
The Role of RhoH in TCR Signalling and Its Involvement in Diseases

 , and
, and
Abstract

1. Introduction
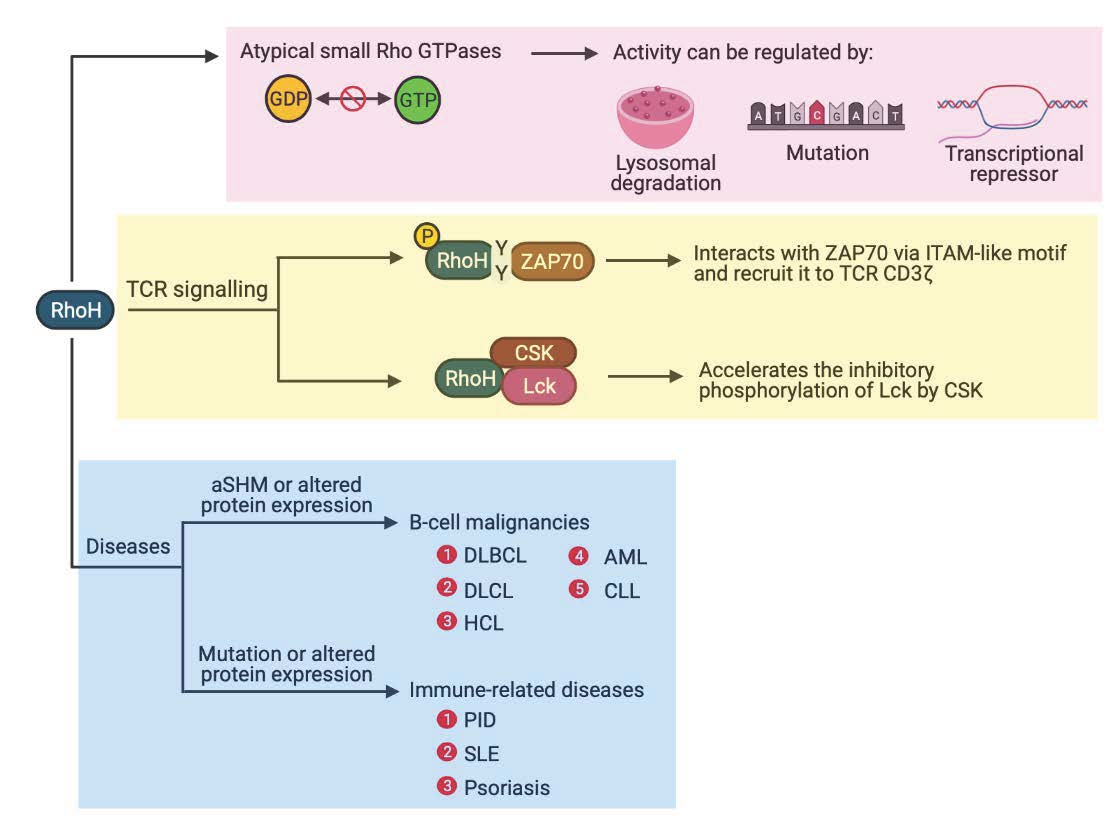
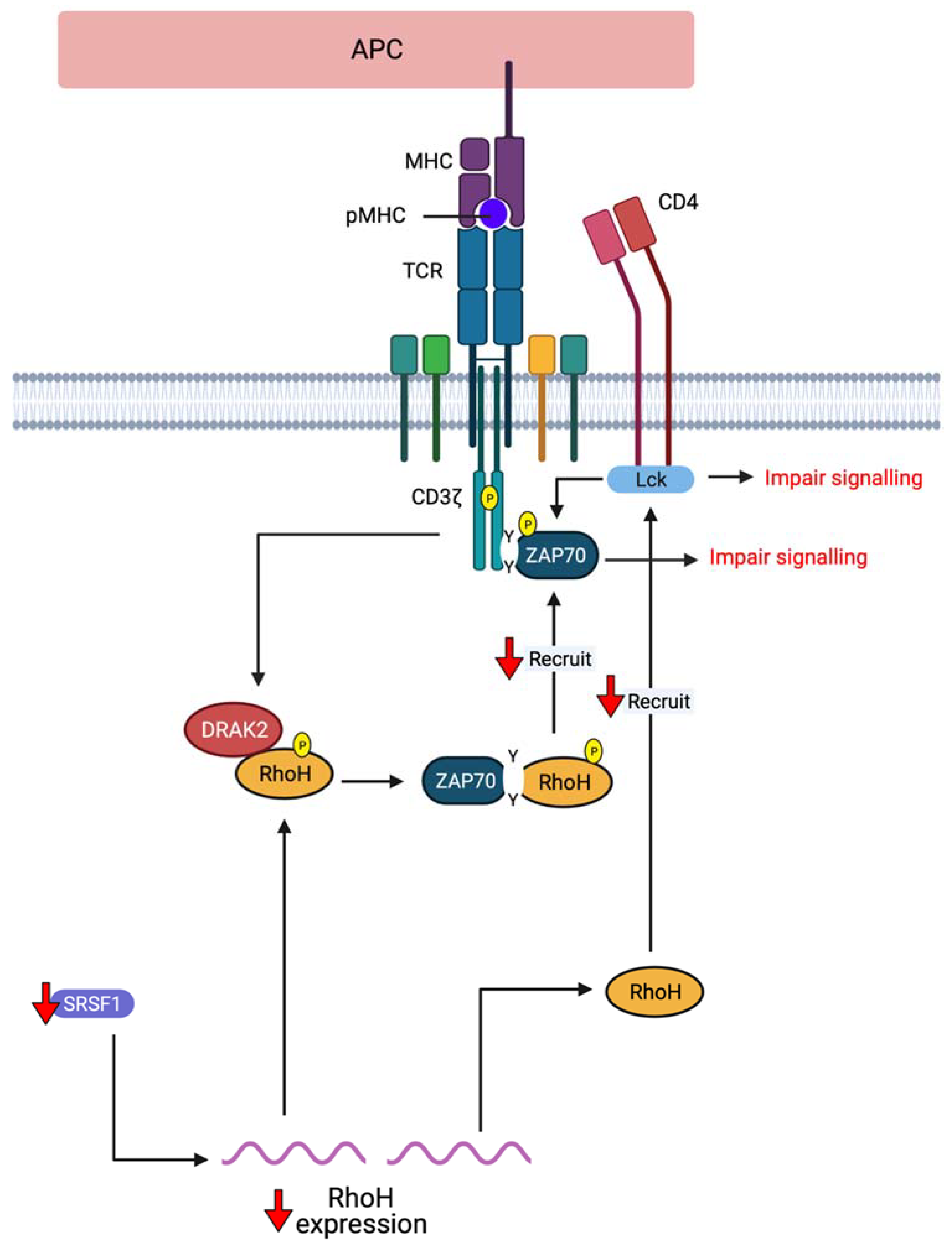
2. RhoH, An Atypical Rho Family Small GTPase
3. Deregulation of RhoH in Diseases
3.1. RhoH in B-Cell Malignancies
3.2. Immune-Related Diseases
3.2.1. Primary Immunodeficiencies (PIDs)
3.2.2. Autoimmune-Related Diseases
4. RhoH as a Therapeutic Target
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TCR | T cell receptor |
| CMA | chaperone-mediated autophagy |
| LAT | linker of activated T cells |
| DLBCL | diffuse large B-cell lymphoma |
| aSHM | aberrant somatic hypermutation |
| AID | activation-induced deaminase |
| CLL | Chronic lymphocytic leukaemia |
| FL | follicular lymphoma |
| CSK | C-terminal Src kinase |
| HCL | hairy cell leukaemia |
| AML | acute myeloid leukaemia |
| PIDs | primary immunodeficiencies |
| SLE | systemic lupus erythematosus |
| BCR | B cell receptor |
| LCK | lymphocyte-specific protein tyrosine kinase |
| SRSF1 | serine/arginine-rich splicing factor 1 |
| GEF | guanine nucleotide exchange factors |
| GDI | Guanosine nucleotide dissociation inhibitor |
References
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Fransson, Å.; Ruusala, A.; Aspenström, P. Atypical Rho GTPases Have Roles in Mitochondrial Homeostasis and Apoptosis. J. Biol. Chem. 2003, 278, 6495–6502. [Google Scholar] [CrossRef] [PubMed]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho Family of Ras-Like GTPases in Eukaryotes. Mol. Biol. Evol. 2006, 24, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Aspenström, P.; Ruusala, A.; Pacholsky, D. Taking Rho GTPases to the next level: The cellular functions of atypical Rho GTPases. Exp. Cell Res. 2007, 313, 3673–3679. [Google Scholar] [CrossRef]
- Valencia, A.; Chardin, P.; Wittinghofer, A.; Sander, C. The ras protein family: Evolutionary tree and role of conserved amino acids. Biochemistry 1991, 30, 4637–4648. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef]
- Citalán-Madrid, A.F.; García-Ponce, A.; Vargas-Robles, H.; Betanzos, A.; Schnoor, M. Small GTPases of the Ras superfamily regulate intestinal epithelial homeostasis and barrier function via common and unique mechanisms. Tissue Barriers 2013, 1, e26938. [Google Scholar] [CrossRef]
- Michaelson, D.; Silletti, J.; Murphy, G.; D’Eustachio, P.; Rush, M.; Philips, M.R. Differential localization of Rho GTPases in live cells: Regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 2001, 152, 111–126. [Google Scholar] [CrossRef]
- Choy, E.; Chiu, V.K.; Silletti, J.; Feoktistov, M.; Morimoto, T.; Michaelson, D.; Philips, M.R. Endomembrane Trafficking of Ras: The CAAX Motif Targets Proteins to the ER and Golgi. Cell 1999, 98, 69–80. [Google Scholar] [CrossRef]
