
The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy

 and
and 
Abstract
1. Introduction
2. Classification
3. Thyroid Cancer Pathogenesis
4. Genetics of Thyroid Cancers
5. Tumour Initiation
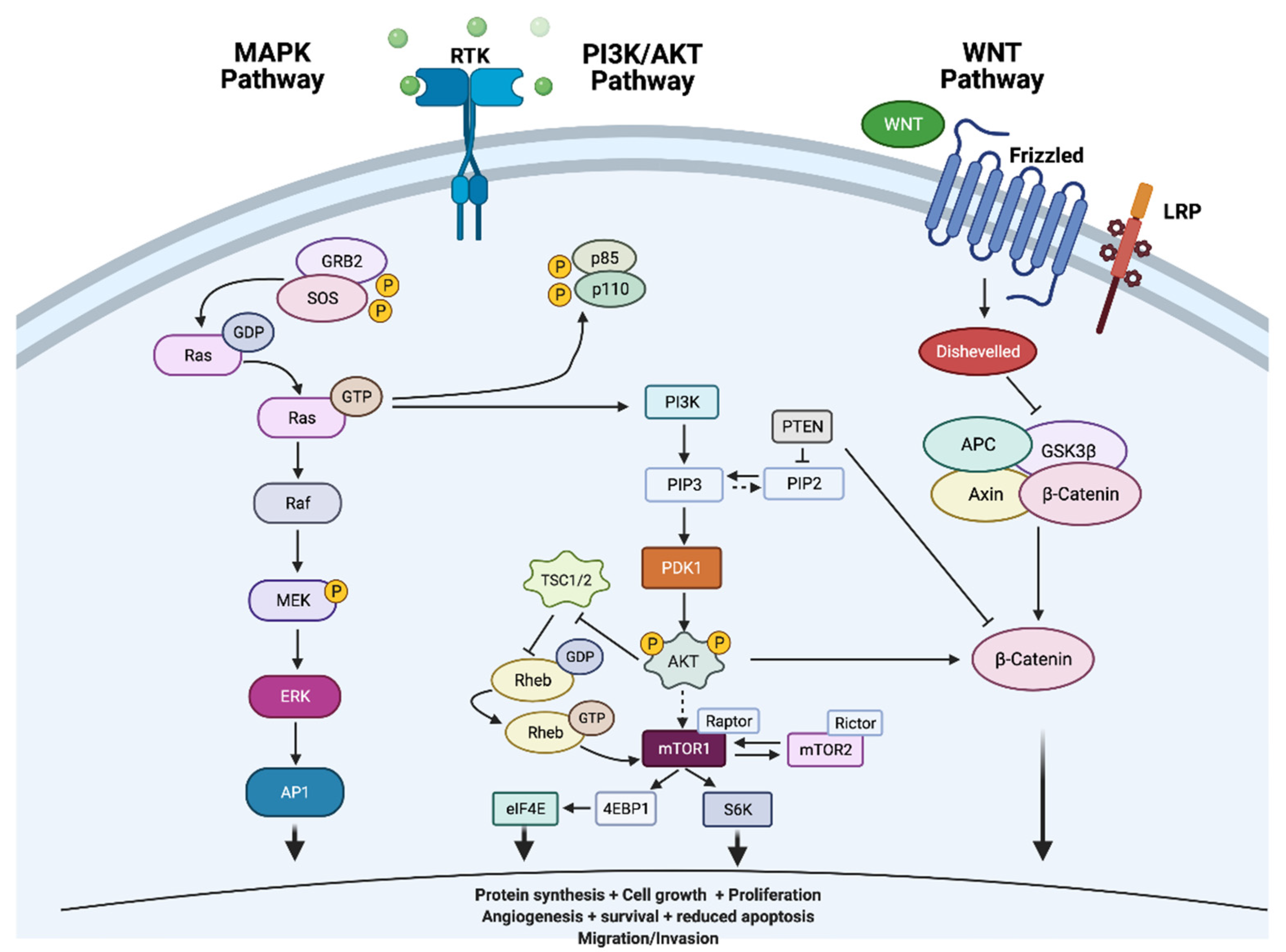
5.1. The Mitogen-Activated Protein Kinase (MAPK) Pathway
5.2. RAS Mutations
5.3. BRAF Mutations
5.4. RET/PTC Rearrangements
5.5. EIF1AX Mutations
6. Tumour Progression
6.1. The Phosphatidylinositol 3-Kinase (PI3K)/Ak Strain Transforming (AKT)/Mammalian Target of Rapamycin (mTOR) Pathway
6.2. PAX8/PPARγ, ALK and NTRK Rearrangements
6.3. Telomerase Reverse Transcriptase (TERT) Promoter Mutations
6.4. TP53 Mutations
6.5. CDKN2A
6.6. Mismatch Repair Gene Deficiency
6.7. SWI/SNF Chromatin Remodelling Complex
6.8. Wnt Signalling Pathway
6.9. Epigenetic Modifications in Thyroid Neoplasms
6.10. Copy Number Variations
6.11. Other Genetic Aberrations–Mitochondrial DNA and Genomic Haploidisation

{kind=link}
{kind=link}
| Genetic Aberration | Benign/Borderline | Follicular Thyroid Carcinoma (FTC)/Hürthle Cell Carcinoma (HCC) | Papillary Thyroid Carcinoma (PTC) | Poorly Differentiated Thyroid Carcinoma (PDTC) | Anaplastic Thyroid Carcinoma (ATC) | Clinical Implication |
|---|---|---|---|---|---|---|
| RAS Point Mutations | 28.1–30% (Follicular adenoma) [33,35] 5.6% (Hyperplastic nodule [HN]) [33,35] 7–25% (Goitres [G]) [33,35] 0–4% (Hürthle cell adenoma [HCA])) [32] 29.6–55.6% (Non-invasive follicular thyroid neoplasm with papillary like nuclear features [NIFTP]) [10,13,22] | 20–57% (Follicular thyroid carcinoma [FTC]) [33] 15–25% (Hürthle cell carcinoma [HCC]) [32,38] | 1.7–52% (follicular variant of papillary thyroid carcinoma [FVPTC] [19,32,36] 13% (classic variant of papillary thyroid carcinoma [CVPTC]) [14] | 28–55% [22,40] | 23–52% [17,19,40] | Downstream Mitogen-activated protein kinase (MEK)1/2 inhibitor (selumetinib) [42] Higher metastasis risk with N-Rat sarcoma (RAS) codon 61 mutation147 Follicular morphology |
| V-raf murine sarcoma viral oncogene homolog B1(BRAF) activating mutations (most common is p.V600E; others are p.K601E and small deletions) and fusion (AKAP9-BRAF) | 3.7% NIFTP [10] | Up to 62% (mostly CVPTC) [20,45,46,47,48,49,50,51] | 12–33% [20,45,46,47,48,49,50,51] | 25–29% [20,51] | Selective MEK inhibitors (dabrafenib and trametinib) and BRAF inhibitors (vemurafenib and dabrafenib) Classic and tall cell morphology [20,53,54,55] Refractiveness to radioactive iodine (RAI) [52,53,55] Increased unfavourable prognostic Factors [47,52,53,55] | |
| Rearranged during transfection (RET)-PTC rearrangements | 17–63.2% (HT) | 6.8–32.9% [69,70,71,72,73] | 12.9% [69,70,71,72,73] | Selective RET kinase inhibitors (e.g., selpercatinib) [79] | ||
| Eukaryotic translation initiation factor 1A X-(E1F1AX) activating mutations | 5–10% (FA) [81,82] 0–5% (HN) [81,82] | 17% (FTC) [81,82] 11% (HCC) [38] | 1–2% (mostly FVPTC) [18,80,82] | 5–15% [17,80,81,82] | 9–30% [17,80,81,82]2 | Co-expression with RAS mutations to drive tumourigenesis [82] Co-expression with tumour portein (TP) 53/Telomerase reverse transcriptase (TERT) mutations in biologically aggressive tumours [82] |
