altmannsberger, H.M.; Rübsamen-Waigmann, H.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997, 14, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Kristiansen, G.; Winzer, K.-J.; Schmidt, M.; Gekeler, V.; Noske, A.; Müller, B.-M.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase isoforms in breast cancer: Expression patterns and prognostic implications. Virchows Arch. 2005, 446, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, H.; Sun, Z.; Guo, Q.; Shi, H.; Jia, Y. The clinical and prognostic value of polo-like kinase 1 in lung squamous cell carcinoma patients: Immunohistochemical analysis. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, Y.; Mori, M.; Tanaka, S.; Akazawa, K.; Nakano, S.; Niho, Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int. J. Oncol. 1999, 15, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Lan, B.; Liu, B.-Y.; Chen, X.-H.; Qu, Y.; Zhang, X.-Q.; Cai, Q.; Zhu, Z.-G. Polo like kinase 1 expression and prognostic value in gastric carcinomas. Zhonghua Wei Chang. Wai Ke Za Zhi 2007, 10, 70–72. [Google Scholar] [PubMed]
- Feng, Y.-B.; Lin, D.-C.; Shi, Z.-Z.; Wang, X.-C.; Shen, X.-M.; Zhang, Y.; Du, X.-L.; Luo, M.-L.; Xu, X.; Han, Y.-L.; et al. Overexpression of PLK1 is associated with poor survival by inhibiting apoptosis via enhancement of survivin level in esophageal squamous cell carcinoma. Int. J. Cancer 2009, 124, 578–588. [Google Scholar] [CrossRef]
- Knecht, R.; Elez, R.; Oechler, M.; Solbach, C.; von Ilberg, C.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. 1999, 59, 2794–2797. [Google Scholar]
- Knecht, R.; Oberhauser, C.; Strebhardt, K. PLK (polo-like kinase), a new prognostic marker for oropharyngeal carcinomas. Int. J. Cancer 2000, 89, 535–536. [Google Scholar] [CrossRef]
- King, S.I.; Purdie, C.A.; Bray, S.E.; Quinlan, P.R.; Jordan, L.B.; Thompson, A.M.; Meek, D.W. Immunohistochemical detection of Polo-like kinase-1 (PLK1) in primary breast cancer is associated with TP53 mutation and poor clinical outcom. Breast Cancer Res. 2012, 14, R40. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Hua, Y.; Yu, B.; Ye, X.; He, Z.; Li, C.; Wang, J.; Mo, Y.; Wei, X.; Chen, Y.; et al. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol. Cancer 2020, 19, 19. [Google Scholar] [CrossRef]
- Strebhardt, K.; Kneisel, L.; Linhart, C.; Bernd, A.; Kaufmann, R. Prognostic value of pololike kinase expression in melanomas. JAMA 2000, 283, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Amani, V.; Prince, E.W.; Alimova, I.; Balakrishnan, I.; Birks, D.; Donson, A.M.; Harris, P.; Levy, J.M.M.; Handler, M.; Foreman, N.K.; et al. Polo-like Kinase 1 as a potential therapeutic target in Diffuse Intrinsic Pontine Glioma. BMC Cancer 2016, 16, 647. [Google Scholar] [CrossRef] [PubMed]
- Schmit, T.L.; Zhong, W.; Setaluri, V.; Spiegelman, V.S.; Ahmad, N. Targeted depletion of Polo-like kinase (Plk) 1 through lentiviral shRNA or a small-molecule inhibitor causes mitotic catastrophe and induction of apoptosis in human melanoma cells. J. Investig. Dermatol. 2009, 129, 2843–2853. [Google Scholar] [CrossRef] [PubMed]
- Jalili, A.; Moser, A.; Pashenkov, M.; Wagner, C.; Pathria, G.; Borgdorff, V.; Gschaider, M.; Stingl, G.; Ramaswamy, S.; Wagner, S.N. Polo-like kinase 1 is a potential therapeutic target in human melanoma. J. Investig. Dermatol. 2011, 131, 1886–1895. [Google Scholar] [CrossRef]
- Schmit, T.L.; Zhong, W.; Nihal, M.; Ahmad, N. Polo-like kinase 1 (Plk1) in non-melanoma skin cancers. Cell Cycle 2009, 8, 2697–2702. [Google Scholar] [CrossRef]
- Takahashi, T.; Sano, B.; Nagata, T.; Kato, H.; Sugiyama, Y.; Kunieda, K.; Kimura, M.; Okano, Y.; Saji, S. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. 2003, 94, 148–152. [Google Scholar] [CrossRef]
- Han, D.-P.; Zhu, Q.-L.; Cui, J.-T.; Wang, P.-X.; Qu, S.; Cao, Q.-F.; Zong, Y.-P.; Feng, B.; Zheng, M.-H.; Lu, A.-G. Polo-like kinase 1 is overexpressed in colorectal cancer and participates in the migration and invasion of colorectal cancer cells. Med. Sci. Monit. 2012, 18, BR237–BR246. [Google Scholar] [CrossRef]
- Tut, T.G.; Lim, S.H.S.; Dissanayake, I.U.; Descallar, J.; Chua, W.; Ng, W.; de Souza, P.; Shin, J.-S.; Lee, C.S. Upregulated Polo-Like Kinase 1 Expression Correlates with Inferior Survival Outcomes in Rectal Cancer. PLoS ONE 2015, 10, e0129313. [Google Scholar] [CrossRef]
- Rödel, F.; Keppner, S.; Capalbo, G.; Bashary, R.; Kaufmann, M.; Rödel, C.; Strebhardt, K.; Spänkuch, B. Polo-like kinase 1 as predictive marker and therapeutic target for radiotherapy in rectal cancer. Am. J. Pathol. 2010, 177, 918–929. [Google Scholar] [CrossRef]
- Triscott, J.; Lee, C.; Foster, C.; Manoranjan, B.; Pambid, M.R.; Berns, R.; Fotovati, A.; Venugopal, C.; O’Halloran, K.; Narendran, A.; et al. Personalizing the treatment of pediatric medulloblastoma: Polo-like kinase 1 as a molecular target in high-risk children. Cancer Res. 2013, 73, 6734–6744. [Google Scholar] [CrossRef]
- Linton, A.; Cheng, Y.Y.; Griggs, K.; Schedlich, L.; Kirschner, M.B.; Gattani, S.; Srikaran, S.; Chuan-Hao Kao, S.; McCaughan, B.C.; Klebe, S.; et al. An RNAi-based screen reveals PLK1, CDK1 and NDC80 as potential therapeutic targets in malignant pleural mesothelioma. Br. J. Cancer 2014, 110, 510–519. [Google Scholar] [CrossRef]
- Dietzmann, K.; Kirches, E.; Mawrin, C. Effects of phospholipase Cgamma on Polo-like kinase 1 expression in human glioma cells. J. Cancer Res. Clin. Oncol. 2002, 128, 265–270. [Google Scholar] [CrossRef]
- Li, Z.; Yang, C.; Li, X.; Du, X.; Tao, Y.; Ren, J.; Fang, F.; Xie, Y.; Li, M.; Qian, G.; et al. The dual role of BI 2536, a small-molecule inhibitor that targets PLK1, in induction of apoptosis and attenuation of autophagy in neuroblastoma cells. J. Cancer 2020, 11, 3274–3287. [Google Scholar] [CrossRef]
- Ackermann, S.; Goeser, F.; Schulte, J.H.; Schramm, A.; Ehemann, V.; Hero, B.; Eggert, A.; Berthold, F.; Fischer, M. Polo-like kinase 1 is a therapeutic target in high-risk neuroblastoma. Clin. Cancer Res. 2011, 17, 731–741. [Google Scholar] [CrossRef]
- Takai, N.; Hamanaka, R.; Yoshimatsu, J.; Miyakawa, I. Polo-like kinases (Plks) and cancer. Oncogene 2005, 24, 287–291. [Google Scholar] [CrossRef]