- Adamson, P.; Paterson, H.F.; Hall, A. Intracellular localization of the P21rho proteins. J. Cell Biol. 1992, 119, 617–627. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed]
- Aspenström, P. Fast-cycling Rho GTPases. Small Gtpases 2020, 11, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Dovas, A.; Couchman, J.R. RhoGDI: Multiple functions in the regulation of Rho family GTPase activities. Biochem. J. 2005, 390, 1–9. [Google Scholar] [CrossRef]
- Jaiswal, M.; Fansa, E.K.; Dvorský, R.; Ahmadian, M.R.; Communication, S.; Jaiswal, M.; Fansa, E.K.; Dvorsky, R.; Ahmadian, M.R. New insight into the molecular switch mechanism of human Rho family proteins: Shifting a paradigm. Biol. Chem. 2012, 394, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Berzat, A.C.; Cox, A.D.; Der, C.J. Atypical Mechanism of Regulation of the Wrch-1 Rho Family Small GTPase. Curr. Biol. 2004, 14, 2052–2056. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Berzat, A.C.; Chenette, E.J.; Cox, A.D.; Der, C.J.B.T.-M. Biochemical Analyses of the Wrch Atypical Rho Family GTPases. Regul. Eff. Small Gtpases Rho Fam. 2006, 406, 11–26. [Google Scholar]
- Lin, R.; Bagrodia, S.; Cerione, R.; Manor, D. A novel Cdc42Hs mutant induces cellular transformation. Curr. Biol. 1997, 7, 794–797. [Google Scholar] [CrossRef]
- Aspenström, P. Activated Rho GTPases in Cancer-The Beginning of a New Paradigm. Int. J. Mol. Sci. 2018, 19, 3949. [Google Scholar] [CrossRef]
- Prive, G.G.; Milburn, M.V.; Tong, L.; de Vos, A.M.; Yamaizumi, Z.; Nishimura, S.; Kim, S.H. X-ray crystal structures of transforming p21 ras mutants suggest a transition-state stabilization mechanism for GTP hydrolysis. Proc. Natl. Acad. Sci. USA 1992, 89, 3649–3653. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Maldonado, C.; Zimmer, Y.; Medová, M. A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front. Oncol. 2019, 9, 1088. [Google Scholar] [CrossRef]
- Fueller, F.; Kubatzky, K.F. The small GTPase RhoH is an atypical regulator of haematopoietic cells. Cell Commun. Signal. 2008, 6, 6. [Google Scholar] [CrossRef]
- Chae, H.-D.; Siefring, J.E.; Hildeman, D.A.; Gu, Y.; Williams, D.A. RhoH regulates subcellular localization of ZAP-70 and Lck in T cell receptor signaling. PLoS ONE 2010, 5, e13970. [Google Scholar] [CrossRef]
- Tamehiro, N.; Oda, H.; Shirai, M.; Suzuki, H. Overexpression of RhoH Permits to Bypass the Pre-TCR Checkpoint. PLoS ONE 2015, 10, e0131047. [Google Scholar] [CrossRef]
- Sunshine, H.; Iruela-Arispe, M.L. Membrane lipids and cell signaling. Curr. Opin. Lipidol. 2017, 28, 408–413. [Google Scholar] [CrossRef]
