
Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy

 , ,
, ,
Abstract
1. Introduction
2. Monoclonal Antibodies—Immune Checkpoint Inhibitors
2.1. Approved Checkpoint Inhibitors
2.2. Combination Strategies
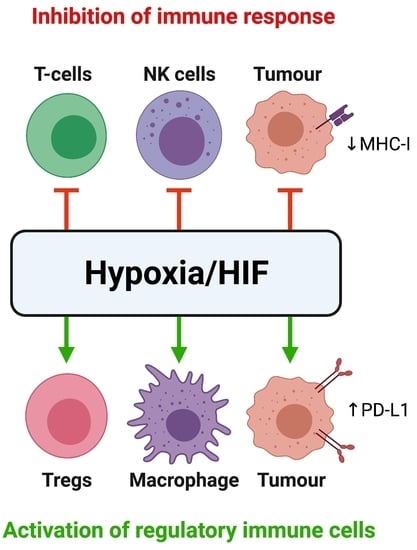
3. Tumour Hypoxia and Hypoxia-Mediated Immunosuppression
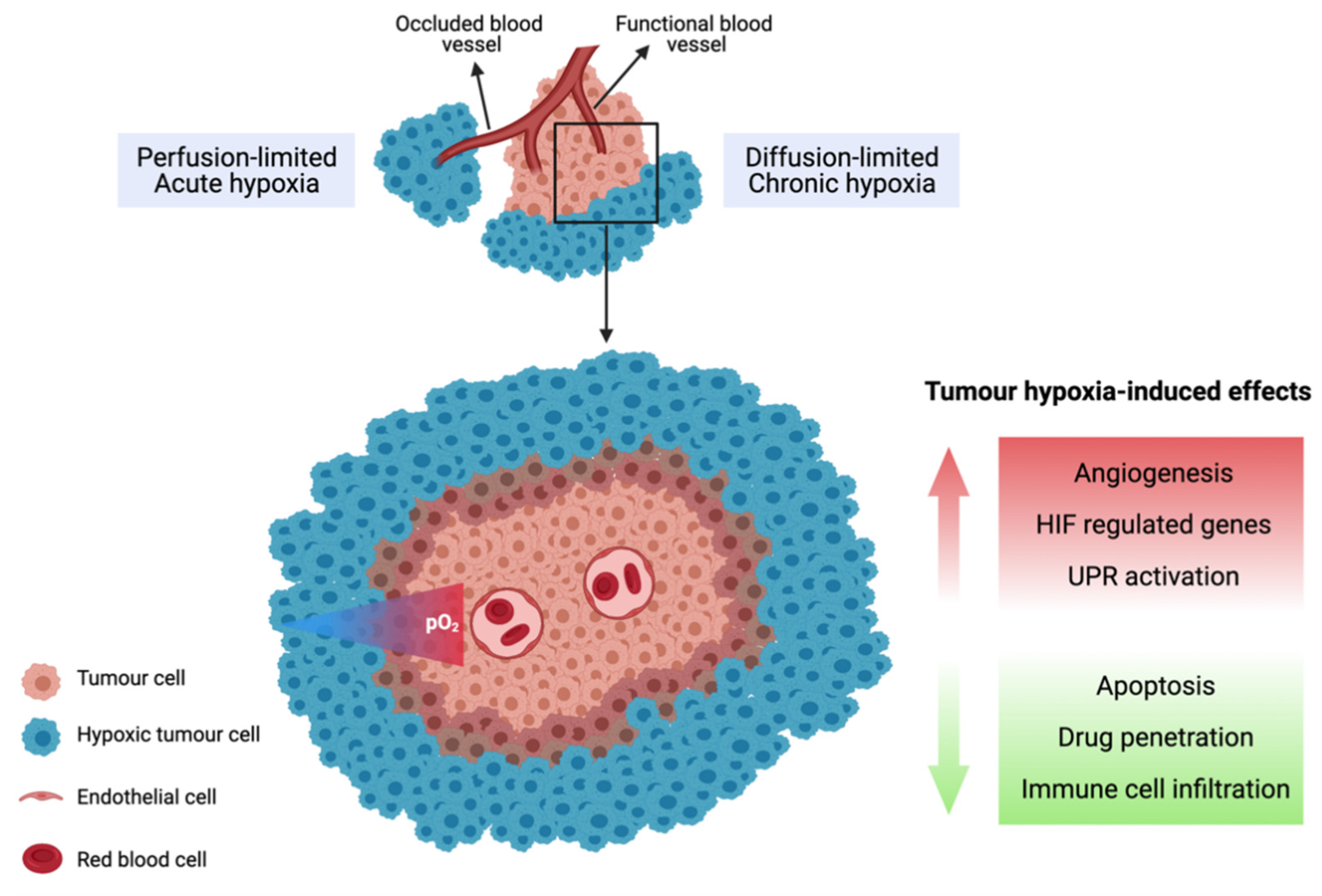
3.1. Tumour Hypoxia
3.2. Biological Response to Tumour Hypoxia
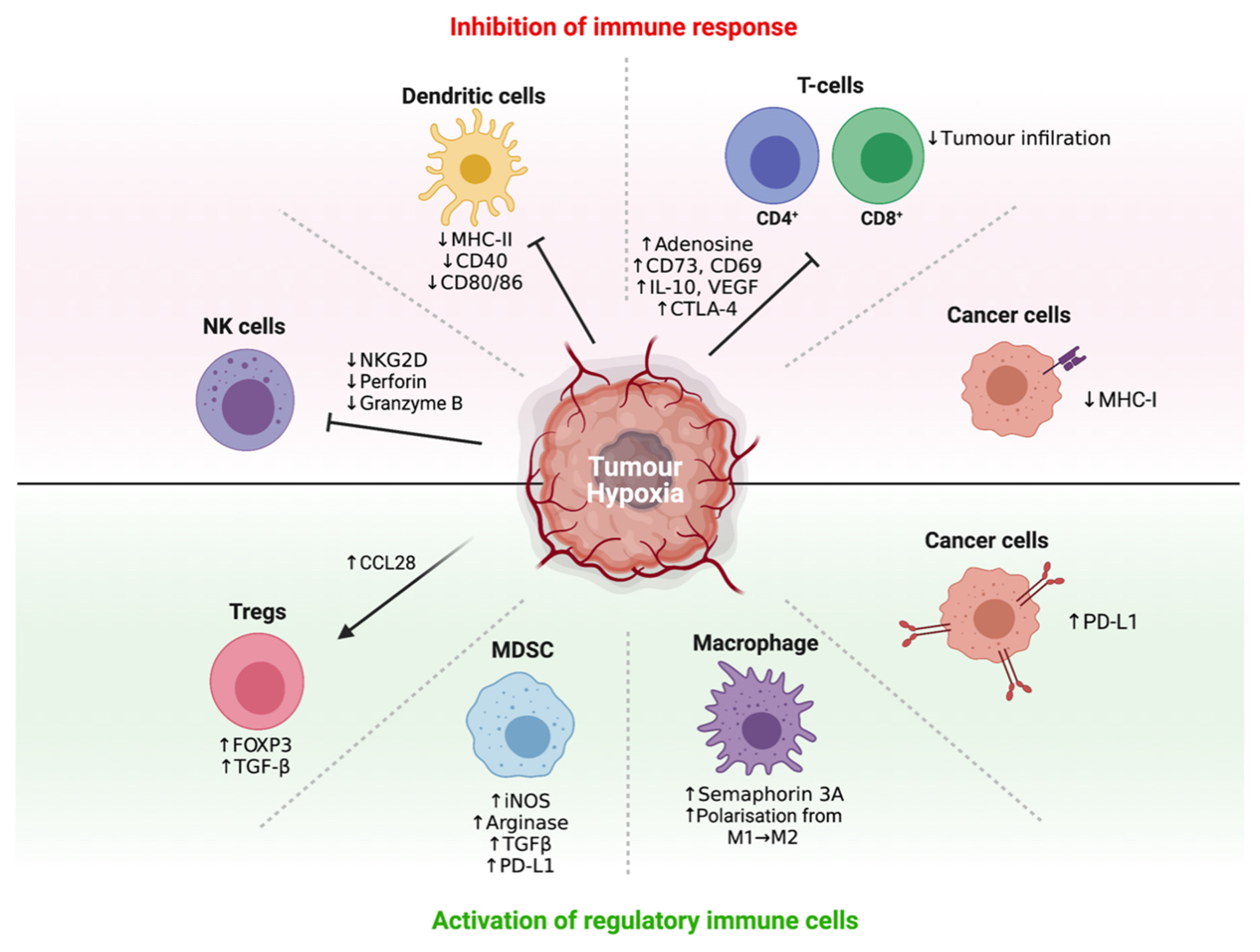
3.3. Hypoxia Promotes Immune Tolerance through Multiple Mechanisms
3.4. Hypoxia Recruits Immunosuppressive Cells to Promote Immune Tolerance
3.5. Hypoxia Interferes with and Suppresses Effector T-Cell, DC and NK Cell Function
4. Therapeutic Approaches That Can Modulate Tumour Hypoxia to Improve Immunotherapy Response
4.1. Tumour Reoxygenation as a Proof-of-Principle
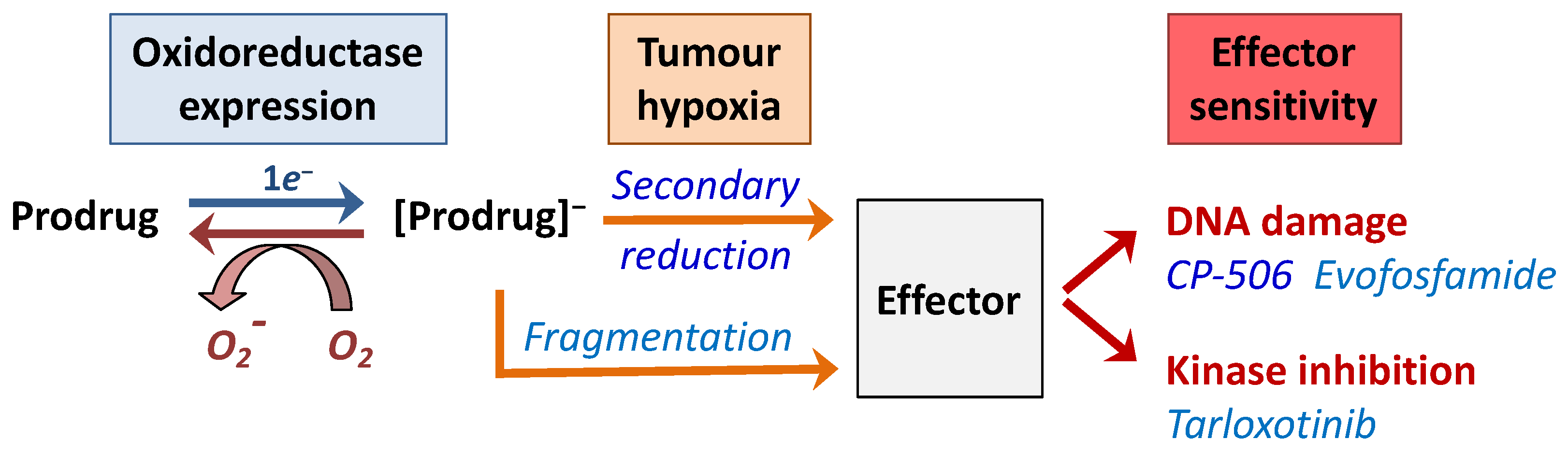
4.2. Hypoxia-Activated Prodrugs
4.2.1. Evofosfamide
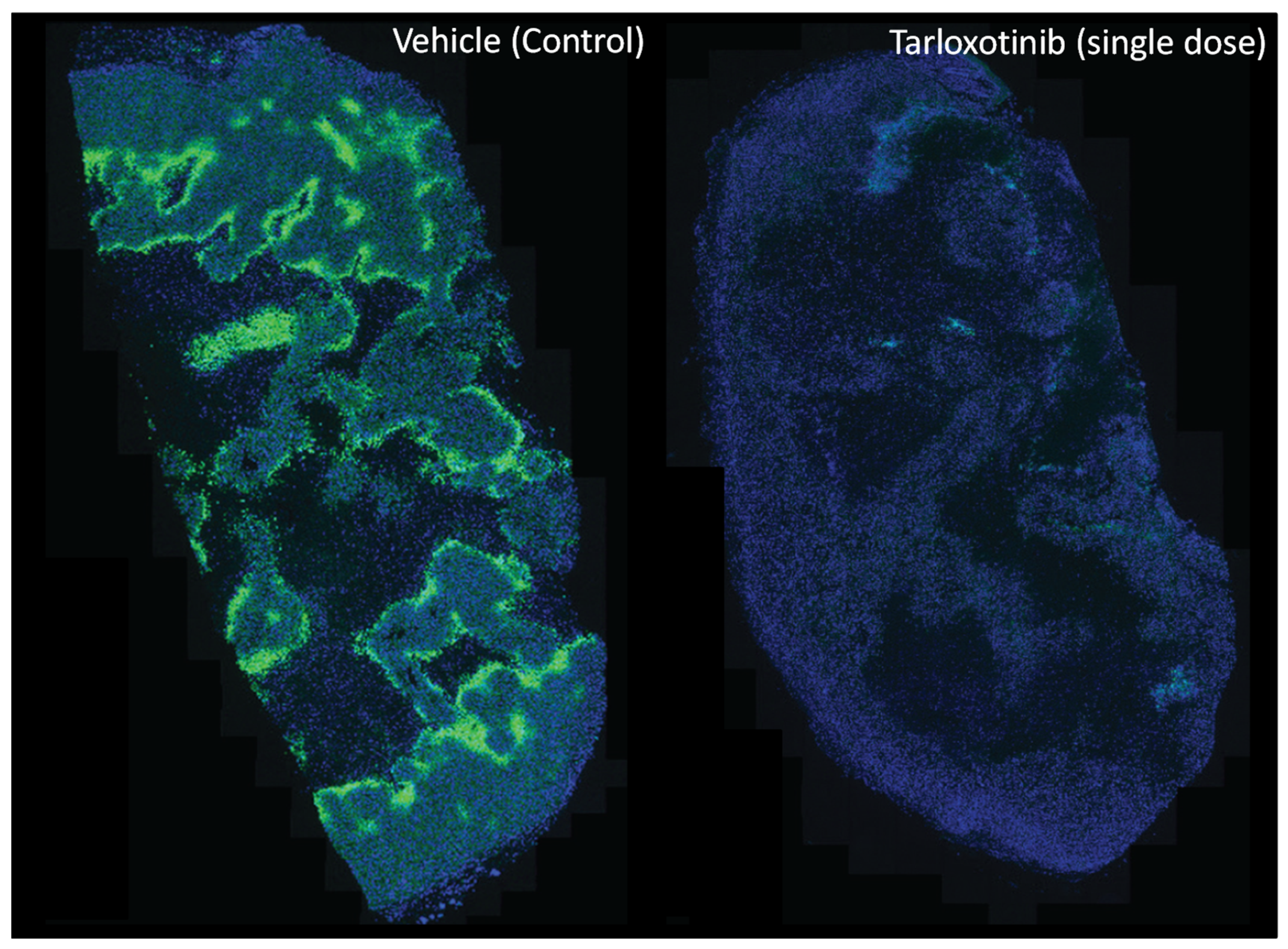
4.2.2. Tarloxotinib
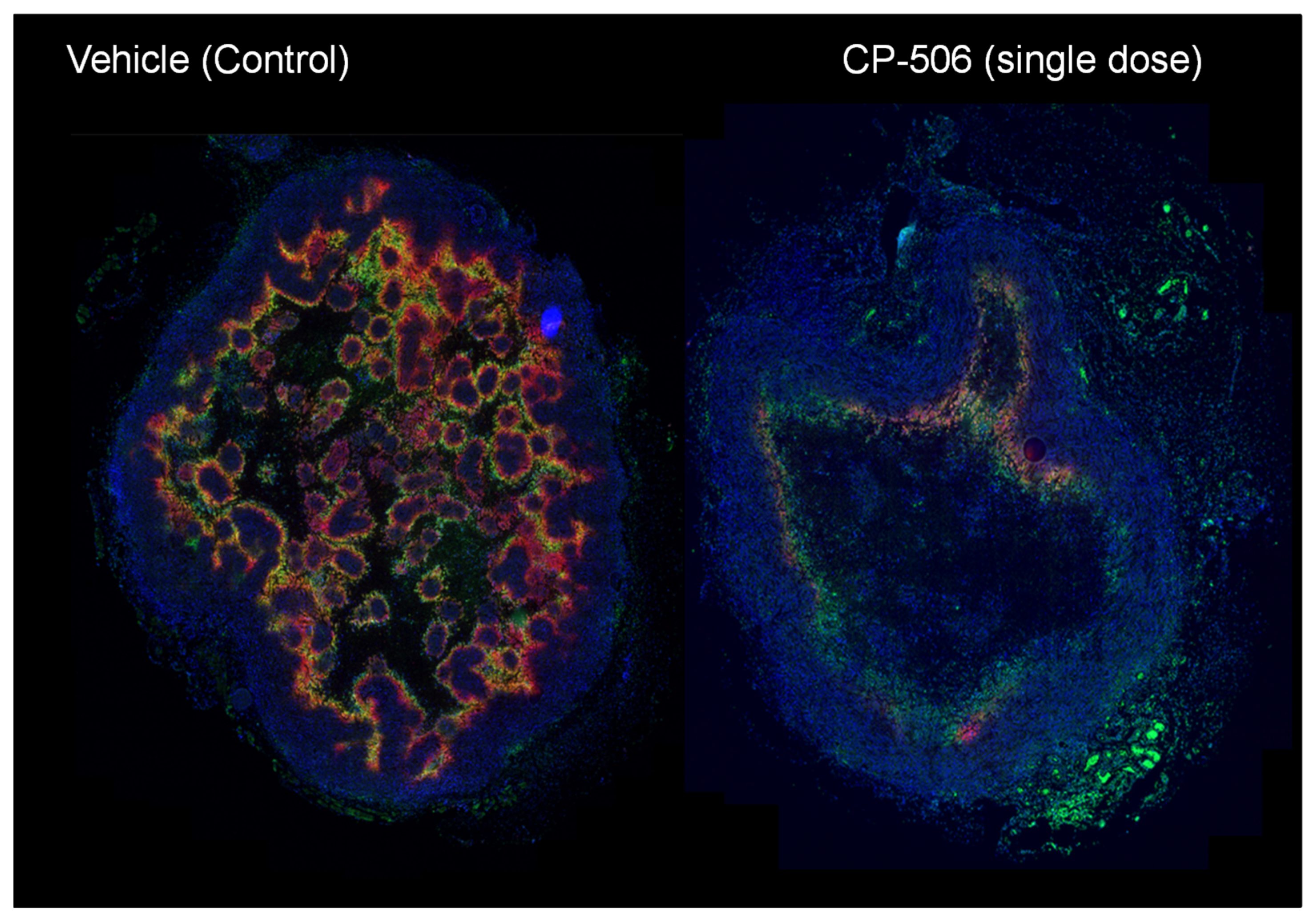
4.2.3. CP-506
4.3. Targeting of the HIF-1 Pathway
4.4. Hypoxia Targeted Biologicals
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Yan, Y.; Kumar, A.B.; Finnes, H.; Markovic, S.N.; Park, S.; Dronca, R.S.; Dong, H. Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76. [Google Scholar] [CrossRef]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef]
- Patel, S.P. Immune checkpoint blockade for lung cancer: State of the art. Transl. Cancer Res. 2015, 4, 415–422. [Google Scholar]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular Pathways: Next-Generation Immunotherapy—Inhibiting Programmed Death-Ligand 1 and Programmed Death-1. Clin. Cancer Res. 2012, 18, 6580. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Houot, R.; Schultz, L.M.; Marabelle, A.; Kohrt, H. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Weaver, C. Janeway’s Immunobiology, 9th ed.; Garland Science/Taylor & Francis Group, LLC: Brighton, UK, 2016. [Google Scholar]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- De Sousa Linhares, A.; Leitner, J.; Grabmeier-Pfistershammer, K.; Steinberger, P. Not All Immune Checkpoints Are Created Equal. Front. Immunol 2018, 9, 1909. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, Y.; Chen, Z. Tim-3 expression and its role in hepatocellular carcinoma. J. Hematol. Oncol. 2018, 11, 126. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Melero, I.; Grimaldi, A.M.; Perez-Gracia, J.L.; Ascierto, P.A. Clinical Development of Immunostimulatory Monoclonal Antibodies and Opportunities for Combination. Clin. Cancer Res. 2013, 19, 997–1008. [Google Scholar] [CrossRef]
- Chin, C.D.; Fares, C.M.; Campos, M.; Chen, H.-W.; Shintaku, I.P.; Konecny, G.E.; Rao, J. Association of PD-L1 expression by immunohistochemistry and gene microarray with molecular subtypes of ovarian tumors. Mod. Pathol. 2020, 33, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Velcheti, V.; Carvajal, D.; Wimberly, H.; Brown, J.; Pusztai, L.; Rimm, D.L. In situ tumor PD-L1 mRNA expression is associated with increased TILs and better outcome in breast carcinomas. Clin. Cancer Res. 2014, 20, 2773–2782. [Google Scholar] [CrossRef]