- Ito, Y.; Yoshida, H.; Matsuzuka, F.; Matsuura, N.; Nakamura, Y.; Nakamine, H.; Kakudo, K.; Kuma, K.; Miyauchi, A. Polo-like kinase 1 (PLK1) expression is associated with cell proliferative activity and cdc2 expression in malignant lymphoma of the thyroid. Anticancer Res. 2004, 24, 259–263. [Google Scholar]
- Gray, P.J.; Bearss, D.J.; Han, H.; Nagle, R.; Tsao, M.-S.; Dean, N.; von Hoff, D.D. Identification of human polo-like kinase 1 as a potential therapeutic target in pancreatic cancer. Mol. Cancer Ther. 2004, 3, 641–646. [Google Scholar]
- Weichert, W.; Schmidt, M.; Gekeler, V.; Denkert, C.; Stephan, C.; Jung, K.; Loening, S.; Dietel, M.; Kristiansen, G. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate 2004, 60, 240–245. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Ahmad, N. Polo-like kinase (Plk) 1 as a target for prostate cancer management. IUBMB Life 2005, 57, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.-i.; Ohira, M.; Horie, H.; Ando, K.; Takayasu, H.; Suzuki, Y.; Sugano, S.; Hirata, T.; Goto, T.; Matsunaga, T.; et al. Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: Identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene 2004, 23, 5901–5911. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Zhu, Y.Q.; Lui, K.S.; Cai, Q.; Lu, P.; Poon, R.T. Aberrant Polo-like kinase 1-Cdc25A pathway in metastatic hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 6813–6820. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Wen, D.-Y.; Dang, Y.-W.; He, Y.; Yang, H.; Chen, G. Comprehensive and Integrative Analysis Reveals the Diagnostic, Clinicopathological and Prognostic Significance of Polo-Like Kinase 1 in Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2018, 47, 925–947. [Google Scholar] [CrossRef]
- Yousef, E.H.; El-Mesery, M.E.; Habeeb, M.R.; Eissa, L.A. Polo-like kinase 1 as a promising diagnostic biomarker and potential therapeutic target for hepatocellular carcinoma. Tumor Biol. 2020, 42. [Google Scholar] [CrossRef]
- Mito, K.; Kashima, K.; Kikuchi, H.; Daa, T.; Nakayama, I.; Yokoyama, S. Expression of Polo-Like Kinase (PLK1) in non-Hodgkin’s lymphomas. Leuk. Lymphoma 2005, 46, 225–231. [Google Scholar] [CrossRef]
- Stutz, N.; Nihal, M.; Wood, G.S. Polo-like kinase 1 (Plk1) in cutaneous T-cell lymphoma. Br. J. Dermatol. 2011, 164, 814–821. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Z.; Liu, Z. Polo-like kinase 1 is overexpressed in renal cancer and participates in the proliferation and invasion of renal cancer cells. Tumor Biol. 2013, 34, 1887–1894. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, G.; Kong, C. High expression of polo-like kinase 1 is associated with the metastasis and recurrence in urothelial carcinoma of bladder. Urol. Oncol. 2013, 31, 1222–1230. [Google Scholar] [CrossRef]
- Lake, R.J.; Jelinek, W.R. Cell cycle- and terminal differentiation-associated regulation of the mouse mRNA encoding a conserved mitotic protein kinase. Mol. Cell. Biol. 1993, 13, 7793–7801. [Google Scholar] [CrossRef][Green Version]
- Golsteyn, R.M.; Schultz, S.J.; Bartek, J.; Ziemiecki, A.; Ried, T.; Nigg, E.A. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J. Cell Sci. 1994, 107 Pt 6, 1509–1517. [Google Scholar] [CrossRef]
- Matsubara, N.; Yanagisawa, M.; Nishimune, Y.; Obinata, M.; Matsui, Y. Murine polo like kinase 1 gene is expressed in meiotic testicular germ cells and oocytes. Mol. Reprod. Dev. 1995, 41, 407–415. [Google Scholar] [CrossRef]
- Takai, N.; Yoshimatsu, J.; Nishida, Y.; Narahara, H.; Miyakawa, I.; Hamanaka, R. Expression of polo-like kinase (PLK) in the mouse placenta and ovary. Reprod. Fertil. Dev. 1999, 11, 31–35. [Google Scholar] [CrossRef]
- Rödel, F.; Zhou, S.; Győrffy, B.; Raab, M.; Sanhaji, M.; Mandal, R.; Martin, D.; Becker, S.; Strebhardt, K. The Prognostic Relevance of the Proliferation Markers Ki-67 and Plk1 in Early-Stage Ovarian Cancer Patients With Serous, Low-Grade Carcinoma Based on mRNA and Protein Expression. Front. Oncol. 2020, 10, 558932. [Google Scholar] [CrossRef]
- Kaczorowski, M.; Borowiec, T.; Donizy, P.; Pagacz, K.; Fendler, W.; Lipinski, A.; Halon, A.; Matkowski, R. Polo-like kinase-1 immunoreactivity is associated with metastases in cutaneous melanoma. J. Cutan. Pathol. 2017, 44, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Cholewa, B.D.; Liu, X.; Ahmad, N. The role of polo-like kinase 1 in carcinogenesis: Cause or consequence? Cancer Res. 2013, 73, 6848–6855. [Google Scholar] [CrossRef]
- Fu, Z.; Wen, D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers 2017, 9, 131. [Google Scholar] [CrossRef]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef]
- De Cárcer, G. The Mitotic Cancer Target Polo-Like Kinase 1: Oncogene or Tumor Suppressor? Genes 2019, 10, 208. [Google Scholar] [CrossRef]
- Louwen, F.; Yuan, J. Battle of the eternal rivals: Restoring functional p53 and inhibiting Polo-like kinase 1 as cancer therapy. Oncotarget 2013, 4, 958–971. [Google Scholar] [CrossRef]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Bi, C.; Zhao, X.; Lwin, T.; Wang, C.; Yuan, J.; Silva, A.S.; Shah, B.D.; Fang, B.; Li, T.; et al. PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. J. Clin. Investig. 2018, 128, 5517–5530. [Google Scholar] [CrossRef]
- Zhang, R.; Shi, H.; Ren, F.; Liu, H.; Zhang, M.; Deng, Y.; Li, X. Misregulation of polo-like protein kinase 1, P53 and P21WAF1 in epithelial ovarian cancer suggests poor prognosis. Oncol. Rep. 2015, 33, 1235–1242. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Bi, P.; Lu, Y.; Burcham, G.; Elzey, B.D.; Ratliff, T.; Konieczny, S.F.; Ahmad, N.; Kuang, S.; et al. Plk1 phosphorylation of PTEN causes a tumor-promoting metabolic state. Mol. Cell. Biol. 2014, 34, 3642–3661. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Sim, W.; Kim, J.-H. Differential Dependency of Human Pancreatic Cancer Cells on Targeting PTEN via PLK 1 Expression. Cancers 2020, 12, 277. [Google Scholar] [CrossRef]
- Gutteridge, R.E.A.; Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Targeted knockdown of polo-like kinase 1 alters metabolic regulation in melanoma. Cancer Lett. 2017, 394, 13–21. [Google Scholar] [CrossRef]
- Ma, X.; Wang, L.; Huang, D.; Li, Y.; Yang, D.; Li, T.; Li, F.; Sun, L.; Wei, H.; He, K.; et al. Polo-like kinase 1 coordinates biosynthesis during cell cycle progression by directly activating pentose phosphate pathway. Nat. Commun. 2017, 8, 1506. [Google Scholar] [CrossRef]