- Troeger, A.; Chae, H.-D.; Senturk, M.; Wood, J.; Williams, D.A. A Unique Carboxyl-terminal Insert Domain in the Hematopoietic-specific, GTPase-deficient Rho GTPase RhoH Regulates Post-translational Processing. J. Biol. Chem. 2013, 288, 36451–36462. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bu, X.; Lu, B.; Avraham, H.; Flavell, R.A.; Lim, B. The Hematopoiesis-Specific GTP-Binding Protein RhoH Is GTPase Deficient and Modulates Activities of Other Rho GTPases by an Inhibitory Function. Mol. Cell. Biol. 2002, 22, 1158–1171. [Google Scholar] [CrossRef]
- Horiguchi, H.; Ciuculescu, M.F.; Troeger, A.; Xu, H.; Brendel, C.; Williams, D.A. Deletion of Murine Rhoh induces More Aggressive Diffuse Large B Cell Lymphoma (DLBCL) Via Interaction with Kaiso and Regulation of BCL-6 Expression. Blood 2018, 132, 1574. [Google Scholar] [CrossRef]
- Gu, Y.; Chae, H.-D.; Siefring, J.E.; Jasti, A.C.; Hildeman, D.A.; Williams, D.A. RhoH GTPase recruits and activates Zap70 required for T cell receptor signaling and thymocyte development. Nat. Immunol. 2006, 7, 1182. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Tajadura-Ortega, V.; Garg, R.; Allen, R.; Owczarek, C.; Bright, M.D.; Kean, S.; Mohd-Noor, A.; Grigoriadis, A.; Elston, T.C.; Hahn, K.M.; et al. An RNAi screen of Rho signalling networks identifies RhoH as a regulator of Rac1 in prostate cancer cell migration. Bmc Biol. 2018, 16, 1–20. [Google Scholar] [CrossRef]
- Dorn, T.; Kuhn, U.; Bungartz, G.; Stiller, S.; Bauer, M.; Ellwart, J.; Peters, T.; Scharffetter-Kochanek, K.; Semmrich, M.; Laschinger, M.; et al. RhoH is important for positive thymocyte selection and T-cell receptor signaling. Blood 2007, 109, 2346–2355. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aguilera, A.; Rattmann, I.; Drew, D.Z.; Müller, L.U.W.; Summey, V.; Lucas, D.M.; Byrd, J.C.; Croce, C.M.; Gu, Y.; Cancelas, J.A.; et al. Involvement of RhoH GTPase in the development of B-cell chronic lymphocytic leukemia. Leukemia 2010, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Tybulewicz, V.L.J.; Henderson, R.B. Rho family GTPases and their regulators in lymphocytes. Nat. Rev. Immunol. 2009, 9, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Kay, L. Characterisation of Atypical Human GTPases: Elucidation of Molecular Functions and Interactors. Ph.D. Thesis, Northumbria University, Newcastle, UK, 2016. [Google Scholar]
- Yablonski, D. Bridging the Gap: Modulatory Roles of the Grb2-Family Adaptor, Gads, in Cellular and Allergic Immune Responses. Front. Immunol. 2019, 10, 1704. [Google Scholar] [CrossRef]
- Wang, H.; Zeng, X.; Fan, Z.; Lim, B. RhoH modulates pre-TCR and TCR signalling by regulating LCK. Cell. Signal. 2011, 23, 249–258. [Google Scholar] [CrossRef]
- Preudhomme, C.; Roumier, C.; Hildebrand, M.P.; Dallery-Prudhomme, E.; Lantoine, D.; Laï, J.L.; Daudignon, A.; Adenis, C.; Bauters, F.; Fenaux, P.; et al. Nonrandom 4p13 rearrangements of the RhoH/TTF gene, encoding a GTP-binding protein, in non-Hodgkin’s lymphoma and multiple myeloma. Oncogene 2000, 19, 2023–2032. [Google Scholar] [CrossRef]