- Enwere, E.K.; Kornaga, E.N.; Dean, M.; Koulis, T.A.; Phan, T.; Kalantarian, M.; Köbel, M.; Ghatage, P.; Magliocco, A.M.; Lees-Miller, S.P.; et al. Expression of PD-L1 and presence of CD8-positive T cells in pre-treatment specimens of locally advanced cervical cancer. Mod. Pathol. 2017, 30, 577–586. [Google Scholar] [CrossRef]
- Taube, J.M.; Young, G.D.; McMiller, T.L.; Chen, S.; Salas, J.T.; Pritchard, T.S.; Xu, H.; Meeker, A.K.; Fan, J.; Cheadle, C.; et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clin. Cancer Res. 2015, 21, 3969–3976. [Google Scholar] [CrossRef]
- Shan, T.; Chen, S.; Wu, T.; Yang, Y.; Li, S.; Chen, X. PD-L1 expression in colon cancer and its relationship with clinical prognosis. Int. J. Clin. Exp. Path. 2019, 12, 1764–1769. [Google Scholar]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Velcheti, V.; Schalper, K.A.; Carvajal, D.E.; Anagnostou, V.K.; Syrigos, K.N.; Sznol, M.; Herbst, R.S.; Gettinger, S.N.; Chen, L.; Rimm, D.L. Programmed death ligand-1 expression in non-small cell lung cancer. Lab. Investig. 2014, 94, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008, 111, 3635–3643. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Khunger, M.; Rakshit, S.; Pasupuleti, V.; Hernandez, A.V.; Mazzone, P.; Stevenson, J.; Pennell, N.A.; Velcheti, V. Incidence of Pneumonitis With Use of Programmed Death 1 and Programmed Death-Ligand 1 Inhibitors in Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis of Trials. Chest 2017, 152, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Behera, M.; Owonikoko, T.K.; Kamphorst, A.O.; Pakkala, S.; Belani, C.P.; Khuri, F.R.; Ahmed, R.; Ramalingam, S.S. Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer: A systematic analysis of the literature. Cancer 2018, 124, 271–277. [Google Scholar] [CrossRef]
- De Sousa Linhares, A.; Battin, C.; Jutz, S.; Leitner, J.; Hafner, C.; Tobias, J.; Wiedermann, U.; Kundi, M.; Zlabinger, G.J.; Grabmeier-Pfistershammer, K.; et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci. Rep. 2019, 9, 11472. [Google Scholar] [CrossRef]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Mortensen, L.S.; Petersen, J.B.; Busk, M.; Overgaard, J. Imaging hypoxia to improve radiotherapy outcome. Nat. Rev. Clin. Oncol. 2012, 9, 674–687. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Hu, C.J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol. Biol. Cell 2007, 18, 4528–4542. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, Q.; Zhang, Y.; Liu, Z.; Zheng, Z.; Liu, S.; Meng, L.; Xin, Y.; Jiang, X. Targeting hypoxia in the tumor microenvironment: A potential strategy to improve cancer immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 24. [Google Scholar] [CrossRef]
- Pietrobon, V.; Marincola, F.M. Hypoxia and the phenomenon of immune exclusion. J. Transl. Med. 2021, 19, 9. [Google Scholar] [CrossRef]
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Transl. Med. 2007, 117, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell. Mol. Biol. Lett. 2020, 25, 18. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. All roads lead to ATF4. Dev. Cell 2003, 4, 442–444. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. ATF6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell Biol. 2000, 20, 5096–5106. [Google Scholar] [CrossRef]