| Paired box gene 8-peroxisome proliferator-activated receptor (PAX8-PPARγ) rearrangement | 4–33% (FA) [19,32,91,92,93,94] 22% NIFTP [10] | 30–58% (FTC) [19,32,91,92,93,94] 0–3% (HCC) [38] | 37.5% (FVPTC), <1% (CVPTC) [19,32,92,93,94] | Follicular phenotype | ||
| TERT promoter | 1–35% [19] | 9–15% [19] | 40% [17] | 73% [17] | Usually aggressive biology | |
| TP53 | 8% [109] | 13% [109] | 8–35% [17,19,20] | Up to 73% [17,19,20] | Usually aggressive biology | |
| Cyclin-dependent kinase inhibitor 2A/2B (CDKN2A/2B) | 15–23% [89] | Aggressive biology. Possible utilisation of Cyclin dependent kinases (CDK) 4/6 inhibitor (palbociclib) [61] | ||||
| Catenin beta 1 (CTNNB1) activating mutations | Up to 25% [127,128,129] | Up to 65% [127,128,129] | Usually aggressive biology | |||
| Anaplastic lymphoma kinase (ALK) fusions (STRN or EML4) or activating mutations | 0.8% [20] | Up to 16% [95,96] | 0–10% [95,96] | ALK inhibitors | ||
| Tyrosine kinase (NTRK)1/3 fusions | 0–5% [19] | 1.3–26% [19,90] | Targeted therapies (entrectinib or larotrectinib) [61] | |||
| Others | Phosphatase and tensin homolog (PTEN) loss of heterozygosity (7% FAs) [85,86] THADA (22% NIFTP) [11] | Phosphatidylinositol-4,5-bisphophate 3-kinase catalytic subunit alpha (PIK3C) (0–11% FTC) [19] PTEN (0–27%) [85,86] Thyroid stimulating hormone receptor (TSHR) BRAFK601E Copy number variations (CNVs) Mismatch repair (MMR) genes mtDNA and diploidies (HCC) | PIK3CA (3%) [19,90] PTEN (2%) [90] BRAFK601E Thyroid adenoma-associated protein (THADA) (5%) [19] TSHR MMR genes [115,116] Copy Number variations (CNVs) | PIK3CA (0–11%) [17,90] PTEN (5–20%) [17,89] Ak strain transforming (AKT)1 (0–13%) [17,89] Switch/sucrose non-fermentable (SWI/SNF) complex subunit mutations CNVs | PIK3CA (5–25%) [17,89] PTEN (10–15%) [17,89] AKT1 (0–8%) [17,89] Ataxia telangiectasia mutated (ATM), retinoblastoma 1 (RB1), Multiple endocrine neoplasia (MEN1) Neurofibromatosis (NF1), NF2, AT-rich interacting domain containing protein 2 (ARID2), MMR genes, V-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT), SWI-SNF complex subunit mutations CNVs | AKT1 mutation is present in metastatic or recurrent RAI-refractory tumours [89] |
7. Tumour Microenvironment and Programmed Cell Death 1 Ligand 1 (PD-L1) Expression
8. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kitahara, C.M.; Sosa, J.A. The changing incidence of thyroid cancer. Nat. Rev. Endocrinol. 2016, 12, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Seib, C.D.; Sosa, J.A. Evolving understanding of the epidemiology of thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Australian Institute of Health and Welfare 2019 Australian Government. Canberra, AIHW. Available online: http://www.aihw.gov.au/reports/cancer/cancer-in-australia-2019/contents/summary (accessed on 28 August 2020).