- Hiraga, J.; Katsumi, A.; Iwasaki, T.; Abe, A.; Kiyoi, H.; Matsushita, T.; Kinoshita, T.; Naoe, T. Prognostic analysis of aberrant somatic hypermutation of RhoH gene in diffuse large B cell lymphoma. Leukemia 2007, 21, 1846–1847. [Google Scholar] [CrossRef]
- Deutsch, A.J.A.; Frühwirth, M.; Aigelsreiter, A.; Cerroni, L.; Neumeister, P. Primary Cutaneous Marginal Zone B-Cell Lymphomas Are Targeted by Aberrant Somatic Hypermutation. J. Investig. Dermatol. 2009, 129, 476–479. [Google Scholar] [CrossRef]
- Voena, C.; Chiarle, R. RHO Family GTPases in the Biology of Lymphoma. Cells 2019, 8, 646. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.K.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346. [Google Scholar] [CrossRef]
- Reiniger, L.; Bödör, C.; Bognár, Á.; Balogh, Z.; Csomor, J.; Szepesi, Á.; Kopper, L.; Matolcsy, A. Richter’s and prolymphocytic transformation of chronic lymphocytic leukemia are associated with high mRNA expression of activation-induced cytidine deaminase and aberrant somatic hypermutation. Leukemia 2006, 20, 1089–1095. [Google Scholar] [CrossRef]
- Rossi, D.; Berra, E.; Cerri, M.; Deambrogi, C.; Barbieri, C.; Franceschetti, S.; Lunghi, M.; Conconi, A.; Paulli, M.; Matolcsy, A.; et al. Aberrant somatic hypermutation in transformation of follicular lymphoma and chronic lymphocytic leukemia to diffuse large B-cell lymphoma. Haematologica 2006, 91, 1405–1409. [Google Scholar]
- Cattoretti, G.; Pasqualucci, L.; Ballon, G.; Tam, W.; Nandula, S.V.; Shen, Q.; Mo, T.; Murty, V.V.; Dalla-Favera, R. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell 2005, 7, 445–455. [Google Scholar] [CrossRef]
- Gruber, T.A.; Chang, M.S.; Sposto, R.; Müschen, M. Activation-induced cytidine deaminase accelerates clonal evolution in BCR-ABL1-driven B-cell lineage acute lymphoblastic leukemia. Cancer Res. 2010, 70, 7411–7420. [Google Scholar] [CrossRef]
- Perona, R.; Montaner, S.; Saniger, L.; Sánchez-Pérez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Gastonguay, A.; Berg, T.; Hauser, A.D.; Schuld, N.; Lorimer, E.; Williams, C.L. The role of Rac1 in the regulation of NF-κB activity, cell proliferation, and cell migration in non-small cell lung carcinoma. Cancer Biol. Ther. 2012, 13, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Tergaonkar, V. Rho protein GTPases and their interactions with NFκB: Crossroads of inflammation and matrix biology. Biosci. Rep. 2014, 34, e00115. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Katsumi, A.; Kiyoi, H.; Tanizaki, R.; Ishikawa, Y.; Ozeki, K.; Kobayashi, M.; Abe, A.; Matsushita, T.; Watanabe, T.; et al. Prognostic implication and biological roles of RhoH in acute myeloid leukaemia. Eur. J. Haematol. 