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 2017, 7, 40. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 2007, 1773, 642–652. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef]
- Wu, J.; Ivanov, A.I.; Fisher, P.B.; Fu, Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.P.; Chen, L.D.; Song, H.B.; Zhang, C.X.; Yuan, Z.W.; Xiang, Z.X. PLK1 promotes epithelial-mesenchymal transition and metastasis of gastric carcinoma cells. Am. J. Transl. Res. 2016, 8, 4172–4183. [Google Scholar]
- De Cárcer, G.; Wachowicz, P.; Martínez-Martínez, S.; Oller, J.; Méndez-Barbero, N.; Escobar, B.; González-Loyola, A.; Takaki, T.; El Bakkali, A.; Cámara, J.A.; et al. Plk1 regulates contraction of postmitotic smooth muscle cells and is required for vascular homeostasis. Nat. Med. 2017, 23, 964–974. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, Y.; Li, Q.; Chen, J. Polo-like kinase 2 promotes chemoresistance and predicts limited survival benefit from adjuvant chemotherapy in colorectal cancer. Int. J. Oncol. 2018, 52, 1401–1414. [Google Scholar] [CrossRef]
- Alafate, W.; Xu, D.; Wu, W.; Xiang, J.; Ma, X.; Xie, W.; Bai, X.; Wang, M.; Wang, J. Loss of PLK2 induces acquired resistance to temozolomide in GBM via activation of notch signaling. J. Exp. Clin. Cancer Res. 2020, 39, 239. [Google Scholar] [CrossRef]
- Pellegrino, R.; Calvisi, D.F.; Ladu, S.; Ehemann, V.; Staniscia, T.; Evert, M.; Dombrowski, F.; Schirmacher, P.; Longerich, T. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology 2010, 51, 857–868. [Google Scholar] [CrossRef]
- Benetatos, L.; Dasoula, A.; Hatzimichael, E.; Syed, N.; Voukelatou, M.; Dranitsaris, G.; Bourantas, K.L.; Crook, T. Polo-like kinase 2 (SNK/PLK2) is a novel epigenetically regulated gene in acute myeloid leukemia and myelodysplastic syndromes: Genetic and epigenetic interactions. Ann. Hematol. 2011, 90, 1037–1045. [Google Scholar] [CrossRef][Green Version]
- Syed, N.; Smith, P.; Sullivan, A.; Spender, L.C.; Dyer, M.; Karran, L.; O’Nions, J.; Allday, M.; Hoffmann, I.; Crawford, D.; et al. Transcriptional silencing of Polo-like kinase 2 (SNK/PLK2) is a frequent event in B-cell malignancies. Blood 2006, 107, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, S.; Zhao, Z.; Mao, X.; Huang, J.; Wu, Z.; Zheng, L.; Wang, Q. MicroRNA-27b up-regulated by human papillomavirus 16 E7 promotes proliferation and suppresses apoptosis by targeting polo-like kinase2 in cervical cancer. Oncotarget 2016, 7, 19666–19679. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Wang, W.; Zhao, L.Y.; Guo, B.; Yang, J.; Zhao, X.G.; Song, T.S.; Huang, C.; Xu, J.R. Silencing of polo-like kinase 2 increases cell proliferation and decreases apoptosis in SGC-7901 gastric cancer cells. Mol. Med. Rep. 2015, 11, 3033–3038. [Google Scholar] [CrossRef]
- Gee, H.E.; Buffa, F.M.; Harris, A.L.; Toohey, J.M.; Carroll, S.L.; Cooper, C.L.; Beith, J.; McNeil, C.; Carmalt, H.; Mak, C.; et al. MicroRNA-Related DNA Repair/Cell-Cycle Genes Independently Associated With Relapse After Radiation Therapy for Early Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 1104–1114. [Google Scholar] [CrossRef]
- Matthew, E.M.; Yang, Z.; Peri, S.; Andrake, M.; Dunbrack, R.; Ross, E.; El-Deiry, W.S. Plk2 Loss Commonly Occurs in Colorectal Carcinomas but not Adenomas: Relationship to mTOR Signaling. Neoplasia 2018, 20, 244–255. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. p53 function and dysfunction. Cell 1992, 70, 523–526. [Google Scholar] [CrossRef]
- Ou, B.; Zhao, J.; Guan, S.; Wangpu, X.; Zhu, C.; Zong, Y.; Ma, J.; Sun, J.; Zheng, M.; Feng, H.; et al. Plk2 promotes tumor growth and inhibits apoptosis by targeting Fbxw7/Cyclin E in colorectal cancer. Cancer Lett. 2016, 380, 457–466. [Google Scholar] [CrossRef]
- Han, T.; Lin, J.; Wang, Y.; Fan, Q.; Sun, H.; Tao, Y.; Sun, C. Forkhead box D1 promotes proliferation and suppresses apoptosis via regulating polo-like kinase 2 in colorectal cancer. Biomed. Pharmacother. 2018, 103, 1369–1375. [Google Scholar] [CrossRef]
- Shen, T.; Li, Y.; Yang, L.; Xu, X.; Liang, F.; Liang, S.; Ba, G.; Xue, F.; Fu, Q. Upregulation of Polo-like kinase 2 gene expression by GATA-1 acetylation in human osteosarcoma MG-63 cells. Int. J. Biochem. Cell Biol. 2012, 44, 423–429. [Google Scholar] [CrossRef]
- Fingas, C.D.; Mertens, J.C.; Razumilava, N.; Sydor, S.; Bronk, S.F.; Christensen, J.D.; Rizvi, S.H.; Canbay, A.; Treckmann, J.W.; Paul, A.; et al. Polo-like kinase 2 is a mediator of hedgehog survival signaling in cholangiocarcinoma. Hepatology 2013, 58, 1362–1374. [Google Scholar] [CrossRef]
- Hu, Z.; Xu, Z.; Liao, X.; Yang, X.; Dong, C.; Luk, K.; Jin, A.; Lu, H. Polo-like kinase 2 acting as a promoter in human tumor cells with an abundance of TAp73. OncoTargets Ther. 2015, 8, 3475–3488. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Xi, X.; Li, Y.; Quan, H.; Liu, S.; Wu, L.; Wu, P.; Lan, W.; Shao, Y.; et al. PLK2 modulation of enriched TAp73 affects osteogenic differentiation and prognosis in human osteosarcoma. Cancer Med. 2020, 9, 4371–4385. [Google Scholar] [CrossRef] [PubMed]
- Kothari, V.; Wei, I.; Shankar, S.; Kalyana-Sundaram, S.; Wang, L.; Ma, L.W.; Vats, P.; Grasso, C.S.; Robinson, D.R.; Wu, Y.-M.; et al. Outlier kinase expression by RNA sequencing as targets for precision therapy. Cancer Discov. 2013, 3, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, Y.; André, E.A.; Lee, K.J.; Nguyen, T.; Feng, Y.; Jia, N.; Harris, B.T.; Burns, M.P.; Pak, D.T.S. Inhibition of Polo-like kinase 2 ameliorates pathogenesis in Alzheimer’s disease model mice. PLoS ONE 2019, 14, e0219691. [Google Scholar] [CrossRef]
- Li, B.; Ouyang, B.; Pan, H.; Reissmann, P.T.; Slamon, D.J.; Arceci, R.; Lu, L.; Dai, W. Prk, a cytokine-inducible human protein serine/threonine kinase whose expression appears to be down-regulated in lung carcinomas. J. Biol. Chem. 1996, 271, 19402–19408. [Google Scholar] [CrossRef]
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. 2004, 279, 25549–25561. [Google Scholar] [CrossRef]
- Dai, W.; Li, Y.; Ouyang, B.; Pan, H.; Reissmann, P.; Li, J.; Wiest, J.; Stambrook, P.; Gluckman, J.L.; Noffsinger, A.; et al. PRK, a cell cycle gene localized to 8p21, is downregulated in head and neck cancer. Genes Chromosomes Cancer 2000, 27, 332–336. [Google Scholar] [CrossRef]