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in thyroid cancer incidence and mortality in the United States, 1974–2013. JAMA 2017, 317, 1338. [Google Scholar] [CrossRef]
- La Vecchia, C.; Malvezzi, M.; Bosetti, C.; Garavello, W.; Bertuccio, P.; Levi, F.; Negri, E. Thyroid cancer mortality and incidence: A global overview. Int. J. Cancer 2015, 136, 2187–2195. [Google Scholar] [CrossRef]
- Vaccarella, S.; Franceschi, S.; Bray, F.; Wild, C.; Plummer, M.; Dal Maso, L. The increase in thyroid cancer may be due to an increase in medical surveillance and the introduction of new diagnostic techniques, such as neck ultrasonography. N. Engl. J. Med. 2016, 375, 614–617. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M. American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, R.Y.; Klöppel, G.; Rosai, J. World Health Organization Classification of Tumours of Endocrine Organs; IARC Press: Lyon, France, 2017. [Google Scholar]
- Nikiforov, Y.E.; Seethala, R.R.; Tallini, G.; Baloch, Z.W.; Basolo, F.; Thompson, L.D.; Barletta, J.A.; Wenig, B.M.; Al Ghuzlan, A.; Kakudo, K.; et al. Nomenclature revision for encapsulated follicular variant of papillary thyroid carcinoma: A paradigm shift to reduce overtreatment of indolent tumors. JAMA Oncol. 2016, 2, 1023–1029. [Google Scholar] [CrossRef]
- Bychkov, A.; Jung, C.K.; Liu, Z.; Kakudo, K. Noninvasive follicular thyroid neoplasm with papillary-like nuclear features in Asian practice: Perspectives for surgical pathology and cytopathology. Endocr. Pathol. 2018, 29, 276–288. [Google Scholar] [CrossRef]
- Golding, A.; Shively, D.; Bimston, D.N.; Harrell, R.M. Noninvasive encapsulated follicular variant of papillary thyroid cancer: Clinical lessons from a community-based endocrine surgical practice. Int. J. Surg. Pract. 2017, 2017, 4689465. [Google Scholar] [CrossRef]
- Seo, J.Y.; Park, J.H.; Pyo, J.Y.; Cha, Y.J.; Jung, C.K.; Song, D.E.; Kwak, J.J.; Park, S.Y.; Na, Y.N.; Kim, J.-H.; et al. A multi-institutional study of prevalence and clinicopathologic features of non-invasive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP) in Korea. J. Pathol. Transl. Med. 2019, 53, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.J. Papillary and follicular thyroid carcinoma. N. Engl. J. Med. 1998, 338, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Haymart, M.R.; Esfandiari, N.H.; Stang, M.T.; Sosa, J.A. Controversies in the management of low-risk differentiated thyroid cancer. Endocr. Rev. 2017, 38, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Lamartina, L.; Grani, G.; Durante, C.; Filetti, S. Recent advances in managing differentiated thyroid cancer. F1000Research 2018, 7, 86. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.-C.; et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.S.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Hsiao, S.J.; Nikiforov, Y. Molecular approaches to thyroid cancer diagnosis. Endocr. Relat. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Adeniran, A.J.; Zhu, Z.; Gandhi, M.; Steward, D.L.; Fidler, J.P.; Giordano, T.J.; Biddinger, P.W.; Nikiforov, Y.E. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006, 30, 216–222. [Google Scholar] [CrossRef]
- Rivera, M.; Ricarte-Filho, J.; Knauf, J.; Shaha, A.; Tuttle, M.; Fagin, J.A.; Ghossein, R.A. Molecular genotyping of papillary thyroid carcinoma follicular variant according to its histological subtypes (encapsulated vs infiltrative) reveals distinct BRAF and RAS mutation patterns. Mod. Pathol. 2010, 23, 1191–1200. [Google Scholar] [CrossRef]
- Guerra, A.; Sapio, M.R.; Marotta, V.; Campanile, E.; Rossi, S.; Forno, I.; Fugazzola, L.; Budillon, A.; Moccia, T.; Fenzi, G.; et al. The primary occurrence of BRAFV600E is a rare clonal event in papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, 517–524. [Google Scholar] [CrossRef]
- Guerra, A.; Zeppa, P.; Bifulco, M.; Vitale, M. Concomitant BRAFV600E mutation and RET/PTC rearrangement is a frequent occurrence in papillary thyroid carcinoma. Thyroid 2014, 24, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harbor Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Castellano, E.; Santos, E. Functional specificity of RAS isoforms: So similar but so different. Genes Cancer 2011, 2, 216–231. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of RAS mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Shirokawa, J.M.; Elisei, R.; Knauf, J.A.; Hara, T.; Wang, J.; Saavedra, H.I.; Fagin, J.A. Conditional apoptosis induced by oncogenic rats in thyroid cells. Mol. Endocrinol. 2000, 14, 1725–1738. [Google Scholar] [CrossRef]
- De Vita, G.; Bauer, L.; da Costa, V.M.; De Felice, M.; Baratta, M.G.; De Menna, M.; Di Lauro, R. Dose-dependent inhibition of thyroid differentiation by RAS oncogenes. Mol. Endocrinol. 2005, 19, 76–89. [Google Scholar] [CrossRef]
- Vitagliano, D.; Portella, G.; Troncone, G.; Francione, A.; Rossi, C.; Bruno, A.; Giorgini, A.; Coluzzi, S.; Nappi, T.C.; Rothstein, J.L.; et al. Thyroid targeting of the N-ras (Gln61Lys) oncogene in transgenic mice results in follicular tumors that progress to poorly differentiated carcinomas. Oncogene 2006, 25, 5467–5474. [Google Scholar] [CrossRef] [PubMed]
- Vasko, V.; Ferrand, M.; Di Cristofaro, J.; Carayon, P.; Henry, J.F.; de Micco, C. Specific pattern of RAS oncogene mutations in follicular thyroid tumours. J. Clin. Endocrinol. Metab. 2003, 88, 2745–2752. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W.; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPARγ rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef]
- Fukahori, M.; Yoshida, A.; Hayashi, H.; Yoshihara, M.; Matsukuma, S.; Sakuma, Y.; Koizume, S.; Okamoto, N.; Kondo, T.; Masuda, M.; et al. The associations between RAS mutations and clinical characteristics in follicular thyroid tumors: New insights from a single center and a large patient cohort. Thyroid 2012, 22, 683–689. [Google Scholar] [CrossRef]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Hans-Juergen, S.; Salama, S.; Al-Ahmadi, A.; Al-Mansouri, Z.; Mirza, Z.; Al-Ghamdi, K.; Al-Hamour, O.A.; Huwait, E.; Gari, M.; Al-Qahtani, M.H.; et al. Comprehensive survey of HRAS, KRAS, and NRAS mutations in proliferative thyroid lesions from an ethinically diverse population. Anticancer Res. 2013, 33, 4779–4784. [Google Scholar]