- Rzymski, T.; Milani, M.; Singleton, D.C.; Harris, A.L. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 2009, 8, 3838–3847. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.P.; Bristow, R.G.; Fyles, A.; Koritzinsky, M.; Milosevic, M.; Wouters, B.G. Hypoxia and Predicting Radiation Response. Semin. Radiat. Oncol. 2015, 25, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Dhani, N.; Fyles, A.; Hedley, D.; Milosevic, M. The Clinical Significance of Hypoxia in Human Cancers. Semin. Nucl. Med. 2015, 45, 110–121. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef]
- Vuillefroy de Silly, R.; Dietrich, P.Y.; Walker, P.R. Hypoxia and antitumor CD8(+) T cells: An incompatible alliance? Oncoimmunology 2016, 5, e1232236. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Lunt, S.J.; Chaudary, N.; Hill, R.P. The tumor microenvironment and metastatic disease. Clin. Exp. Metastasis 2009, 26, 19–34. [Google Scholar] [CrossRef]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential Regulatory Function of Human Dendritic Cells Expressing Indoleamine 2,3-Dioxygenase. Science 2002, 297, 1867. [Google Scholar] [CrossRef] [PubMed]
- Boutard, V.; Havouis, R.; Fouqueray, B.; Philippe, C.; Moulinoux, J.P.; Baud, L. Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. J. Immunol. 1995, 155, 2077–2084. [Google Scholar] [PubMed]
- Staveley-O’Carroll, K.; Sotomayor, E.; Montgomery, J.; Borrello, I.; Hwang, L.; Fein, S.; Pardoll, D.; Levitsky, H. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA 1998, 95, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Maeurer, M.J.; Gollin, S.M.; Martin, D.; Swaney, W.; Bryant, J.; Castelli, C.; Robbins, P.; Parmiani, G.; Storkus, W.J.; Lotze, M.T. Tumor escape from immune recognition: Lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen. J. Clin. Investig. 1996, 98, 1633–1641. [Google Scholar] [CrossRef]
- Garrido, F.; Ruiz-Cabello, F.; Cabrera, T.; Pérez-Villar, J.J.; López-Botet, M.; Duggan-Keen, M.; Stern, P.L. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol. Today 1997, 18, 89–95. [Google Scholar] [CrossRef]
- Johnsen, A.K.; Templeton, D.J.; Sy, M.; Harding, C.V. Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J. Immunol. 1999, 163, 4224–4231. [Google Scholar]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.L.; Smothers, J.; Srinivasan, R.; Hoos, A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2015, 14, 603–622. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Chornoguz, O.; Ecker, C. Regulating the suppressors: Apoptosis and inflammation govern the survival of tumor-induced myeloid-derived suppressor cells (MDSC). Cancer Immunol. Immunother. 2012, 61, 1319–1325. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Sitkovsky, M. Oxygenation to improve cancer vaccines, adoptive cell transfer and blockade of immunological negative regulators. Oncoimmunology 2015, 4, e1052934. [Google Scholar] [CrossRef]
- Corthay, A. How do Regulatory T Cells Work? Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef]
- Ren, L.; Yu, Y.; Wang, L.; Zhu, Z.; Lu, R.; Yao, Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget 2016, 7, 75763–75773. [Google Scholar] [CrossRef]
- Yan, M.; Jene, N.; Byrne, D.; Millar, E.K.; O’Toole, S.A.; McNeil, C.M.; Bates, G.J.; Harris, A.L.; Banham, A.H.; Sutherland, R.L.; et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. BCR 2011, 13, R47. [Google Scholar] [CrossRef]