- Dai, W.; Liu, T.; Wang, Q.; Rao, C.V.; Reddy, B.S. Down-regulation of PLK3 gene expression by types and amount of dietary fat in rat colon tumors. Int. J. Oncol. 2002, 20, 121–126. [Google Scholar] [CrossRef]
- Lin, C.; Bai, S.; Du, T.; Lai, Y.; Chen, X.; Peng, S.; Ma, X.; Wu, W.; Guo, Z.; Huang, H. Polo-like kinase 3 is associated with poor prognosis and regulates proliferation and metastasis in prostate cancer. Cancer Manag. Res. 2019, 11, 1517–1524. [Google Scholar] [CrossRef]
- Fleischmann, M.; Martin, D.; Peña-Llopis, S.; Oppermann, J.; von der Grün, J.; Diefenhardt, M.; Chatzikonstantinou, G.; Fokas, E.; Rödel, C.; Strebhardt, K.; et al. Association of Polo-Like Kinase 3 and PhosphoT273 Caspase 8 Levels With Disease-Related Outcomes Among Cervical Squamous Cell Carcinoma Patients Treated With Chemoradiation and Brachytherapy. Front. Oncol. 2019, 9, 742. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yao, Y.; Lu, L.; Costa, M.; Dai, W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J. Biol. Chem. 2010, 285, 38944–38950. [Google Scholar] [CrossRef]
- Xu, D.; Dai, W.; Li, C. Polo-like kinase 3, hypoxic responses, and tumorigenesis. Cell Cycle 2017, 16, 2032–2036. [Google Scholar] [CrossRef] [PubMed]
- Jen, K.-Y.; Cheung, V.G. Identification of novel p53 target genes in ionizing radiation response. Cancer Res. 2005, 65, 7666–7673. [Google Scholar] [CrossRef]
- Li, Z.; Niu, J.; Uwagawa, T.; Peng, B.; Chiao, P.J. Function of polo-like kinase 3 in NF-kappaB-mediated proapoptotic response. J. Biol. Chem. 2005, 280, 16843–16850. [Google Scholar] [CrossRef]
- Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Chabannon, C.; Charpin, C.; Chaffanet, M.; Jacquemier, J.; Viens, P.; Birnbaum, D.; Bertucci, F. Sixteen-kinase gene expression identifies luminal breast cancers with poor prognosis. Cancer Res. 2008, 68, 767–776. [Google Scholar] [CrossRef]
- Marina, M.; Saavedra, H.I. Nek2 and Plk4: Prognostic markers, drivers of breast tumorigenesis and drug resistance. Front. Biosci. 2014, 19, 352–365. [Google Scholar] [CrossRef]
- Denu, R.A.; Zasadil, L.M.; Kanugh, C.; Laffin, J.; Weaver, B.A.; Burkard, M.E. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 2016, 16, 47. [Google Scholar] [CrossRef]
- Li, Z.; Dai, K.; Wang, C.; Song, Y.; Gu, F.; Liu, F.; Fu, L. Expression of Polo-Like Kinase 4(PLK4) in Breast Cancer and Its Response to Taxane-Based Neoadjuvant Chemotherapy. J. Cancer 2016, 7, 1125–1132. [Google Scholar] [CrossRef]
- Kawakami, M.; Mustachio, L.M.; Zheng, L.; Chen, Y.; Rodriguez-Canales, J.; Mino, B.; Kurie, J.M.; Roszik, J.; Villalobos, P.A.; Thu, K.L.; et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 1913–1918. [Google Scholar] [CrossRef]
- Zhou, Q.; Fan, G.; Dong, Y. Polo-like kinase 4 correlates with greater tumor size, lymph node metastasis and confers poor survival in non-small cell lung cancer. J. Clin. Lab. Anal. 2020, 34, e23152. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, N.; Hohenfellner, M.; Duensing, S. CAND1 promotes PLK4-mediated centriole overduplication and is frequently disrupted in prostate cancer. Neoplasia 2012, 14, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Kurabe, N.; Goto, M.; Yamada, H.; Natsume, H.; Konno, H.; Sugimura, H. PLK4 overexpression and its effect on centrosome regulation and chromosome stability in human gastric cancer. Mol. Biol. Rep. 2014, 41, 6635–6644. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Yi, S.; Yang, X.; Wu, Q. Clinical Significance of Polo-Like Kinase 4 as a Marker for Advanced Tumor Stage and Dismal Prognosis in Patients With Surgical Gastric Cancer. Technol. Cancer Res. Treat. 2020, 19, 1533033820935531. [Google Scholar] [CrossRef]
- Tian, X.; Zhou, D.; Chen, L.; Tian, Y.; Zhong, B.; Cao, Y.; Dong, Q.; Zhou, M.; Yan, J.; Wang, Y.; et al. Polo-like kinase 4 mediates epithelial-mesenchymal transition in neuroblastoma via PI3K/Akt signaling pathway. Cell Death Dis. 2018, 9, 54. [Google Scholar] [CrossRef]
- Wang, J.; Zuo, J.; Wang, M.; Ma, X.; Gao, K.; Bai, X.; Wang, N.; Xie, W.; Liu, H. Polo-like kinase 4 promotes tumorigenesis and induces resistance to radiotherapy in glioblastoma. Oncol. Rep. 2019, 41, 2159–2167. [Google Scholar] [CrossRef]
- Goroshchuk, O.; Kolosenko, I.; Vidarsdottir, L.; Azimi, A.; Palm-Apergi, C. Polo-like kinases and acute leukemia. Oncogene 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Goroshchuk, O.; Vidarsdottir, L.; Björklund, A.-C.; Hamil, A.S.; Kolosenko, I.; Dowdy, S.F.; Palm-Apergi, C. Targeting Plk1 with siRNNs in primary cells from pediatric B-cell acute lymphoblastic leukemia patients. Sci. Rep. 2020, 10, 2688. [Google Scholar] [CrossRef]
- Coelho, P.A.; Bury, L.; Shahbazi, M.N.; Liakath-Ali, K.; Tate, P.H.; Wormald, S.; Hindley, C.J.; Huch, M.; Archer, J.; Skarnes, W.C.; et al. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 2015, 5, 150209. [Google Scholar] [CrossRef]
- Serçin, Ö.; Larsimont, J.-C.; Karambelas, A.E.; Marthiens, V.; Moers, V.; Boeckx, B.; Le Mercier, M.; Lambrechts, D.; Basto, R.; Blanpain, C. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat. Cell Biol. 2016, 18, 100–110. [Google Scholar] [CrossRef]
- Denu, R.A.; Shabbir, M.; Nihal, M.; Singh, C.K.; Longley, B.J.; Burkard, M.E.; Ahmad, N. Centriole Overduplication is the Predominant Mechanism Leading to Centrosome Amplification in Melanoma. Mol. Cancer Res. 2018, 16, 517–527. [Google Scholar] [CrossRef]
- Macmillan, J.C.; Hudson, J.W.; Bull, S.; Dennis, J.W.; Swallow, C.J. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann. Surg. Oncol. 2001, 8, 729–740. [Google Scholar] [CrossRef]
- Bao, J.; Yu, Y.; Chen, J.; He, Y.; Chen, X.; Ren, Z.; Xue, C.; Liu, L.; Hu, Q.; Li, J.; et al. MiR-126 negatively regulates PLK-4 to impact the development of hepatocellular carcinoma via ATR/CHEK1 pathway. Cell Death Dis. 2018, 9, 1045. [Google Scholar] [CrossRef]
- Liao, Z.; Zhang, H.; Fan, P.; Huang, Q.; Dong, K.; Qi, Y.; Song, J.; Chen, L.; Liang, H.; Chen, X.; et al. High PLK4 expression promotes tumor progression and induces epithelial-mesenchymal transition by regulating the Wnt/β-catenin signaling pathway in colorectal cancer. Int. J. Oncol. 2019, 54, 479–490. [Google Scholar] [CrossRef]
- Ko, M.A.; Rosario, C.O.; Hudson, J.W.; Kulkarni, S.; Pollett, A.; Dennis, J.W.; Swallow, C.J. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat. Genet. 2005, 37, 883–888. [Google Scholar] [CrossRef]