- Horie, H.; Yokogoshi, Y.; Tsuyuguchi, M.; Saito, S. Point mutations of RAS and Gs alpha subunit genes in thyroid tumors. Jpn. J. Cancer Res. 1995, 86, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Makarov, V.; Deraje, S.; Dong, Y.; Reznik, E.; Seshan, V.; Nanjangud, G.; Eng, S.; Bose, P.; Kuo, F.; et al. Integrated genomic analysis of Hürthle cell cancer reveals oncogenic drivers, recurrent mitochondrial mutations, and unique chromosomal landscapes. Cancer Cell 2018, 34, 256–270.e5. [Google Scholar] [CrossRef]
- Moura, M.M.; Cavaco, B.M.; Pinto, A.E.; Leite, V. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J. Clin. Endocrinol. Metab. 2011, 96, E863–E868. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. Ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235. [Google Scholar] [CrossRef]
- Costa, A.M.; Herrero, A.; Fresno, M.F.; Heymann, J.; Alvarez, J.A.; Cameselle-Teijeiro, J.; García-Rostán, G. BRAF mutation associated with other genetic events identifies a subset of aggressive papillary thyroid carcinoma. Clin. Endocrinol. 2008, 68, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, N.; Miyagishi, M.; Mitsutake, S.; Akeno, N.; Mesa, C., Jr.; Knauf, J.A.; Zhang, L.; Taira, K.; Fagin, J.A. BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: Functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology 2006, 147, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Cohen, Y.; Xing, M.; Mambo, E.; Guo, Z.; Wu, G.; Trink, B.; Beller, U.; Westra, W.H.; Ladenson, P.W.; Sidransky, D. BRAF mutation in papillary thyroid carcinoma. JNCI J. Natl. Cancer Inst. 2003, 95, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Suzuki, S.; Mashiko, M.; Ohtake, T.; Endo, Y.; Takebayashi, Y.; Sekikawa, K.; Hagiwara, K.; Takenoshita, S. BRAF mutations in papillary carcinomas of the thyroid. Oncogene 2003, 22, 6455–6457. [Google Scholar] [CrossRef]
- Namba, H.; Nakashima, M.; Hayashi, T.; Hayashida, N.; Maeda, S.; Rogounovitch, T.I.; Ohtsuru, A.; Saenko, V.A.; Kanematsu, T.; Yamashita, S. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J. Clin. Endocrinol. Metab. 2003, 88, 4393–4397. [Google Scholar] [CrossRef]
- Soares, P.; Trovisco., V.; Rocha, A.S.; Lima, J.; Castro, P.; Preto, A.; Maximo, V.; Botelho, T.; Seruca, R.; Sobrinho-Simoes, M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003, 22, 4578–4580. [Google Scholar] [CrossRef]
- Xu, X.; Quiros, R.M.; Gattuso, P.; Ain, K.B.; Prinz, R.A. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res. 2003, 63, 4561–4567. [Google Scholar]
- Xing, M.; Westra, W.H.; Tufano, R.P.; Cohen, Y.; Rosenbaum, E.; Rhoden, K.J.; Carson, K.A.; Vasko, V.; Larin, A.; Tallini, G. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2005, 90, 6373–6379. [Google Scholar] [CrossRef]
- Ciampi, R.; Knauf, J.A.; Kerler, R.; Gandhi, M.; Zhu, Z.; Nikiforova, M.N.; Rabes, H.M.; Fagin, J.A.; Nikiforov, Y.E. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J. Clin. Investig. 2005, 115, 94–101. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Kimura, E.T.; Gandhi, M.; Biddinger, P.W.; Knauf, J.A.; Basolo, F.; Zhu, Z.; Giannini, R.; Salvatore, G.; Fusco, A.; et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wei, S.; Han, Y.; Li, Y.; Yu, Y.; Yun, X.; Ren, X.; Gao, M. Papillary microcarcinoma of the thyroid: Clinical characteristics and BRAFV600E mutational status of 977 cases. Ann. Surg. Oncol. 2013, 20, 2266–2273. [Google Scholar] [CrossRef]
- Walczyk, A.; Kowalska, A.; Kowalik, A.; Sygut, J.; Wypiórkiewicz, E.; Chodurska, R.; Pieciak, L.; Gozdz, S. The BRAFV600E mutation in papillary thyroid microcarcinoma: Does the mutation have an impact on clinical outcome? Clin. Endocrinol. 2014, 80, 899–904. [Google Scholar] [CrossRef]
- Romei, C.; Ciampi, R.; Faviana, P.; Agate, L.; Molinaro, E.; Bottici, V.; Basolo, F.; Miccoli, P.; Pacini, F.; Pinchera, A.; et al. BRAFV600E mutation, but not RET/PTC rearrangements, is correlated with a lower expression of both thyroperoxidase and sodium iodide symporter genes in papillary thyroid cancer. Endocr. Relat. Cancer 2008, 15, 511–520. [Google Scholar] [CrossRef]
- Xing, M. BRAF mutation in papillary thyroid cancer: Pathogenic role, molecular bases, and clinical implications. Endocr. Rev. 2007, 28, 742–762. [Google Scholar] [CrossRef]
- Kim, T.H.; Park, Y.J.; Lim, J.A.; Ahn, H.Y.; Lee, E.K.; Lee, Y.J.; Kim, K.W.; Hahn, S.K.; Youn, Y.K.; Kim, K.H.; et al. The association of the BRAFV600E mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer. Cancer 2012, 118, 1764–1773. [Google Scholar] [CrossRef]
- Li, C.; Lee, K.C.; Schneider, E.B.; Zeiger, M.A. BRAFV600E mutation and its association with clinicopathological features of papillary thyroid cancer: A meta-analysis. J. Clin. Endocrinol. Metab. 2012, 97, 4559–4570. [Google Scholar] [CrossRef]
- Riesco-Eizaguirre, G.; Gutiérrez-Martínez, P.; García-Cabezas, M.A.; Nistal, M.; Santisteban, P. The oncogene BRAF V600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I- targeting to the membrane. Endocr. Relat. Cancer 2006, 13, 257–269. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Ryder, M.; Jimenez, C. Targeted therapy for advanced thyroid cancer: Kinase inhibitors and beyond. Endocr. Rev. 2019, 40, 1573–1604. [Google Scholar] [CrossRef] [PubMed]
- Grieco, M.; Santoro, M.; Berlingieri, M.T.; Melillo, R.M.; Donghi, R.; Bongarzone, I.; Pierotti, M.A.; Della Porta, G.; Fusco, A.; Vecchio, G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 1990, 60, 557–563. [Google Scholar] [CrossRef]
- Bongarzone, I.; Monzini, N.; Borrello, M.G.; Carcano, C.; Ferraresi, G.; Arighi, E.; Mondellini, P.; Della Porta, G.; Pierotti, M.A. Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subuniy RI alpha of cyclic AMP-dependent protein kinase A. Mol. Cell. Biol. 1993, 13, 358–366. [Google Scholar] [CrossRef]