2008, 81, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-R.; Byeon, Y.; Kim, D.; Park, S.-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Lambe, T.; Crawford, G.; Johnson, A.L.; Crockford, T.L.; Bouriez-Jones, T.; Smyth, A.M.; Pham, T.H.M.; Zhang, Q.; Freeman, A.F.; Cyster, J.G.; et al. DOCK8 is essential for T-cell survival and the maintenance of CD8+ T-cell memory. Eur. J. Immunol. 2011, 41, 3423–3435. [Google Scholar] [CrossRef]
- Rodríguez-Fdez, S.; Bustelo, X.R. The Vav GEF Family: An Evolutionary and Functional Perspective. Cells 2019, 8, 465. [Google Scholar] [CrossRef]
- Mino, A.; Troeger, A.; Brendel, C.; Cantor, A.; Harris, C.; Ciuculescu, M.F.; Williams, D.A. RhoH participates in a multi-protein complex with the zinc finger protein kaiso that regulates both cytoskeletal structures and chemokine-induced T cells. Small Gtpases 2018, 9, 260–273. [Google Scholar] [CrossRef]
- Antoni, A.; Ray, C.; Kohn, R.; Andreyko, D.; Levine, J. Analysis of the misregulation of RhoA and RhoH in autoimmune mice. (HUM7P.302). J. Immunol. 2014, 192. [Google Scholar]
- Stoeckle, C.; Geering, B.; Yousefi, S.; Rožman, S.; Andina, N.; Benarafa, C.; Simon, H.-U. RhoH is a negative regulator of eosinophilopoiesis. Cell Death Differ. 2016, 23, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Salloum, G.; Jaafar, L.; El-Sibai, M. Rho A and Rac1: Antagonists moving forward. Tissue Cell 2020, 65, 101364. [Google Scholar] [CrossRef]
- Galiègue-Zouitina, S.; Delestré, L.; Dupont, C.; Troussard, X.; Shelley, C.S. Underexpression of RhoH in Hairy Cell Leukemia. Cancer Res. 2008, 68, 4531–4540. [Google Scholar] [CrossRef] [PubMed]
- Troeger, A.; Johnson, A.J.; Wood, J.; Blum, W.G.; Andritsos, L.A.; Byrd, J.C.; Williams, D.A. RhoH is critical for cell-microenvironment interactions in chronic lymphocytic leukemia in mice and humans. Blood 2012, 119, 4708–4718. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Mende, J.; Geering, B.; Yousefi, S.; Simon, H.-U. Lysosomal degradation of RhoH protein upon antigen receptor activation in T but not B cells. Eur. J. Immunol. 2010, 40, 525–529. [Google Scholar] [CrossRef]
- Delestré, L.; Berthon, C.; Quesnel, B.; Figeac, M.; Kerckaert, J.-P.; Galiègue-Zouitina, S.; Shelley, C.S. Repression of the RHOH gene by JunD. Biochem. J. 2011, 437, 75–88. [Google Scholar] [CrossRef]
- Gazon, H.; Lemasson, I.; Polakowski, N.; Césaire, R.; Matsuoka, M.; Barbeau, B.; Mesnard, J.-M.; Peloponese, J.-M. Human T-Cell Leukemia Virus Type 1 (HTLV-1) bZIP Factor Requires Cellular Transcription Factor JunD To Upregulate HTLV-1 Antisense Transcription from the 3′ Long Terminal Repeat. J. Virol. 2012, 86, 9070–9078. [Google Scholar] [CrossRef] [PubMed]