- Hsu, T.-S.; Lin, Y.-L.; Wang, Y.-A.; Mo, S.-T.; Chi, P.-Y.; Lai, A.C.-Y.; Pan, H.-Y.; Chang, Y.-J.; Lai, M.-Z. HIF-2α is indispensable for regulatory T cell function. Nat. Commun. 2020, 11, 5005. [Google Scholar] [CrossRef]
- Chiu, D.K.-C.; Tse, A.P.-W.; Xu, I.M.-J.; Di Cui, J.; Lai, R.K.-H.; Li, L.L.; Koh, H.-Y.; Tsang, F.H.-C.; Wei, L.L.; Wong, C.-M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef]
- Chiu, D.K.; Xu, I.M.; Lai, R.K.; Tse, A.P.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef]
- Díaz-Bulnes, P.; Saiz, M.L.; López-Larrea, C.; Rodríguez, R.M. Crosstalk Between Hypoxia and ER Stress Response: A Key Regulator of Macrophage Polarization. Front. Immunol. 2019, 10, 2951. [Google Scholar] [CrossRef] [PubMed]
- Labiano, S.; Palazon, A.; Melero, I. Immune Response Regulation in the Tumor Microenvironment by Hypoxia. Semin. Oncol. 2015, 42, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Kawanaka, T.; Kubo, A.; Ikushima, H.; Sano, T.; Takegawa, Y.; Nishitani, H. Prognostic significance of HIF-2alpha expression on tumor infiltrating macrophages in patients with uterine cervical cancer undergoing radiotherapy. J. Med. Investig. 2008, 55, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Giannoudis, A.; Corke, K.P.; Gill, D.; Wells, M.; Ziegler-Heitbrock, L.; Lewis, C.E. Hypoxia-induced gene expression in human macrophages: Implications for ischemic tissues and hypoxia-regulated gene therapy. Am. J. Pathol. 2003, 163, 1233–1243. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage Expression of Hypoxia-Inducible Factor-1α Suppresses T-Cell Function and Promotes Tumor Progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef]
- Ohta, A.; Diwanji, R.; Kini, R.; Subramanian, M.; Ohta, A.; Sitkovsky, M. In vivo T Cell Activation in Lymphoid Tissues is Inhibited in the Oxygen-Poor Microenvironment. Front. Immunol. 2011, 2. [Google Scholar] [CrossRef]
- Thiel, M.; Caldwell, C.C.; Kreth, S.; Kuboki, S.; Chen, P.; Smith, P.; Ohta, A.; Lentsch, A.B.; Lukashev, D.; Sitkovsky, M.V. Targeted Deletion of HIF-1α Gene in T Cells Prevents their Inhibition in Hypoxic Inflamed Tissues and Improves Septic Mice Survival. PLoS ONE 2007, 2, e853. [Google Scholar] [CrossRef]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019, 458, 13–20. [Google Scholar] [CrossRef]
- Messai, Y.; Gad, S.; Noman, M.Z.; Le Teuff, G.; Couve, S.; Janji, B.; Kammerer, S.F.; Rioux-Leclerc, N.; Hasmim, M.; Ferlicot, S.; et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel-Lindau Gene Mutation Status. Eur. Urol. 2016, 70, 623–632. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Negishi, Y.; Shimizu, M.; Takahashi, M.; Ichikawa, M.; Takahashi, H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol. Lett. 2015, 167, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. ImmunoTher. Cancer 2018, 6, 57. [Google Scholar] [CrossRef]
- Casanello, P.; Torres, A.; Sanhueza, F.; Gonzalez, M.; Farias, M.; Gallardo, V.; Pastor-Anglada, M.; San Martin, R.; Sobrevia, L. Equilibrative nucleoside transporter 1 expression is downregulated by hypoxia in human umbilical vein endothelium. Circ. Res. 2005, 97, 16–24. [Google Scholar] [CrossRef]