- Santoro, M.; Dathan, N.A.; Berlingieri, M.T.; Bongarzone, I.; Paulin, C.; Grieco, M.; Pierotti, M.A.; Vecchio, G.; Fusco, A. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene 1994, 9, 509–516. [Google Scholar] [PubMed]
- Santoro, M.; Melillo, R.M.; Fusco, A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur. J. Endocrinol. 2006, 155, 645–653. [Google Scholar] [CrossRef]
- Gertz, R.J.; Nikiforov, Y.; Rehrauer, W.; McDaniel, L.; Lloyd, R.V. Mutation in BRAF and other members of the MAPK pathway in papillary thyroid carcinoma in the pediatric population. Arch. Pathol. Lab. Med. 2016, 140, 134–139. [Google Scholar] [CrossRef]
- Hamatani, K.; Eguchi, H.; Ito, R.; Mukai, M.; Takahashi, K.; Taga, M.; Imai, K.; Cologne, J.; Soda, M.; Arihiro, K.; et al. RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer Res. 2008, 68, 7176–7182. [Google Scholar] [CrossRef]
- Rabes, H.M.; Demidchik, E.P.; Sidorow, J.D.; Lengfelder, E.; Beimfohr, C.; Hoelzel, D.; Klugbauer, S. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: Biological, phenotypic, and clinical implications. Clin. Cancer Res. 2000, 6, 1093–1103. [Google Scholar]
- Rhoden, K.J.; Johnson, C.; Brandao, G.; Howe, J.G.; Smith, B.R.; Tallini, G. Real-time quantitative RT-PCR identifies disticnt c-RET, RET/PTC1 and RET/PTC3 expression patterns in papillary thyroid carcinoma. Lab. Investig. 2004, 84, 1557–1570. [Google Scholar] [CrossRef]
- Kang, D.Y.; Kim, K.H.; Kim, J.M.; Kim, S.H.; Kim, J.Y.; Bail, H.W.; Kim, Y.S. High prevalence of RET, RAS, and ERK expression in Hashimoto’s thyroiditis and in papillary thyroid carcinoma in the Korean population. Thyroid 2007, 17, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Baitei, E.Y.; Alzahrani, A.S.; Binhumaid, F.S.; Alkhafaji, D.; Al-Rijjal, R.A.; Meyer, B.F.; Shi, Y. Concomitant RAS, RET/PTC, or BRAF mutations in advanced stage of papillary thyroid carcinoma. Thyroid 2014, 24, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ciampi, R.; Nikiforova, M.N.; Gandhi, M.; Nikiforov, Y.E. Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: Effects of the detection methods and genetic heterogeneity. J. Clin. Endocrinol. Metab. 2006, 91, 3603–3610. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Papotti, M.; Chiappetta, G.; Garcia-Rostan, G.; Volante, M.; Johnson, C.; Camp, R.L.; Pentimalli, F.; Monaco, C.; Herrero, A.; et al. RET activation and clinicopathologic features in poorly differentiated thyroid tumors. J. Clin. Endocrinol. Metab. 2002, 87, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Rhoden, K.J.; Unger, K.; Salvatore, G.; Yilmaz, Y.; Vovk, V.; Chiappetta, G.; Qumsiyeh, M.B.; Rothstein, J.L.; Fusco, A.; Santoro, M.; et al. RET/papillary thyroid cancer rearrangement in nonneoplastic thyrocytes: Follicular cells of Hashimoto’s thyroiditis share low-level recombination events with a subset of papillary carcinoma. J. Clin. Endocrinol. Metab. 2006, 91, 2414–2423. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nikiforov, Y.E. RET/PTC rearragment in thyroid tumours. Endocr. Pathol. 2002, 13, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Sapio, M.R.; Guerra, A.; Marotta, V.; Campanile, E.; Formisano, R.; Deandrea, M.; Motta, M.; Limone, P.P.; Fenzi, G.; Rossi, G.; et al. High growth rate of benign thyroid nodules BearingRET/PTCRearrangements. J. Clin. Endocrinol. Metab. 2011, 96, E916–E919. [Google Scholar] [CrossRef] [PubMed]
- Melillo, R.M.; Castellone, M.D.; Guarino, V.; De Falco, V.; Cirafici, A.M.; Salvatore, G.; Caiazzo, F.; Basolo, F.; Giannini, R.; Kruhoffer, M.; et al. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J. Clin. Investig. 2005, 115, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Castellone, M.D.; Guarino, V.; De Falco, V.; Carlomagno, F.; Basolo, F.; Faviana, P.; Kruhoffer, M.; Orntoft, T.; Russell, J.P.; Rothstein, J.L.; et al. Functional expression of the CXCR4 chemokine receptor is induced by RET/PTC oncogenes and is a common event in human papillary thyroid carcinomas. Oncogene 2004, 23, 5958–5967. [Google Scholar] [CrossRef] [PubMed]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef]
- Karunamurthy, A.; Panebianco, F.; Hsiao, S.J.; Vorhauer, J.; Nikiforova, M.N.; Chiosea, S.; Nikiforov, Y.E. Prevalence and phenotypic correlations of EIF1AX mutations in thyroid nodules. Endocr. Relat. Cancer 2016, 23, 295–301. [Google Scholar] [CrossRef]
- Nicolson, N.G.; Murtha, T.D.; Dong, W.; Paulsson, J.O.; Choi, J.; Barbieri, A.L.; Brown, T.C.; Kunstman, J.W.; Larsson, C.; Prasad, M.L.; et al. Comprehensive genetic analysis of follicular thyroid carcinoma predicts prognosis independent of histology. J. Clin. Endocrinol. Metab. 2018, 103, 2640–2650. [Google Scholar] [CrossRef]
- Krishnamoorthy, G.P.; Davidson, N.R.; Leach, S.D.; Zhao, Z.; Lowe, S.W.; Lee, G.; Landa, I.; Nagarajah, J.; Saqcena, M.; Singh, K.; et al. EIF1AX and RAS mutations cooperate to drive thyroid tumorigenesis through ATF4 and c-MYC. Cancer Discov. 2019, 9, 264–281. [Google Scholar] [CrossRef]
- Hou, P.; Liu, D.; Shan, Y.; Hu, S.; Studeman, K.; Condouris, S.; Wang, Y.; Trink, A.; El-Naggar, A.K.; Tallini, G.; et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/akt pathway in thyroid cancer. Clin. Cancer Res. 2007, 13, 1161–1170. [Google Scholar] [CrossRef]
- Eng, C. Genetics of Cowden syndrome: Through the looking glass of oncology. Int. J. Oncol. 1998, 12, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Halachmi, N.; Halachmi, S.; Evron, E.; Cairns, P.; Okami, K.; Saji, M.; Westra, W.H.; Zeiger, M.A.; Jen, J.; Sidransky, D. Somatic mutations of the PTEN tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosomes Cancer 1998, 23, 239–243. [Google Scholar] [CrossRef]
- Yeager, N.; Klein-Szanto, A.; Kimura, S.; Di Cristofano, A. Pten loss in the mouse thyroid causes goitre and follicular adenomas: Insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007, 67, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.P.; Silva, J.; Iezza, G.; Phillips, W.A.; McMahon, M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol. Cancer Res. 2014, 12, 979–986. [Google Scholar] [CrossRef]