- Galiègue-Zouitina, S.; Fu, Q.; Carton-Latreche, C.; Poret, N.; Cheok, M.; Leprêtre, F.; Figeac, M.; Quesnel, B.; El Bouazzati, H.; Shelley, C.S. Bimodal expression of RHOH during myelomonocytic differentiation: Implications for the expansion of AML differentiation therapy. EJHaem 2021. [Google Scholar] [CrossRef]
- Nicolaou, F.; Teodoridis, J.M.; Park, H.; Georgakis, A.; Farokhzad, O.C.; Böttinger, E.P.; Da Silva, N.; Rousselot, P.; Chomienne, C.; Ferenczi, K.; et al. CD11c gene expression in hairy cell leukemia is dependent upon activation of the proto-oncogenes ras andjunD. Blood 2003, 101, 4033–4041. [Google Scholar] [CrossRef]
- Umit, E.G.; Baysal, M.; Durmus, Y.; Demir, A.M. CD11c expression in chronic lymphocytic leukemia revisited, related with complications and survival. Int. J. Lab. Hematol. 2017, 39, 552–556. [Google Scholar] [CrossRef]
- Park, H.; Shelley, C.S.; Arnaout, M.A. The zinc finger transcription factor ZBP-89 is a repressor of the human β2-integrin CD11b gene. Blood 2003, 101, 894–902. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pilling, D.; Fan, T.; Huang, D.; Kaul, B.; Gomer, R.H. Identification of Markers that Distinguish Monocyte-Derived Fibrocytes from Monocytes, Macrophages, and Fibroblasts. PLoS ONE 2009, 4, e7475. [Google Scholar] [CrossRef]
- Wagner, M.; Oelsner, M.; Moore, A.; Götte, F.; Kuhn, P.-H.; Haferlach, T.; Fiegl, M.; Bogner, C.; Baxter, E.J.; Peschel, C.; et al. Integration of innate into adaptive immune responses in ZAP-70–positive chronic lymphocytic leukemia. Blood 2016, 127, 436–448. [Google Scholar] [CrossRef]
- Burger, J.A.; Chiorazzi, N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013, 34, 592–601. [Google Scholar] [CrossRef]
- Kohlhaas, V.; Blakemore, S.J.; Al-Maarri, M.; Nickel, N.; Pal, M.; Roth, A.; Hövelmeyer, N.; Schäfer, S.C.; Knittel, G.; Lohneis, P.; et al. Active Akt signaling triggers CLL toward Richter transformation via overactivation of Notch1. Blood 2021, 137, 646–660. [Google Scholar] [CrossRef]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Al-Herz, W.; Ailal, F.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 2020, 40, 66–81. [Google Scholar] [CrossRef]
- Crequer, A.; Troeger, A.; Patin, E.; Ma, C.S.; Picard, C.; Pedergnana, V.; Fieschi, C.; Lim, A.; Abhyankar, A.; Gineau, L.; et al. Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J. Clin. Investig. 2012, 122, 3239–3247. [Google Scholar] [CrossRef]
- Gaidano, G.; Pasqualucci, L.; Capello, D.; Berra, E.; Deambrogi, C.; Rossi, D.; Larocca, L.M.; Gloghini, A.; Carbone, A.; Dalla-Favera, R. Aberrant somatic hypermutation in multiple subtypes of AIDS-associated non-Hodgkin lymphoma. Blood 2003, 102, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Vakiani, E.; Basso, K.; Klein, U.; Mansukhani, M.M.; Narayan, G.; Smith, P.M.; Murty, V.V.; Dalla-Favera, R.; Pasqualucci, L.; Bhagat, G. Genetic and phenotypic analysis of B-cell post-transplant lymphoproliferative disorders provides insights into disease biology. Hematol. Oncol. 2008, 26, 199–211. [Google Scholar] [CrossRef]