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 42, 101304. [Google Scholar] [CrossRef] [PubMed]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Mancino, A.; Schioppa, T.; Larghi, P.; Pasqualini, F.; Nebuloni, M.; Chen, I.-H.; Sozzani, S.; Austyn, J.M.; Mantovani, A.; Sica, A. Divergent effects of hypoxia on dendritic cell functions. Blood 2008, 112, 3723–3734. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huang, B.; Shi, J. Fas ligand and lytic granule differentially control cytotoxic dynamics of natural killer cell against cancer target. Oncotarget 2016, 7, 47163–47172. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Hamilton, T.K.; Li, X.; Cotechini, T.; Miles, E.A.; Siemens, D.R.; Graham, C.H. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: Role of nitric oxide. Cancer Res. 2011, 71, 7433–7441. [Google Scholar] [CrossRef]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486. [Google Scholar] [CrossRef]
- Martinez-Zaguilan, R.; Seftor, E.A.; Seftor, R.E.; Chu, Y.W.; Gillies, R.J.; Hendrix, M.J. Acidic pH enhances the invasive behavior of human melanoma cells. Clin. Exp. Metastasis 1996, 14, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, C.; Dai, H.; Hou, J.; Dong, Y.; Cui, X.; Xu, L.; Zhang, M.; Xia, Q. Hypoxia-Inducible Factor-2α Limits Natural Killer T Cell Cytotoxicity in Renal Ischemia/Reperfusion Injury. JASN 2016, 27, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Keck, M.K.; Zuo, Z.; Khattri, A.; Stricker, T.P.; Brown, C.D.; Imanguli, M.; Rieke, D.; Endhardt, K.; Fang, P.; Brägelmann, J.; et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin. Cancer Res. 2015, 21, 870–881. [Google Scholar] [CrossRef]
- Duechler, M.; Peczek, L.; Zuk, K.; Zalesna, I.; Jeziorski, A.; Czyz, M. The heterogeneous immune microenvironment in breast cancer is affected by hypoxia-related genes. Immunobiology 2014, 219, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Zhu, J.-M.; Wang, Y.; Liu, T.-T.; Ding, Y.-B.; Xiao, W.-M.; Lu, G.-T.; Bo, P.; Shen, X.-Z. Intratumor Hypoxia Promotes Immune Tolerance by Inducing Regulatory T Cells via TGF-β1 in Gastric Cancer. PLoS ONE 2013, 8, e63777. [Google Scholar] [CrossRef] [PubMed]
- Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Ioannou, K.; Ziogas, A.C.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br. J. Cancer 2012, 107, 1869–1875. [Google Scholar] [CrossRef]
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front. Immunol. 2012, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Mahiddine, K.; Blaisdell, A.; Ma, S.; Crequer-Grandhomme, A.; Lowell, C.A.; Erlebacher, A. Relief of tumor hypoxia unleashes the tumoricidal potential of neutrophils. J. Clin. Investig. 2019. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2016. [Google Scholar] [CrossRef]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef]
- Skwarski, M.; McGowan, D.R.; Belcher, E.; Di Chiara, F.; Stavroulias, D.; McCole, M.; Derham, J.L.; Chu, K.-Y.; Teoh, E.; Chauhan, J.; et al. Mitochondrial Inhibitor Atovaquone Increases Tumor Oxygenation and Inhibits Hypoxic Gene Expression in Patients with Non–Small Cell Lung Cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Phillips, R.M. Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother. Pharmacol. 2016, 77, 441–457. [Google Scholar] [CrossRef]