- Shimamura, M.; Shibusawa, N.; Kurashige, T.; Mussazhanova, Z.; Matsuzaki, H.; Nakashima, M.; Yamada, M.; Nagayama, Y. Mouse models of sporadic thyroid cancer derived from BRAFV600E alone or in combination with PTEN haploinsufficiency under physiologic TSH levels. PLoS ONE 2018, 13, e0201365. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Kroll, T.G.; Sarraf, P.; Pecciarini, L.; Chen, C.J.; Mueller, E.; Spiegelman, B.M.; Fletcher, J.A. PAX8-PPPAR-gamma 1 fusion oncogene in human thyroid carcinoma. Science 2000, 289, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Biddinger, P.W.; Caudill, C.M.; Kroll, T.G.; Nikiforov, Y.E. PAX8-PPAR gamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am. J. Surg. Pathol. 2002, 26, 1016–1023. [Google Scholar] [CrossRef]
- Castro, P.; Rebocho, A.P.; Soares, R.J.; Mgalhaes, J.; Roque, L.; Trovisco, V.; Vieira de Castro, I.; Cardoso-de-Oliveira, M.; Fonseca, E.; Soares, P.; et al. PAX8-PPAR gamma rearrangement is frequently detected in the follicular variant of papillary thyroid carcioma. J. Clin. Endocrinol. Metab. 2006, 92, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Espadinha, C.; Catarino, A.L.; Moniz, S.; Pereira, T.; Sobrinho, L.G.; Leite, V. Expression of PAX-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J. Clin. Endocrinol. Metab. 2002, 87, 3947–3952. [Google Scholar]
- Kelly, L.M.; Barila, G.; Liu, P.; Evdokimova, V.N.; Trivedi, S.; Panebianco, F.; Gandhi, M.; Carty, S.E.; Hodak, S.P.; Luo, J.; et al. Identification of the transforming STRNALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4233–4238. [Google Scholar] [CrossRef]
- Panebianco, F.; Nikitski, A.V.; Nikiforova, M.N.; Kaya, C.; Yip, L.; Condello, V.; Wald, A.I.; Nikiforov, Y.E.; Chiosea, S.I. Characterization of thyroid cancer driven by known and novel ALK fusions. Endocr. Relat. Cancer 2019, 26, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Song, Y.S.; Lim, J.A.; Choi, H.; Won, J.K.; Moon, J.H.; Cho, S.W.; Lee, K.E.; Park, Y.J.; Yi, K.H.; Park, D.J.; et al. Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients. Cancer 2016, 122, 1370–1379. [Google Scholar] [CrossRef]
- Liu, J.; Liu, R.; Shen, X.; Zhu, G.; Li, B.; Xing, M. The genetic duet of BRAF V600E and TERT promoter mutations robustly predicts loss of radioiodine avidity in recurrent papillary thyroid cancer. J. Nucl. Med. 2020, 61, 177–182. [Google Scholar] [CrossRef]
- Melo, M.; Gaspar Da Rocha, A.; Batista, R.; Vinagre, J.; Martins, M.J.; Costa, G.; Ribeiro, C.; Carrilho, F.; Leite, V.; Lobo, C.; et al. TERT, BRAF, and NRAS in primary thyroid cancer and metastatic disease. J. Clin. Endocrinol. Metab. 2017, 102, 1898–1907. [Google Scholar] [CrossRef]
- Melo, M.; da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, S.; Eloy, C.; et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef]
- Song, Y.S.; Yoo, S.K.; Kim, H.H.; Jung, G.; Oh, A.R.; Cha, J.Y.; Kim, S.-J.; Cho, S.W.; Lee, K.E.; Seo, J.S.; et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr. Relat. Cancer 2019, 26, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Song, Y.S.; Kim, Y.A.; Lim, J.A.; Cho, S.W.; Moon, J.H.; Hahn, S.; Park, D.J.; Park, Y.J. Effects of coexistent BRAFV600E and TERT promoter mutations on poor clinical outcomes in papillary thyroid cancer: A meta-analysis. Thyroid 2017, 27, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gong, Y.; Yan, S.; Chen, H.; Qin, S.; Gong, R. Association between TERT promoter mutations and clinical behaviors in differentiated thyroid carcinoma: A systematic review and meta-analysis. Endocrine 2020, 67, 44–57. [Google Scholar] [CrossRef]
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.A.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.J.; Killela, P.J.; Loriaux, D.B.; Lipp, E.S.; et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat. Commun. 2018, 9, 2087. [Google Scholar] [CrossRef]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Kramer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Liang, W.S.; Hendricks, W.; Kiefer, J.; Schmidt, J.; Sekar, S.; Carpten, J.; Craig, D.W.; Adkins, J.; Cuyugan, L.; Manojlovic, Z.; et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017, 27, 524–532. [Google Scholar] [CrossRef]
- Suzuki, K.; Matsubara, H. Recent advances in p53 research and cancer treatment. J. Biomed. Biotechnol. 2011, 3, 978312. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.-J.; Cho, S.W.; Won, J.-K.; et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid. cancer. Nat. Commun. 2019, 10, 2764. [Google Scholar] [CrossRef]
- McFadden, D.G.; Vernon, A.; Santiago, P.M.; Martinez-McFaline, R.; Bhutkar, A.; Crowley, D.M.; McMahon, M.; Sadow, P.M.; Jacks, T. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E1600–E1609. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Baitei, E.; Al-Rijjal, R.; Parhar, R.; Al-Mohanna, F.; Kimura, S.; Pritchard, C.; Binessa, H.A.; Alzahrani, A.S.; Al-Khalaf, H.H.; et al. TSH overcomes Braf V600E-induced senescence to promote tumor progression via downregulation of p53 expression in papillary thyroid cancer. Oncogene 2016, 35, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Ravi, N.; Yang, M.; Gretarsson, S.; Jansson, C.; Mylona, N.; Sydow, S.R.; Woodward, E.L.; Ekblad, L.; Wennerberg, J.; Paulsson, K. Identification of targetable lesions in anaplastic thyroid cancer by genome profiling. Cancers 2019, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.O.; Backman, S.; Wang, N.; Stenman, A.; Crona, J.; Thutkawkorapin, J.; Ghaderi, M.; Tham, E.; Stalberg, P.; Zedenius, J.; et al. Whole-genome sequencing of synchronous thyroid carcinomas identifies aberrant DNA repair in thyroid cancer dedifferentiation. J. Pathol. 2020, 250, 183–194. [Google Scholar] [CrossRef]