- Finn, O.J. Immuno-oncology: Understanding the function and dysfunction of the immune system in cancer. Ann. Oncol. 2012, 23, viii6–viii9. [Google Scholar] [CrossRef] [PubMed]
- Satgé, D. A Tumor Profile in Primary Immune Deficiencies Challenges the Cancer Immune Surveillance Concept. Front. Immunol. 2018, 9, 1149. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef]
- Chae, H.-D.; Lee, K.E.; Williams, D.A.; Gu, Y. Cross-talk between RhoH and Rac1 in regulation of actin cytoskeleton and chemotaxis of hematopoietic progenitor cells. Blood 2008, 111, 2597–2605. [Google Scholar] [CrossRef]
- Troeger, A.; Williams, D.A. Hematopoietic-specific Rho GTPases Rac2 and RhoH and human blood disorders. Exp. Cell Res. 2013, 319, 2375–2383. [Google Scholar] [CrossRef]
- Shaverdashvili, K.; Padlo, J.; Weinblatt, D.; Jia, Y.; Jiang, W.; Rao, D.; Laczkó, D.; Whelan, K.A.; Lynch, J.P.; Muir, A.B.; et al. KLF4 activates NFκB signaling and esophageal epithelial inflammation via the Rho-related GTP-binding protein RHOF. PLoS ONE 2019, 14, e0215746. [Google Scholar] [CrossRef]
- Itan, Y.; Casanova, J.-L. Novel Primary Immunodeficiency Candidate Genes Predicted by the Human Gene Connectome. Front. Immunol. 2015, 6, 142. [Google Scholar] [CrossRef]
- Côté, J.-F.; Vuori, K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 2007, 17, 383–393. [Google Scholar] [CrossRef]
- Han, J.; Das, B.; Wei, W.; Van Aelst, L.; Mosteller, R.D.; Khosravi-Far, R.; Westwick, J.K.; Der, C.J.; Broek, D. Lck regulates Vav activation of members of the Rho family of GTPases. Mol. Cell. Biol. 1997, 17, 1346–1353. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.-T.; Baek, K.E.; Kim, B.-Y.; Lee, H.G. Regulation of Rho GTPases by RhoGDIs in Human Cancers. Cells 2019, 8, 1037. [Google Scholar] [CrossRef]
- Ahmad Mokhtar, A.M. Investigating the Functional Interaction between RhoGDI Family Proteins and Activated Cdc42 Associated-Kinase (ACK). Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2020. [Google Scholar]
- Tamehiro, N.; Nishida, K.; Sugita, Y.; Hayakawa, K.; Oda, H.; Nitta, T.; Nakano, M.; Nishioka, A.; Yanobu-Takanashi, R.; Goto, M.; et al. Ras homolog gene family H (RhoH) deficiency induces psoriasis-like chronic dermatitis by promoting TH17 cell polarization. J. Allergy Clin. Immunol. 2019, 143, 1878–1891. [Google Scholar] [CrossRef]
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of Psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef]
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef]