- Schukken, K.M.; Foijer, F. CIN and Aneuploidy: Different Concepts, Different Consequences. Bioessays 2018, 40. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Polo-like kinase 4 inhibition: A strategy for cancer therapy? Cancer Cell 2014, 26, 151–153. [Google Scholar] [CrossRef]
- Kops, G.J.P.L.; Weaver, B.A.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef]
- Susini, T.; Olivieri, S.; Molino, C.; Amunni, G.; Rapi, S.; Taddei, G.; Scarselli, G. DNA ploidy is stronger than lymph node metastasis as prognostic factor in cervical carcinoma: 10-year results of a prospective study. Int. J. Gynecol. Cancer 2011, 21, 678–684. [Google Scholar] [CrossRef]
- Braun, M.; Stomper, J.; Kirsten, R.; Shaikhibrahim, Z.; Vogel, W.; Böhm, D.; Wernert, N.; Kristiansen, G.; Perner, S. Landscape of chromosome number changes in prostate cancer progression. World J. Urol. 2013, 31, 1489–1495. [Google Scholar] [CrossRef]
- Xu, J.; Huang, L.; Li, J. DNA aneuploidy and breast cancer: A meta-analysis of 141,163 cases. Oncotarget 2016, 7, 60218–60229. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev. Cell 2017, 40, 313–322.e5. [Google Scholar] [CrossRef] [PubMed]
- Godinho, S.A.; Picone, R.; Burute, M.; Dagher, R.; Su, Y.; Leung, C.T.; Polyak, K.; Brugge, J.S.; Théry, M.; Pellman, D. Oncogene-like induction of cellular invasion from centrosome amplification. Nature 2014, 510, 167–171. [Google Scholar] [CrossRef]
- Rosario, C.O.; Kazazian, K.; Zih, F.S.W.; Brashavitskaya, O.; Haffani, Y.; Xu, R.S.Z.; George, A.; Dennis, J.W.; Swallow, C.J. A novel role for Plk4 in regulating cell spreading and motility. Oncogene 2015, 34, 3441–3451. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, K.; Go, C.; Wu, H.; Brashavitskaya, O.; Xu, R.; Dennis, J.W.; Gingras, A.-C.; Swallow, C.J. Plk4 Promotes Cancer Invasion and Metastasis through Arp2/3 Complex Regulation of the Actin Cytoskeleton. Cancer Res. 2017, 77, 434–447. [Google Scholar] [CrossRef]
- Liu, Y.; Kim, J.; Philip, R.; Sridhar, V.; Chandrashekhar, M.; Moffat, J.; van Breugel, M.; Pelletier, L. Direct interaction between CEP85 and STIL mediates PLK4-driven directed cell migration. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Luo, Y.; Barrios-Rodiles, M.; Gupta, G.D.; Zhang, Y.Y.; Ogunjimi, A.A.; Bashkurov, M.; Tkach, J.M.; Underhill, A.Q.; Zhang, L.; Bourmoum, M.; et al. Atypical function of a centrosomal module in WNT signalling drives contextual cancer cell motility. Nat. Commun. 2019, 10, 2356. [Google Scholar] [CrossRef]
- Li, Z.; Hao, P.; Wu, Q.; Li, F.; Zhao, J.; Wu, K.; Qu, C.; Chen, Y.; Li, M.; Chen, X.; et al. Genetic mutations associated with metastatic clear cell renal cell carcinoma. Oncotarget 2016, 7, 16172–16179. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, X. PLK4: A promising target for cancer therapy. J. Cancer Res. Clin. Oncol. 2019, 145, 2413–2422. [Google Scholar] [CrossRef]
- Spänkuch-Schmitt, B.; Bereiter-Hahn, J.; Kaufmann, M.; Strebhardt, K. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J. Natl. Cancer Inst. 2002, 94, 1863–1877. [Google Scholar] [CrossRef]
- Spänkuch-Schmitt, B.; Wolf, G.; Solbach, C.; Loibl, S.; Knecht, R.; Stegmüller, M.; von Minckwitz, G.; Kaufmann, M.; Strebhardt, K. Downregulation of human polo-like kinase activity by antisense oligonucleotides induces growth inhibition in cancer cells. Oncogene 2002, 21, 3162–3171. [Google Scholar] [CrossRef]
- Spänkuch, B.; Matthess, Y.; Knecht, R.; Zimmer, B.; Kaufmann, M.; Strebhardt, K. Cancer inhibition in nude mice after systemic application of U6 promoter-driven short hairpin RNAs against PLK1. J. Natl. Cancer Inst. 2004, 96, 862–872. [Google Scholar] [CrossRef][Green Version]
- Cholewa, B.D.; Ndiaye, M.A.; Huang, W.; Liu, X.; Ahmad, N. Small molecule inhibition of polo-like kinase 1 by volasertib (BI 6727) causes significant melanoma growth delay and regression in vivo. Cancer Lett. 2017, 385, 179–187. [Google Scholar] [CrossRef]
- Murugan, R.N.; Ahn, M.; Lee, W.C.; Kim, H.-Y.; Song, J.H.; Cheong, C.; Hwang, E.; Seo, J.-H.; Shin, S.Y.; Choi, S.H.; et al. Exploring the binding nature of pyrrolidine pocket-dependent interactions in the polo-box domain of polo-like kinase 1. PLoS ONE 2013, 8, e80043. [Google Scholar] [CrossRef]
- Schmit, T.L.; Ledesma, M.C.; Ahmad, N. Modulating polo-like kinase 1 as a means for cancer chemoprevention. Pharm. Res. 2010, 27, 989–998. [Google Scholar] [CrossRef]
- Li, J.; Karki, A.; Hodges, K.B.; Ahmad, N.; Zoubeidi, A.; Strebhardt, K.; Ratliff, T.L.; Konieczny, S.F.; Liu, X. Cotargeting Polo-Like Kinase 1 and the Wnt/β-Catenin Signaling Pathway in Castration-Resistant Prostate Cancer. Mol. Cell. Biol. 2015, 35, 4185–4198. [Google Scholar] [CrossRef]
- Liu, X. Targeting Polo-Like Kinases: A Promising Therapeutic Approach for Cancer Treatment. Transl. Oncol. 2015, 8, 185–195. [Google Scholar] [CrossRef]
- Palmisiano, N.D.; Kasner, M.T. Polo-like kinase and its inhibitors: Ready for the match to start? Am. J. Hematol. 2015, 90, 1071–1076. [Google Scholar] [CrossRef]
- Kumar, S.; Kim, J. PLK-1 Targeted Inhibitors and Their Potential against Tumorigenesis. BioMed Res. Int. 2015, 2015, 705745. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Burke, T.R.; Park, J.-E.; Bang, J.K.; Lee, E. Recent Advances and New Strategies in Targeting Plk1 for Anticancer Therapy. Trends Pharmacol. Sci. 2015, 36, 858–877. [Google Scholar] [CrossRef]
- Hu, C.-K.; Ozlü, N.; Coughlin, M.; Steen, J.J.; Mitchison, T.J. Plk1 negatively regulates PRC1 to prevent premature midzone formation before cytokinesis. Mol. Biol. Cell 2012, 23, 2702–2711. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Krämer, A.; Hehlgans, S.; Sanhaji, M.; Kurunci-Csacsko, E.; Dötsch, C.; Bug, G.; Ottmann, O.; Becker, S.; Pachl, F.; et al. Mitotic arrest and slippage induced by pharmacological inhibition of Polo-like kinase 1. Mol. Oncol. 2015, 9, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Kreis, N.-N.; Sommer, K.; Sanhaji, M.; Krämer, A.; Matthess, Y.; Kaufmann, M.; Strebhardt, K.; Yuan, J. Long-term downregulation of Polo-like kinase 1 increases the cyclin-dependent kinase inhibitor p21(WAF1/CIP1). Cell Cycle 2009, 8, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Lénárt, P.; Petronczki, M.; Steegmaier, M.; Di Fiore, B.; Lipp, J.J.; Hoffmann, M.; Rettig, W.J.; Kraut, N.; Peters, J.-M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 2007, 17, 304–315. [Google Scholar] [CrossRef]