- Genutis, L.K.; Tomsic, J.; Bundschuh, R.A.; Brock, P.L.; Williams, M.D.; Roychowdhury, S.; Reeser, J.W.; Frankel, W.L.; Alsomali, M.; Routbort, M.J.; et al. Microsatellite instability occurs in a subset of follicular thyroid cancers. Thyroid 2019, 29, 523–529. [Google Scholar] [CrossRef]
- Santos, J.C.; Bastos, A.U.; Cerutti, J.M.; Ribeiro, M.L. Correlation of MLH1 and MGMT expression and promoter methylation with genomic instability in patients with thyroid carcinoma. BMC Cancer 2013, 13, 79. [Google Scholar] [CrossRef]
- Mitmaker, E.; Alvarado, C.; Bégin, L.R.; Trifiro, M. Microsatellite instability in benign and malignant thyroid neoplasms. J. Surg. Res. 2008, 150, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.; Fagin, J.A. Prevalence of minisatellite and microsatellite instability in radiation-induced post-Chernobyl pediatric thyroid carcinomas. Oncogene 1998, 17, 1983–1988. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lengfelder, E.; Bauchinger, M. Microsatellite instability and loss of heterozygosity in radiation-associated thyroid carcinomas of Belarussian children and adults. Carcinogenesis 1999, 20, 2247–2252. [Google Scholar] [CrossRef][Green Version]
- <named-content content-type="background:white">Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Roberts, C.W.M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef] [PubMed]
- <named-content content-type="background:white">Wang, X.; Haswell, J.R.; Roberts, C.W.M. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer-mechanisms and potential therapeutic insights. Clin. Cancer Res. 2014, 20, 21–27. [Google Scholar]
- Fagin, J.A.; Mitsiades, N. Molecular pathology of thyroid cancer: Diagnostic and clinical implications. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Gavert, N.; Ben-Ze’ev, A. Beta-catenin signaling in biological control and cancer. J. Cell Biochem. 2007, 102, 820–828. [Google Scholar] [CrossRef]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Cetta, F.; Montalto, G.; Gori, M.; Curia, M.C.; Cama, A.; Olschwang, S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J. Clin. Endocrinol Metab. 2000, 85, 286–292. [Google Scholar] [PubMed]
- Von, W.R.; Rhein, A.; Werner, M.; Scheumann, G.F.; Dralle, H.; Potter, E.; Brabant, G.; Georgii, A. Immunohistochemical detection of E-cadherin in differentiated thyroid carcinomas correlates with clinical outcome. Cancer Res. 1997, 57, 2501–2507. [Google Scholar]
- Garcia-Rostan, G.; Camp, R.L.; Herrero, A.; Carcangiu, M.L.; Rimm, D.L.; Tallini, G. Beta-catenin dysregulation in thyroid neoplasms: Down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am. J. Pathol. 2001, 158, 987–996. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Tallini, G.; Herrero, A.; D’Aquila, T.G.; Carcangiu, M.L.; Rimm, D.L. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999, 59, 1811–1815. [Google Scholar] [PubMed]
- Baylin, S.B. Tying it all together: Epigenetics, genetics, cell cycle, and cancer. Science 1997, 277, 1948–1949. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Usadel, H.; Cohen, Y.; Tokumaru, Y.; Guo, Z.; Westra, W.B.; Tong, B.C.; Tallinni, G.; Udelsman, R.; Califano, J.A.; et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: A marker of malignancy and a cause of gene silencing. Cancer Res. 2003, 63, 2316–2321. [Google Scholar]
- Kartal, K.O.S. Methylation status of TSHr in well-differentiated thyroid cancer by using cytologic material. BMC Cancer 2015, 15, 824–830. [Google Scholar] [CrossRef]
- Kim, G.W.; Zhu, X.; Kim, W.D.; Zhang, L.; Kebew, E.; Cheng, S. Reactivation of the silenced thyroid hormone receptor β gene expression delays thyroid tumor progression. Endocrinology 2013, 154, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Ji, M.; Liu, D.; Hou, P.; Xing, M. Lack of mutations in the thyroid hormone receptor (TR) α and β genes but frequent hypermethylation of theTRβ gene in differentiated thyroid tumors. J. Clin. Endocrinol. Metab. 2007, 92, 4766–4770. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Sunaga, N.; Tomizawa, Y.; Imai, H.; Iijima, H.; Yanagitani, N.; Horiguchi, K.; Yamada, M.; Mori, M. Epigenetic inactivation of the thyroid hormone receptor β1 gene at 3p24.2 in lung cancer. Ann. Surg. Oncol. 2010, 17, 2222–2228. [Google Scholar] [CrossRef]
- Li, Z.; Meng, Z.H.; Chandrasekaran, R.; Kuo, W.L.; Collins, C.C.; Gray, J.W.; Dairkee, S.H. Biallelic inactivation of the thyroid hormone receptor β 1 gene in early stage breast cancer. Cancer Res. 2002, 62, 1939–1943. [Google Scholar] [PubMed]
- Hörkkö, T.T.; Tuppurainen, K.; George, S.M.; Jernvall, P.; Karttunen, T.J.; Mäkinen, M.J. Thyroid hormone receptor β 1 in normal colon and colorectal cancer-association with differentiation, polypoid growth type and K-ras mutations. Int. J. Cancer 2006, 118, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Dunwell, T.L.; Hesson, L.B.; Pavlova., T.; Zabarovska., V.; Kashuba, V.; Catchpoole, D.; Chiaramonte, R.; Brini, A.T.; Griffiths, M.; Maher, E.R.; et al. Epigenetic analysis of childhood acute lymphoblastic leukemia. Epigenetics 2009, 4, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Boltze, C.; Zack, S.; Quednow, C.; Bettge, S.; Roessner, A.; Schneider-Stock, R. Hypermethylation of the CDKN2/p16INK4A promoter in thyroid carcinogenesis. Pathol. Res. Pract. 2003, 199, 399–404. [Google Scholar] [CrossRef]
- Shou, F.; Xu, F.; Li, G.; Zhao, Z.; Mao, Y.; Yang, F.; Wang, H.; Guo, H. RASSF1A promoter methylation is associated with incresaed risk of thyroid cancer: A meta-analysis. Onco Targets Ther. 2017, 10, 247–257. [Google Scholar] [CrossRef]
- Hu, S.; Liu, D.; Tufano, R.P.; Carson, K.A.; Rosenbaum, E.; Cohen, Y.; Holy, E.H.; Kiseljak-Vassiliades, K.; Rhoden, K.J.; Tolaney, S.; et al. Association of aberrant methyrlation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int J. Cancer 2006, 119, 2322–2329. [Google Scholar] [CrossRef]
- Xing, M.; Tokumaru, Y.; Wu, G.; Westra, W.B.; Ladenson, P.W.; Sidransky, D. Hypermethylation of the Pendred syndrome gene SLC26A is an early event in thyroid tumorigenesis. Cancer Res. 2003, 63, 2312–2315. [Google Scholar]