- McKeown, S.R.; Cowen, R.L.; Williams, K.J. Bioreductive drugs: From concept to clinic. Clin. Oncol. 2007, 19, 427–442. [Google Scholar] [CrossRef]
- Guise, C.P.; Mowday, A.M.; Ashoorzadeh, A.; Yuan, R.; Lin, W.H.; Wu, D.H.; Smaill, J.B.; Patterson, A.V.; Ding, K. Bioreductive prodrugs as cancer therapeutics: Targeting tumor hypoxia. Chin. J. Cancer 2014, 33, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Baran, N.; Konopleva, M. Molecular Pathways: Hypoxia-Activated Prodrugs in Cancer Therapy. Clin. Cancer Res. 2017, 23, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Evans, J.W.; Bhupathi, D.; Banica, M.; Lan, L.; Lorente, G.; Duan, J.-X.; Cai, X.; Mowday, A.M.; Guise, C.P.; et al. Molecular and Cellular Pharmacology of the Hypoxia-Activated Prodrug TH-302. Mol. Cancer Ther. 2012, 11, 740. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.D.; Liu, Q.; Wang, J.; Ahluwalia, D.; Ferraro, D.; Wang, Y.; Duan, J.-X.; Ammons, W.S.; Curd, J.G.; Matteucci, M.D.; et al. Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin. Cancer Res. 2012, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sun, J.D.; Wang, J.; Ahluwalia, D.; Baker, A.F.; Cranmer, L.D.; Ferraro, D.; Wang, Y.; Duan, J.X.; Ammons, W.S.; et al. TH-302, a hypoxia-activated prodrug with broad in vivo preclinical combination therapy efficacy: Optimization of dosing regimens and schedules. Cancer Chemother. Pharmacol. 2012, 69, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Cutsem, E.V.; Lenz, H.-J.; Furuse, J.; Tabernero, J.; Heinemann, V.; Ioka, T.; Bazin, I.; Ueno, M.; Csõszi, T.; Wasan, H.; et al. MAESTRO: A randomized, double-blind phase III study of evofosfamide (Evo) in combination with gemcitabine (Gem) in previously untreated patients (pts) with metastatic or locally advanced unresectable pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2016, 34, 4007. [Google Scholar] [CrossRef]
- Spiegelberg, L.; Houben, R.; Niemans, R.; de Ruysscher, D.; Yaromina, A.; Theys, J.; Guise, C.P.; Smaill, J.B.; Patterson, A.V.; Lambin, P.; et al. Hypoxia-activated prodrugs and (lack of) clinical progress: The need for hypoxia-based biomarker patient selection in phase III clinical trials. Clin. Transl. Radiat. Oncol. 2019, 15, 62–69. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Jamieson, S.M.F.; Tsai, P.; Kondratyev, M.K.; Budhani, P.; Liu, A.; Senzer, N.N.; Chiorean, E.G.; Jalal, S.I.; Nemunaitis, J.J.; Kee, D.; et al. Evofosfamide for the treatment of human papillomavirus-negative head and neck squamous cell carcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Hegde, A.; Jayaprakash, P.; Couillault, C.A.; Piha-Paul, S.; Karp, D.; Rodon, J.; Pant, S.; Fu, S.; Ileana Dumbrava, E.E.; Yap, T.A.; et al. A Phase I Dose Escalation Study to Evaluate the Safety and Tolerability of Evofosfamide in Combination with Ipilimumab in Advanced Solid Malignancies. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- ImmunoGenesis: Acquisition Strengthens “Cold” Tumor Targeting Pipeline. Available online: https://www.globenewswire.com/news-release/2020/12/09/2142162/0/en/ImmunoGenesis-Acquisition-Strengthens-Cold-Tumor-Targeting-Pipeline.html (accessed on 30 March 2021).