- Reindl, W.; Yuan, J.; Krämer, A.; Strebhardt, K.; Berg, T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem. Biol. 2008, 15, 459–466. [Google Scholar] [CrossRef]
- Reindl, W.; Strebhardt, K.; Berg, T. A high-throughput assay based on fluorescence polarization for inhibitors of the polo-box domain of polo-like kinase 1. Anal. Biochem. 2008, 383, 205–209. [Google Scholar] [CrossRef]
- Scharow, A.; Raab, M.; Saxena, K.; Sreeramulu, S.; Kudlinzki, D.; Gande, S.; Dötsch, C.; Kurunci-Csacsko, E.; Klaeger, S.; Kuster, B.; et al. Optimized Plk1 PBD Inhibitors Based on Poloxin Induce Mitotic Arrest and Apoptosis in Tumor Cells. ACS Chem. Biol. 2015, 10, 2570–2579. [Google Scholar] [CrossRef]
- Yuan, J.; Sanhaji, M.; Krämer, A.; Reindl, W.; Hofmann, M.; Kreis, N.-N.; Zimmer, B.; Berg, T.; Strebhardt, K. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am. J. Pathol. 2011, 179, 2091–2099. [Google Scholar] [CrossRef]
- Baxter, M.; Chapagai, D.; Craig, S.; Hurtado, C.; Varghese, J.; Nurmemmedov, E.; Wyatt, M.D.; McInnes, C. Peptidomimetic Polo-Box-Targeted Inhibitors that Engage PLK1 in Tumor Cells and Are Selective against the PLK3 Tumor Suppressor. ChemMedChem 2020, 15, 1058–1066. [Google Scholar] [CrossRef]
- Matthess, Y.; Kappel, S.; Spänkuch, B.; Zimmer, B.; Kaufmann, M.; Strebhardt, K. Conditional inhibition of cancer cell proliferation by tetracycline-responsive, H1 promoter-driven silencing of PLK1. Oncogene 2005, 24, 2973–2980. [Google Scholar] [CrossRef][Green Version]
- Kolosenko, I.; Edsbäcker, E.; Björklund, A.-C.; Hamil, A.S.; Goroshchuk, O.; Grandér, D.; Dowdy, S.F.; Palm-Apergi, C. RNAi prodrugs targeting Plk1 induce specific gene silencing in primary cells from pediatric T-acute lymphoblastic leukemia patients. J. Control. Release 2017, 261, 199–206. [Google Scholar] [CrossRef]
- Mason, J.M.; Wei, X.; Fletcher, G.C.; Kiarash, R.; Brokx, R.; Hodgson, R.; Beletskaya, I.; Bray, M.R.; Mak, T.W. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3127–3132. [Google Scholar] [CrossRef]
- Lohse, I.; Mason, J.; Cao, P.M.; Pintilie, M.; Bray, M.; Hedley, D.W. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget 2017, 8, 3064–3071. [Google Scholar] [CrossRef]
- Veitch, Z.W.; Cescon, D.W.; Denny, T.; Yonemoto, L.-M.; Fletcher, G.; Brokx, R.; Sampson, P.; Li, S.-W.; Pugh, T.J.; Bruce, J.; et al. Safety and tolerability of CFI-400945, a first-in-class, selective PLK4 inhibitor in advanced solid tumours: A phase 1 dose-escalation trial. Br. J. Cancer 2019, 121, 318–324. [Google Scholar] [CrossRef]
- Lei, Q.; Xiong, L.; Xia, Y.; Feng, Z.; Gao, T.; Wei, W.; Song, X.; Ye, T.; Wang, N.; Peng, C.; et al. YLT-11, a novel PLK4 inhibitor, inhibits human breast cancer growth via inducing maladjusted centriole duplication and mitotic defect. Cell Death Dis. 2018, 9, 1066. [Google Scholar] [CrossRef]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef]
- Suri, A.; Bailey, A.W.; Tavares, M.T.; Gunosewoyo, H.; Dyer, C.P.; Grupenmacher, A.T.; Piper, D.R.; Horton, R.A.; Tomita, T.; Kozikowski, A.P.; et al. Evaluation of Protein Kinase Inhibitors with PLK4 Cross-Over Potential in a Pre-Clinical Model of Cancer. Int. J. Mol. Sci. 2019, 20, 2112. [Google Scholar] [CrossRef]
- Li, F.; Jo, M.; Curry, T.E.; Liu, J. Hormonal induction of polo-like kinases (Plks) and impact of Plk2 on cell cycle progression in the rat ovary. PLoS ONE 2012, 7, e41844. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Kaku, T.; Ogawa, S.; Kawano, Y.; Ohishi, Y.; Kobayashi, H.; Hirakawa, T.; Nakano, H. Histological classification of ovarian cancer. Med. Electron Microsc. 2003, 36, 9–17. [Google Scholar] [CrossRef]
- Prat, J. Ovarian carcinomas: Five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012, 460, 237–249. [Google Scholar] [CrossRef]
- Kuhn, E.; Tisato, V.; Rimondi, E.; Secchiero, P. Current Preclinical Models of Ovarian Cancer. J. Carcinog. Mutagen. 2015, 06, 1–9. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef]
- Milea, A.; George, S.H.L.; Matevski, D.; Jiang, H.; Madunic, M.; Berman, H.K.; Gauthier, M.L.; Gallie, B.; Shaw, P.A. Retinoblastoma pathway deregulatory mechanisms determine clinical outcome in high-grade serous ovarian carcinoma. Mod. Pathol. 2014, 27, 991–1001. [Google Scholar] [CrossRef]
- Cheasley, D.; Wakefield, M.J.; Ryland, G.L.; Allan, P.E.; Alsop, K.; Amarasinghe, K.C.; Ananda, S.; Anglesio, M.S.; Au-Yeung, G.; Böhm, M.; et al. The molecular origin and taxonomy of mucinous ovarian carcinoma. Nat. Commun. 2019, 10, 3935. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Tamayo, P.; Yang, J.-Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, K.; Xu, Z.; Chen, Z.; Wang, Q.; Wang, C.; Cui, J. MicroRNA-545 suppresses progression of ovarian cancer through mediating PLK1 expression by a direct binding and an indirect regulation involving KDM4B-mediated demethylation. BMC Cancer 2021, 21, 163. [Google Scholar] [CrossRef]
- Zhang, S.; Jing, Y.; Zhang, M.; Zhang, Z.; Ma, P.; Peng, H.; Shi, K.; Gao, W.-Q.; Zhuang, G. Stroma-associated master regulators of molecular subtypes predict patient prognosis in ovarian cancer. Sci. Rep. 2015, 5, 16066. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Rong, X.; Gao, F.; Yang, Y.; Wei, L. TPX2 promotes cell proliferation and migration via PLK1 in OC. Cancer Biomark. 2018, 22, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Sanhaji, M.; Zhou, S.; Rödel, F.; El-Balat, A.; Becker, S.; Strebhardt, K. Blocking Mitotic Exit of Ovarian Cancer Cells by Pharmaceutical Inhibition of the Anaphase-Promoting Complex Reduces Chromosomal Instability. Neoplasia 2019, 21, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Noack, S.; Raab, M.; Matthess, Y.; Sanhaji, M.; Krämer, A.; Győrffy, B.; Kaderali, L.; El-Balat, A.; Becker, S.; Strebhardt, K. Synthetic lethality in CCNE1-amplified high grade serous ovarian cancer through combined inhibition of Polo-like kinase 1 and microtubule dynamics. Oncotarget 2018, 9, 25842–25859. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Selle, F.; Weber, B.; Ray-Coquard, I.-L.; Vergote, I.; Sufliarsky, J.; Del Campo, J.M.; Lortholary, A.; Lesoin, A.; Follana, P.; et al. Volasertib Versus Chemotherapy in Platinum-Resistant or -Refractory Ovarian Cancer: A Randomized Phase II Groupe des Investigateurs Nationaux pour l’Etude des Cancers de l’Ovaire Study. J. Clin. Oncol. 2016, 34, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Valsasina, B.; Beria, I.; Alli, C.; Alzani, R.; Avanzi, N.; Ballinari, D.; Cappella, P.; Caruso, M.; Casolaro, A.; Ciavolella, A.; et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol. Cancer Ther. 2012, 11, 1006–1016. [Google Scholar] [CrossRef]