- McKelvey, B.A.; Zeiger, M.A.; Umbricht, C.B. Characterization of TERT and BRAF copy number variation in papillary thyroid carcinoma: An analysis of the cancer genome atlas study. Genes Chromosomes Cancer 2021, 60, 403–409. [Google Scholar] [CrossRef]
- Máximo, V.; Soares, P.; Lima, J.; Cameselle-Teijeiro, J.; Sobrinho-Simões, M. Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: A study with emphasis on Hürthle cell tumors. Am. J. Pathol. 2002, 160, 1857–1865. [Google Scholar] [CrossRef]
- Gopal, R.K.; Kübler, K.; Calvo, S.E.; Polak, P.; Livitz, D.; Rosebrock, D.; Sadow, P.M.; Campbell, B.; Donovan, S.E.; Amin, S.; et al. Widespread chromosomal losses and mitochondrial DNA alterations as genetic drivers in Hürthle cell carcinoma. Cancer Cell 2018, 34, 242–255.e5. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Corrales, L.; Williams, J.; Horton, B.; Sivan, A.; Spranger, S. Cancer immunotherapy targets based on understanding the T-cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 19–31. [Google Scholar] [PubMed]
- Yu, H.; Huang, X.; Liu, X.; Jin, H.; Zhang, G.; Zhang, Q.; Yu, J. Regulatory T cells and plasmacytoid dendritic cells contribute to the immune escape of papillary thyroid cancer coexisting with multinodular non-toxic goiter. Endocrine 2013, 44, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.L.; Morari, E.C.; Nonogaki, S.; Soarees, F.A.; Vassallo, J.; Ward, L.S. Foxp3 expression is associated with aggressiveness in differentiated thyroid carcinomas. Clinics 2012, 67, 483–488. [Google Scholar] [CrossRef]
- Liu, Y.; Yun, X.; Gao, M.; Yu, Y.; Li, X. Analysis of regulatory T cells frequency in peripheral blood and tumor tissues in papillary thyroid carcinoma with and without Hashimoto’s thyroiditis. Clin. Transl. Oncol. 2015, 17, 274–280. [Google Scholar] [CrossRef]
- Coates, P.J.; Rundle, J.K.; Lorimore, S.A.; Wright, E.G. Indirect macrophage responses to ionizing radiation: Implications for genotype-dependent bystander signaling. Cancer Res. 2008, 68, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Giannini, R.; Moretti, S.; Ugolini, C.; Macerola, E.; Menicali, E.; Nucci, N.; Morelli, S.; Colella, R.; Mandarano, M.; Sidoni, A.; et al. Immune profiling of thyroid carcinomas suggests the existence of two major phenotypes: An ATC-like and a PDTC-like. J. Clin. Endocrinol. Metab. 2019, 104, 3557–3575. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 pathway in the immune response. Am. J. Transpl. 2012, 12, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (N7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Kim, T.H.; Kim, S.W.; Ki, C.S.; Jang, H.W.; Kim, J.S.; Kim, J.H.; Choe, J.-H.; Shin, J.H.; Hahn, S.Y.; et al. Comprehensive screening for PD-L1 expression in thyroid cancer. Endocr. Relat. Cancer 2017, 24, 97–106. [Google Scholar] [CrossRef]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; Correa, A.J.; Lopresti, J.S.; Epstein, A.L. BRAFV600E in papillary thyroid carcinoma is associated with increased programmed death Ligand 1 expression and suppressive immune cell infiltration. Thyroid 2014, 24, 1385–1393. [Google Scholar] [CrossRef]
- Chowdhury, S.; Veyhl, J.; Jessa, F.; Polyakova, O.; Alenzi, A.; MacMillan, C.; Ralhan, R.; Walfish, P.G. Programmed death-ligand 1 overexpression is a prognostic marker for aggressive papillary thyroid cancer and its variants. Oncotarget 2016, 7, 32318–32328. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.W.; Gigliotti, B.J.; Pai, S.I.; Parangi, S.; Wachtel, H.; Mino-Kenudson, M.; Gunda, V.; Faquin, W.C. PD-L1 and IDO1 are expressed in poorly differentiated thyroid carcinoma. Endocr. Pathol. 2018, 29, 59–67. [Google Scholar] [CrossRef]
- Shi, R.; Qu, N.; Luo, T.; Xiang, J.; Liao, T.; Sun, G.; Wang, Y.; Wang, Y.-L.; Huang, C.-P.; Ji, Q.-H. Programmed death-ligand 1 expression in papillary thyroid cancer and its correlation with clinicopathologic factors and recurrence. Thyroid 2017, 27, 537–545. [Google Scholar] [CrossRef]
- Zwaenepoel, K.; Jacobs, J.; De Meulenaere, A.; Silence, K.; Smits, E.; Siozopoulou, V.; Hauben, E.; Rolfo, C.; Rottey, S.; Pauwels, P. CD 70 and PD-L1 in anaplastic thyroid cancer–promising targets for immunotherapy. Histopathology 2017, 71, 357–365. [Google Scholar] [CrossRef]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; De Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef]
- Cantara, S.; Bertelli, E.; Occhini, R.; Regoli, M.; Brilli, L.; Pacini, F.; Castanga, M.G.; Toti, P. Blockade of the programmed death ligand 1 (PD-L1) as potential therapy for anaplastic thyroid cancer. Endocrine 2019, 64, 122–129. [Google Scholar] [CrossRef]
- Gunda, V.; Gigliotti, B.; Ashry, T.; Ndishabandi, D.; McCarthy, M.; Zhou, Z.; Amin, S.; Lee, K.E.; Stork, T.; Wirth, L.; et al. Anti-PD-1/PD-L1 therapy augments lenvatinib’s efficacy by favorably altering the immune microenvironment of murine anaplastic thyroid cancer. Int. J. Cancer 2019, 144, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Ventura, S.; Pojo, M.; Matias, A.T.; Moura, M.M.; Marques, I.J.; Leite, V.; Cavaco, B.M. The efficacy of HRAS and CDK4/6 inhibitors in anaplastic thyroid cancer cell lines. J. Endocrinol. Investig. 2018, 42, 527–540. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.; Ham, J.; Po, J.W.; Niles, N.; Roberts, T.; Lee, C.S. The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy. Cells 2021, 10, 1082. https://doi.org/10.3390/cells10051082
Singh A, Ham J, Po JW, Niles N, Roberts T, Lee CS. The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy. Cells. 2021; 10(5):1082. https://doi.org/10.3390/cells10051082
Chicago/Turabian StyleSingh, Amandeep, Jeehoon Ham, Joseph William Po, Navin Niles, Tara Roberts, and Cheok Soon Lee. 2021. "The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy" Cells 10, no. 5: 1082. https://doi.org/10.3390/cells10051082
APA StyleSingh, A., Ham, J., Po, J. W., Niles, N., Roberts, T., & Lee, C. S. (2021). The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy. Cells, 10(5), 1082. https://doi.org/10.3390/cells10051082