- Lu, G.-L.; Ashoorzadeh, A.; Anderson, R.F.; Patterson, A.V.; Smaill, J.B. Synthesis of substituted 5-bromomethyl-4-nitroimidazoles and use for the preparation of the hypoxia-selective multikinase inhibitor SN29966. Tetrahedron 2013, 69, 9130–9138. [Google Scholar] [CrossRef]
- Smaill, J.B.; Jaiswal, J.; Abbattista, M.; Lu, G.-L.; Anderson, R.F.; Ashoorzadeh, A.; Denny, W.A.; Donate, F.; Hsu, H.-L.; Lee, H.H.; et al. Abstract A247: Mechanism of action of the hypoxia-activated irreversible pan-HER inhibitor SN29966. Mol. Cancer Ther. 2011, 10, A247. [Google Scholar] [CrossRef]
- Estrada-Bernal, A.; Le, A.T.; Doak, A.E.; Tirunagaru, V.G.; Silva, S.; Bull, M.R.; Smaill, J.B.; Patterson, A.V.; Kim, C.; Liu, S.V.; et al. Tarloxotinib Is a Hypoxia-Activated Pan-HER Kinase Inhibitor Active Against a Broad Range of HER-Family Oncogenes. Clin. Cancer Res. 2021, 27, 1463. [Google Scholar] [CrossRef] [PubMed]
- Jackson, V.; Silva, S.; Abbattista, M.; Guise, C.; Bull, M.; Ashoorzadeh, A.; Hart, C.; Pearce, T.; Smaill, J.; Patterson, A.V. Abstract A66: Preclinical rationale for the ongoing Phase 2 study of the hypoxia-activated EGFR-TKI tarloxotinib bromide (TH-4000) in patients with advanced squamous cell carcinoma of the head and neck (SCCHN) or skin (SCCS). Mol. Cancer Ther. 2015, 14, A66. [Google Scholar] [CrossRef]
- Silva, S.; Jackson, V.; Guise, C.; Abbattista, M.; Bull, M.; Grey, A.; Anderson, R.; Ashoorzadeh, A.; Hart, C.; Pearce, T.; et al. Abstract A67: Preclinical efficacy of tarloxotinib bromide (TH-4000), a hypoxia-activated EGFR/HER2 inhibitor: Rationale for clinical evaluation in EGFR mutant, T790M-negative NSCLC following progression on EGFR-TKI therapy. Mol. Cancer Ther. 2015, 14, A67. [Google Scholar] [CrossRef]
- Liu, S.V.; Villaruz, L.C.; Lee, V.; Zhu, V.W.; Baik, C.; Sacher, A.; McCoach, C.; Nguyen, D.; Li, J.Y.C.; Pacheco, J.M.; et al. LBA61 First analysis of RAIN-701: Study of tarloxotinib in patients with non-small cell lung cancer (NSCLC) EGFR Exon 20 insertion, HER2-activating mutations & other solid tumours with NRG1/ERBB gene fusions. Ann. Oncol. 2020, 31, S1189. [Google Scholar] [CrossRef]
- Fu, Z.; Farrand, K.; Guise, C.; Smaill, J.; Hermans, I.; Patterson, A. Targeting tumour hypoxia with tarloxotinib improves the therapeutic efficacy of checkpoint blockade. In Proceedings of the 30th EORTC-NCI-AACR Symposium “Molecular Targets and Cancer Therapeutics”, Dublin, Ireland, 13–16 November 2018. [Google Scholar]
- Patterson, A.V.; Hermans, I.F.; Smaill, J.B.; Fu, Z. Combination of a Kinase Inhibitor and an Immunotherapeutic Agent, Compositions and Methods Comprising the Same. PCT. International Patent Application WO 2,020,084,347A2, 30 April 2020. [Google Scholar]
- Guise, C.P.; Abbattista, M.R.; Singleton, R.S.; Holford, S.D.; Connolly, J.; Dachs, G.U.; Fox, S.B.; Pollock, R.; Harvey, J.; Guilford, P.; et al. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010, 70, 1573–1584. [Google Scholar] [CrossRef]
- Patterson, A.V.; Ferry, D.M.; Edmunds, S.J.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef] [PubMed]
- Van der Wiel, A.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A.; Lieuwes, N.; Biemans, R.; Lin, X.; et al. Selectively Targeting Tumor Hypoxia with the Hypoxia-Activated Prodrug CP-506. Res. Square 2021. [Google Scholar] [CrossRef]
- Palmer, B.D.; Wilson, W.R.; Atwell, G.J.; Schultz, D.; Xu, X.Z.; Denny, W.A. Hypoxia-selective antitumor agents. 9. Structure-activity relationships for hypoxia-selective cytotoxicity among analogues of 5-[N,N-bis(2-chloroethyl)amino]-2,4-dinitrobenzamide. J. Med. Chem. 1994, 37, 2175–2184. [Google Scholar] [CrossRef]
- Siim, B.G.; Denny, W.A.; Wilson, W.R. Nitro reduction as an electronic switch for bioreductive drug activation. Oncol. Res. 1997, 9, 357–369. [Google Scholar] [PubMed]
- Bergstralh, D.T.; Sekelsky, J. Interstrand crosslink repair: Can XPF-ERCC1 be let off the hook? TIG 2008, 24, 70–76. [Google Scholar] [CrossRef]
- Marcus, D.; Van der Wiel, A.; Biemans, R.; Lieuwes, N.; Heyerick, A.; Smaill, J.; Patterson, A.; Theys, J.; Yaromina, A.; Lambin, P.; et al. OC-0203: Eliminating tumour hypoxia to improve the impact of immunotherapy. Radiother. Oncol. 2020, 152, S102. [Google Scholar] [CrossRef]
- Kheshtchin, N.; Arab, S.; Ajami, M.; Mirzaei, R.; Ashourpour, M.; Mousavi, N.; Khosravianfar, N.; Jadidi-Niaragh, F.; Namdar, A.; Noorbakhsh, F.; et al. Inhibition of HIF-1alpha enhances anti-tumor effects of dendritic cell-based vaccination in a mouse model of breast cancer. Cancer Immunol. Immunother. 2016, 65, 1159–1167. [Google Scholar] [CrossRef]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Phase II study of the oral HIF-2α inhibitor MK-6482 for Von Hippel-Lindau disease–associated renal cell carcinoma. J. Clin. Oncol. 2020, 38, 5003. [Google Scholar] [CrossRef]
- Courtney, K.D.; Ma, Y.; Diaz de Leon, A.; Christie, A.; Xie, Z.; Woolford, L.; Singla, N.; Joyce, A.; Hill, H.; Madhuranthakam, A.J.; et al. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 793. [Google Scholar] [CrossRef]
- Bui, M.H.; Seligson, D.; Han, K.R.; Pantuck, A.J.; Dorey, F.J.; Huang, Y.; Horvath, S.; Leibovich, B.C.; Chopra, S.; Liao, S.Y.; et al. Carbonic anhydrase IX is an independent predictor of survival in advanced renal clear cell carcinoma: Implications for prognosis and therapy. Clin. Cancer Res. 2003, 9, 802–811. [Google Scholar]
- Chafe, S.C.; McDonald, P.C.; Saberi, S.; Nemirovsky, O.; Venkateswaran, G.; Burugu, S.; Gao, D.; Delaidelli, A.; Kyle, A.H.; Baker, J.H.E.; et al. Targeting Hypoxia-Induced Carbonic Anhydrase IX Enhances Immune-Checkpoint Blockade Locally and Systemically. Cancer Immunol. Res. 2019, 7, 1064. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-R.; Deng, W.-W.; Liu, J.-F.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. Blockade of adenosine A2A receptor enhances CD8(+) T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 99. [Google Scholar] [CrossRef]