- Affatato, R.; Carrassa, L.; Chilà, R.; Lupi, M.; Restelli, V.; Damia, G. Identification of PLK1 as a New Therapeutic Target in Mucinous Ovarian Carcinoma. Cancers 2020, 12, 672. [Google Scholar] [CrossRef]
- Fei, H.; Chen, S.; Xu, C. Bioinformatics analysis of gene expression profile of serous ovarian carcinomas to screen key genes and pathways. J. Ovarian Res. 2020, 13, 82. [Google Scholar] [CrossRef]
- Parrilla, A.; Barber, M.; Majem, B.; Castellví, J.; Morote, J.; Sánchez, J.L.; Pérez-Benavente, A.; Segura, M.F.; Gil-Moreno, A.; Santamaria, A. Aurora Borealis (Bora), Which Promotes Plk1 Activation by Aurora A, Has an Oncogenic Role in Ovarian Cancer. Cancers 2020, 12, 886. [Google Scholar] [CrossRef]
- Deb, B.; Uddin, A.; Chakraborty, S. miRNAs and ovarian cancer: An overview. J. Cell. Physiol. 2018, 233, 3846–3854. [Google Scholar] [CrossRef]
- Chen, S.-N.; Chang, R.; Lin, L.-T.; Chern, C.-U.; Tsai, H.-W.; Wen, Z.-H.; Li, Y.-H.; Li, C.-J.; Tsui, K.-H. MicroRNA in Ovarian Cancer: Biology, Pathogenesis, and Therapeutic Opportunities. Int. J. Environ. Res. Public Health 2019, 16, 1510. [Google Scholar] [CrossRef]
- Jia, X.; Liu, X.; Li, M.; Zeng, Y.; Feng, Z.; Su, X.; Huang, Y.; Chen, M.; Yang, X. Potential tumor suppressing role of microRNA-545 in epithelial ovarian cancer. Oncol. Lett. 2018, 15, 6386–6392. [Google Scholar] [CrossRef]
- Syed, N.; Coley, H.M.; Sehouli, J.; Koensgen, D.; Mustea, A.; Szlosarek, P.; McNeish, I.; Blagden, S.P.; Schmid, P.; Lovell, D.P.; et al. Polo-like kinase Plk2 is an epigenetic determinant of chemosensitivity and clinical outcomes in ovarian cancer. Cancer Res. 2011, 71, 3317–3327. [Google Scholar] [CrossRef]
- Coley, H.M.; Hatzimichael, E.; Blagden, S.; McNeish, I.; Thompson, A.; Crook, T.; Syed, N. Polo Like Kinase 2 Tumour Suppressor and cancer biomarker: New perspectives on drug sensitivity/resistance in ovarian cancer. Oncotarget 2012, 3, 78–83. [Google Scholar] [CrossRef]
- Ju, W.; Yoo, B.C.; Kim, I.-J.; Kim, J.W.; Kim, S.C.; Lee, H.P. Identification of genes with differential expression in chemoresistant epithelial ovarian cancer using high-density oligonucleotide microarrays. Oncol. Res. 2009, 18, 47–56. [Google Scholar] [CrossRef]
- Szenajch, J.; Szabelska-Beręsewicz, A.; Świercz, A.; Zyprych-Walczak, J.; Siatkowski, I.; Góralski, M.; Synowiec, A.; Handschuh, L. Transcriptome Remodeling in Gradual Development of Inverse Resistance between Paclitaxel and Cisplatin in Ovarian Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9218. [Google Scholar] [CrossRef]
- He, Y.; Wang, H.; Yan, M.; Yang, X.; Shen, R.; Ni, X.; Chen, X.; Yang, P.; Chen, M.; Lu, X.; et al. High LIN28A and PLK4 co-expression is associated with poor prognosis in epithelial ovarian cancer. Mol. Med. Rep. 2018, 18, 5327–5336. [Google Scholar] [CrossRef]
- Wang, Z.J.; Churchman, M.; Campbell, I.G.; Xu, W.H.; Yan, Z.Y.; McCluggage, W.G.; Foulkes, W.D.; Tomlinson, I.P. Allele loss and mutation screen at the Peutz-Jeghers (LKB1) locus (19p13.3) in sporadic ovarian tumours. Br. J. Cancer 1999, 80, 70–72. [Google Scholar] [CrossRef][Green Version]
- Macintyre, G.; Goranova, T.E.; de Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.-A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2012, 31, 14. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar] [CrossRef]
- Barton, C.A.; Hacker, N.F.; Clark, S.J.; O’Brien, P.M. DNA methylation changes in ovarian cancer: Implications for early diagnosis, prognosis and treatment. Gynecol. Oncol. 2008, 109, 129–139. [Google Scholar] [CrossRef]
- Reibenwein, J.; Krainer, M. Targeting signaling pathways in ovarian cancer. Expert Opin. Ther. Targets 2008, 12, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Smolle, E.; Taucher, V.; Pichler, M.; Petru, E.; Lax, S.; Haybaeck, J. Targeting signaling pathways in epithelial ovarian cancer. Int. J. Mol. Sci. 2013, 14, 9536–9555. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Li, N.; Zhan, X. Signaling pathway network alterations in human ovarian cancers identified with quantitative mitochondrial proteomics. EPMA J. 2019, 10, 153–172. [Google Scholar] [CrossRef]
- Cooke, S.L.; Ng, C.K.Y.; Melnyk, N.; Garcia, M.J.; Hardcastle, T.; Temple, J.; Langdon, S.; Huntsman, D.; Brenton, J.D. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene 2010, 29, 4905–4913. [Google Scholar] [CrossRef]
- Yamada, H.Y.; Gorbsky, G.J. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol. Cancer Ther. 2006, 5, 2963–2969. [Google Scholar] [CrossRef]
- Galimberti, F.; Thompson, S.L.; Ravi, S.; Compton, D.A.; Dmitrovsky, E. Anaphase catastrophe is a target for cancer therapy. Clin. Cancer Res. 2011, 17, 1218–1222. [Google Scholar] [CrossRef]
- Rieder, C.L.; Maiato, H. Stuck in division or passing through: What happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef]
- Zhang, K.; Kong, X.; Feng, G.; Xiang, W.; Chen, L.; Yang, F.; Cao, C.; Ding, Y.; Chen, H.; Chu, M.; et al. Investigation of hypoxia networks in ovarian cancer via bioinformatics analysis. J. Ovarian Res. 2018, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Rentschler, J.; Kaiser, R.; Rouyrre, N.; Trommeshauser, D.; Hoesl, C.E.; Munzert, G. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2008, 26, 5511–5517. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Bisanz, K.M.; Peralta, L.A.; Basu, G.D.; Choudhary, A.; Tibes, R.; Azorsa, D.O. RNAi screening of the kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol. Oncol. 2010, 118, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Z.; Qu, Y.; Zeng, J.; Yang, M.; Li, X.; Wang, Z.; Su, J.; Wang, X.; Yu, L.; et al. YLZ-F5, a novel polo-like kinase 4 inhibitor, inhibits human ovarian cancer cell growth by inducing apoptosis and mitotic defects. Cancer Chemother. Pharmacol. 2020, 86, 33–43. [Google Scholar] [CrossRef]