- Vile, R.; Ando, D.; Kirn, D. The oncolytic virotherapy treatment platform for cancer: Unique biological and biosafety points to consider. Cancer Gene Ther. 2002, 9, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Senzer, N.N.; Stephenson, J., Jr.; Loesch, D.; Burroughs, K.D.; Reddy, P.S.; Hann, C.L.; Hallenbeck, P.L. Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin. Cancer Res. 2011, 17, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, J.; Kurozumi, K.; Chiocca, E.A.; Kaur, B. Oncolytic viruses driven by tumor-specific promoters. Curr. Cancer Drug Targets 2007, 7, 181–189. [Google Scholar] [CrossRef]
- Dmitriev, I.; Krasnykh, V.; Miller, C.R.; Wang, M.; Kashentseva, E.; Mikheeva, G.; Belousova, N.; Curiel, D.T. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J. Virol. 1998, 72, 9706–9713. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Bines, S.D. OPTIM trial: A Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol. 2010, 6, 941–949. [Google Scholar] [CrossRef]
- Pipiya, T.; Sauthoff, H.; Huang, Y.Q.; Chang, B.; Cheng, J.; Heitner, S.; Chen, S.; Rom, W.N.; Hay, J.G. Hypoxia reduces adenoviral replication in cancer cells by downregulation of viral protein expression. Gene Ther. 2005, 12, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Aghi, M.K.; Liu, T.C.; Rabkin, S.; Martuza, R.L. Hypoxia enhances the replication of oncolytic herpes simplex virus. Mol. Ther. 2009, 17, 51–56. [Google Scholar] [CrossRef]
- Connor, J.H.; Naczki, C.; Koumenis, C.; Lyles, D.S. Replication and cytopathic effect of oncolytic vesicular stomatitis virus in hypoxic tumor cells in vitro and in vivo. J. Virol. 2004, 78, 8960–8970. [Google Scholar] [CrossRef] [PubMed]
- Post, D.E.; Van Meir, E.G. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene 2003, 22, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.C.; Li, D.; Bai, S.Y.; Syddall, S.P.; Smaill, J.B.; Shen, Y.; Denny, W.A.; Wilson, W.R.; Patterson, A.V. The nitroreductase prodrug SN 28343 enhances the potency of systemically administered armed oncolytic adenovirus ONYX-411NTR. Cancer Gene Ther. 2007, 14, 953–967. [Google Scholar] [CrossRef]
- Patterson, A.V.; Williams, K.J.; Cowen, R.L.; Jaffar, M.; Telfer, B.A.; Saunders, M.; Airley, R.; Honess, D.; Van der Kogel, A.J.; Wolf, C.R.; et al. Oxygen-sensitive enzyme-prodrug gene therapy for the eradication of radiation-resistant solid tumours. Gene Ther. 2002, 9, 946–954. [Google Scholar] [CrossRef]
- Mowday, A.M.; Guise, C.P.; Ackerley, D.F.; Minton, N.P.; Lambin, P.; Dubois, L.J.; Theys, J.; Smaill, J.B.; Patterson, A.V. Advancing Clostridia to Clinical Trial: Past Lessons and Recent Progress. Cancers 2016, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.O.; Coleman, P.N. Infection of a secondary carcinoma by Salmonella montevideo. Br. Med. J. 1952, 1, 1116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, W.; Wang, Y.; Qin, M.; Zhang, X.; Zhang, Z.; Sun, X.; Gu, Z. Bacteria-Driven Hypoxia Targeting for Combined Biotherapy and Photothermal Therapy. ACS Nano 2018, 12, 5995–6005. [Google Scholar] [CrossRef] [PubMed]
- DeClue, A.E.; Axiak-Bechtel, S.M.; Zhang, Y.; Saha, S.; Zhang, L.; Tung, D.; Bryan, J.N. Immune response to C. novyi-NT immunotherapy. Vet. Res. 2018, 49, 38. [Google Scholar] [CrossRef]
- Chabi, S.; Uzan, B.; Naguibneva, I.; Rucci, J.; Fahy, L.; Calvo, J.; Arcangeli, M.-L.; Mazurier, F.; Pflumio, F.; Haddad, R. Hypoxia Regulates Lymphoid Development of Human Hematopoietic Progenitors. Cell Rep. 2019, 29, 2307–2320.e2306. [Google Scholar] [CrossRef] [PubMed]
- Hale, L.P.; Braun, R.D.; Gwinn, W.M.; Greer, P.K.; Dewhirst, M.W. Hypoxia in the thymus: Role of oxygen tension in thymocyte survival. Am. J. Physiol.-Heart Circ. Physiol. 2002, 282, H1467–H1477. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.M.; Menezes, A.N.; Ibrahim, M.; Archer, L.; Lal, N.; Bagnall, C.J.; von Zeidler, S.V.; Valentine, H.R.; Spruce, R.J.; Batis, N.; et al. Development and Validation of a Combined Hypoxia and Immune Prognostic Classifier for Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 5315–5328. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combination Strategy | Examples | Phase | Clinical Trial Identifier |
|---|---|---|---|
| Chemotherapy | Pembrolizumab + usual chemotherapy for NSCLC | III | NCT04267848 |
| Atezolizumab + combination chemotherapy | III | NCT02912559 | |
| Nivolumab + carboplatin and paclitaxel | III | NCT04444921 | |
| Radiotherapy | Pembrolizumab + stereotactic body radiation therapy | III | NCT03867175 |
| Atezolizumab + radiation therapy | II/III | NCT04402788 | |
| Sintilimab + stereotactic body radiation therapy | II/III | NCT04167293 | |
| Multiple ICI agents | Pembrolizumab + Ipilimumab | II | NCT03873818 |
| Tiragolumab + Atezolizumab | I | NCT02794571 | |
| Molecular targets | Nivolumab + Lenvatinib | II | NCT03841201 |
| Atezolizumab + Vemurafenib and/or Cobimetinib | II | NCT04722575 | |
| Pembrolizumab + Idelalisib | I/II | NCT03257722 | |
| Immunostimulatory agents | Nivolumab + CMP-001 (TLR-9 agonist) | II | NCT04401995 |
| Nivolumab (+ Ipilimumab) + OTSGC-A24 (peptide vaccine) | I | NCT03784040 | |
| Tislelizumab + BGB-A445 (anti-OX40 agonist) | I | NCT04215978 | |
| Replicating biological vectors | Nivolumab + Talimogene laherparepvec Pembrolizumab + Clostridium novyi | III | NCT04330430NCT03435952 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Z.; Mowday, A.M.; Smaill, J.B.; Hermans, I.F.; Patterson, A.V. Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy. Cells 2021, 10, 1006. https://doi.org/10.3390/cells10051006
Fu Z, Mowday AM, Smaill JB, Hermans IF, Patterson AV. Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy. Cells. 2021; 10(5):1006. https://doi.org/10.3390/cells10051006
Chicago/Turabian StyleFu, Zhe, Alexandra M. Mowday, Jeff B. Smaill, Ian F. Hermans, and Adam V. Patterson. 2021. "Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy" Cells 10, no. 5: 1006. https://doi.org/10.3390/cells10051006
APA StyleFu, Z., Mowday, A. M., Smaill, J. B., Hermans, I. F., & Patterson, A. V. (2021). Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy. Cells, 10(5), 1006. https://doi.org/10.3390/cells10051006