- Karakashev, S.; Zhang, R.-G. Mouse models of epithelial ovarian cancer for preclinical studies. Zool. Res. 2021, 1–8. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Magnotti, E.; Marasco, W.A. The latest animal models of ovarian cancer for novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 249–257. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Schwede, M.; Waldron, L.; Mok, S.C.; Wei, W.; Basunia, A.; Merritt, M.A.; Mitsiades, C.S.; Parmigiani, G.; Harrington, D.P.; Quackenbush, J.; et al. The Impact of Stroma Admixture on Molecular Subtypes and Prognostic Gene Signatures in Serous Ovarian Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 509–519. [Google Scholar] [CrossRef]
- Shaw, T.J.; Senterman, M.K.; Dawson, K.; Crane, C.A.; Vanderhyden, B.C. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 2004, 10, 1032–1042. [Google Scholar] [CrossRef]
- Fu, X.; Hoffman, R.M. Human ovarian carcinoma metastatic models constructed in nude mice by orthotopic transplantation of histologically-intact patient specimens. Anticancer Res. 1993, 13, 283–286. [Google Scholar]
- Yi, X.-F.; Yuan, S.-T.; Lu, L.-J.; Ding, J.; Feng, Y.-J. A clinically relevant orthotopic implantation nude mouse model of human epithelial ovarian cancer–based on consecutive observation. Int. J. Gynecol. Cancer 2005, 15, 850–855. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, L.; Zheng, X.; Yu, T. An Advanced Orthotopic Ovarian Cancer Model in Mice for Therapeutic Trials. BioMed Res. Int. 2016, 2016, 2585787. [Google Scholar] [CrossRef]
- Cordero, A.B.; Kwon, Y.; Hua, X.; Godwin, A.K. In vivo imaging and therapeutic treatments in an orthotopic mouse model of ovarian cancer. J. Vis. Exp. 2010. [Google Scholar] [CrossRef]
- Yi, C.; Zhang, L.; Zhang, F.; Li, L.; Ling, S.; Wang, X.; Liu, X.; Liang, W. Methodologies for the establishment of an orthotopic transplantation model of ovarian cancer in mice. Front. Med. 2014, 8, 101–105. [Google Scholar] [CrossRef]
- Guo, J.; Cai, J.; Zhang, Y.; Zhu, Y.; Yang, P.; Wang, Z. Establishment of two ovarian cancer orthotopic xenograft mouse models for in vivo imaging: A comparative study. Int. J. Oncol. 2017, 51, 1199–1208. [Google Scholar] [CrossRef]
- Garcia Ribeiro, R.S.; Belderbos, S.; Danhier, P.; Gallo, J.; Manshian, B.B.; Gallez, B.; Bañobre, M.; de Cuyper, M.; Soenen, S.J.; Gsell, W.; et al. Targeting tumor cells and neovascularization using RGD-functionalized magnetoliposomes. Int. J. Nanomed. 2019, 14, 5911–5924. [Google Scholar] [CrossRef]
- Bochner, F.; Fellus-Alyagor, L.; Ketter, D.; Golani, O.; Biton, I.; Neeman, M. Bimodal magnetic resonance and optical imaging of extracellular matrix remodelling by orthotopic ovarian tumours. Br. J. Cancer 2020, 123, 216–225. [Google Scholar] [CrossRef]
- Maniati, E.; Berlato, C.; Gopinathan, G.; Heath, O.; Kotantaki, P.; Lakhani, A.; McDermott, J.; Pegrum, C.; Delaine-Smith, R.M.; Pearce, O.M.T.; et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep. 2020, 30, 525–540.e7. [Google Scholar] [CrossRef]
- Stuckelberger, S.; Drapkin, R. Precious GEMMs: Emergence of faithful models for ovarian cancer research. J. Pathol. 2018, 245, 129–131. [Google Scholar] [CrossRef]
- Zakarya, R.; Howell, V.M.; Colvin, E.K. Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches. Int. J. Mol. Sci. 2020, 21, 4806. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, P.H.; van der Zee, M.; Heijmans-Antonissen, C.; Jia, Y.; DeMayo, F.J.; Lydon, J.P.; van Deurzen, C.H.M.; Ewing, P.C.; Burger, C.W.; Blok, L.J. A mouse model for endometrioid ovarian cancer arising from the distal oviduct. Int. J. Cancer 2014, 135, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Kuhn, E.; Valle, B.L.; Shih, I.-M.; Kurman, R.J.; Wang, T.-L.; Amano, T.; Ko, M.S.H.; Miyoshi, I.; Araki, Y.; et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J. Pathol. 2014, 233, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.; Schulman, S.; Green, M.; Fearon, E.; Cho, K. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J. Pathol. 2017, 243, 267. [Google Scholar] [CrossRef]
- Kim, O.; Park, E.Y.; Klinkebiel, D.L.; Pack, S.D.; Shin, Y.-H.; Abdullaev, Z.; Emerson, R.E.; Coffey, D.M.; Kwon, S.Y.; Creighton, C.J.; et al. In vivo modeling of metastatic human high-grade serous ovarian cancer in mice. PLoS Genet. 2020, 16, e1008808. [Google Scholar] [CrossRef]
- Perets, R.; Drapkin, R. It’s Totally Tubular … Riding The New Wave of Ovarian Cancer Research. Cancer Res. 2016, 76, 10–17. [Google Scholar] [CrossRef]
- Le Bras, A. A new mouse model of ovarian cancer metastasis. Lab Anim. 2020, 49, 200. [Google Scholar] [CrossRef]
- Roby, K.F.; Taylor, C.C.; Sweetwood, J.P.; Cheng, Y.; Pace, J.L.; Tawfik, O.; Persons, D.L.; Smith, P.G.; Terranova, P.F. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000, 21, 585–591. [Google Scholar] [CrossRef]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. CRISPR/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef]
- Iyer, S.; Zhang, S.; Yucel, S.; Horn, H.; Smith, S.G.; Reinhardt, F.; Hoefsmit, E.; Assatova, B.; Casado, J.; Meinsohn, M.-C.; et al. Genetically Defined Syngeneic Mouse Models of Ovarian Cancer as Tools for the Discovery of Combination Immunotherapy. Cancer Discov. 2021, 11, 384–407. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kressin, M.; Fietz, D.; Becker, S.; Strebhardt, K. Modelling the Functions of Polo-Like Kinases in Mice and Their Applications as Cancer Targets with a Special Focus on Ovarian Cancer. Cells 2021, 10, 1176. https://doi.org/10.3390/cells10051176
Kressin M, Fietz D, Becker S, Strebhardt K. Modelling the Functions of Polo-Like Kinases in Mice and Their Applications as Cancer Targets with a Special Focus on Ovarian Cancer. Cells. 2021; 10(5):1176. https://doi.org/10.3390/cells10051176
Chicago/Turabian StyleKressin, Monika, Daniela Fietz, Sven Becker, and Klaus Strebhardt. 2021. "Modelling the Functions of Polo-Like Kinases in Mice and Their Applications as Cancer Targets with a Special Focus on Ovarian Cancer" Cells 10, no. 5: 1176. https://doi.org/10.3390/cells10051176
APA StyleKressin, M., Fietz, D., Becker, S., & Strebhardt, K. (2021). Modelling the Functions of Polo-Like Kinases in Mice and Their Applications as Cancer Targets with a Special Focus on Ovarian Cancer. Cells, 10(5), 1176. https://doi.org/10.3390/cells10051176