- Oda, H.; Tamehiro, N.; Patrick, M.S.; Hayakawa, K.; Suzuki, H. Differential requirement for RhoH in development of TCRαβ CD8αα IELs and other types of T cells. Immunol. Lett. 2013, 151, 1–9. [Google Scholar] [CrossRef]
- Timlin, H.; Syed, A.; Haque, U.; Adler, B.; Law, G.; Machireddy, K.; Manno, R. Fevers in Adult Lupus Patients. Cureus 2018, 10, e2098. [Google Scholar]
- Anaya, J.-M. Common mechanisms of autoimmune diseases (the autoimmune tautology). Autoimmun. Rev. 2012, 11, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, T.; Li, H.; Krishfield, S.M.; Kyttaris, V.C.; Moulton, V.R. Splicing factor SRSF1 limits IFN-γ production via RhoH and ameliorates experimental nephritis. Rheumatology 2021, 60, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.L.; Guoliu, R.N.; Qin, H.; Shen, Y.Y.; Wang, B.; Zhai, Z.M. Elevated plasma levels of IL-12 and IFN-γ in systemic lupus erythematosus. Int. J. Clin. Exp. Pathol. 2017, 10, 3286–3291. [Google Scholar]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19, 89. [Google Scholar] [CrossRef] [PubMed]
- Harigai, M.; Kawamoto, M.; Hara, M.; Kubota, T.; Kamatani, N.; Miyasaka, N. Excessive Production of IFN-γ in Patients with Systemic Lupus Erythematosus and Its Contribution to Induction of B Lymphocyte Stimulator/B Cell-Activating Factor/TNF Ligand Superfamily-13B. J. Immunol. 2008, 181, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Lowin, T.; Anssar, T.M.; Bäuml, M.; Classen, T.; Schneider, M.; Pongratz, G. Positive and negative cooperativity of TNF and Interferon-γ in regulating synovial fibroblast function and B cell survival in fibroblast/B cell co-cultures. Sci. Rep. 2020, 10, 780. [Google Scholar] [CrossRef]
- Morimoto, S.; Nakano, S.; Watanabe, T.; Tamayama, Y.; Mitsuo, A.; Nakiri, Y.; Suzuki, J.; Nozawa, K.; Amano, H.; Tokano, Y.; et al. Expression of B-cell activating factor of the tumour necrosis factor family (BAFF) in T cells in active systemic lupus erythematosus: The role of BAFF in T cell-dependent B cell pathogenic autoantibody production. Rheumatology 2007, 46, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Jiang, W.-Q.; Rao, H.-L.; Huang, J.-J.; Xia, Y.; Huang, H.-Q.; Lin, T.-Y.; Xia, Z.-J.; Li, S.; Li, Z.-M. Expression of BAFF and BAFF-R in Follicular Lymphoma: Correlation with Clinicopathologic Characteristics and Survival Outcomes. PLoS ONE 2012, 7, e50936. [Google Scholar] [CrossRef]
- Yang, S.; Li, J.-Y.; Xu, W. Role of BAFF/BAFF-R axis in B-cell non-Hodgkin lymphoma. Crit. Rev. Oncol. Hematol. 2014, 91, 113–122. [Google Scholar] [CrossRef]
- Vásquez, A.; Baena, A.; González, L.A.; Restrepo, M.; Muñoz, C.H.; Vanegas-García, A.; Ortiz-Reyes, B.; Abdoel, N.; Rojas, M.; García, L.F.; et al. Altered recruitment of Lyn, Syk and ZAP-70 into lipid rafts of activated B cells in Systemic Lupus Erythematosus. Cell. Signal. 2019, 58, 9–19. [Google Scholar] [CrossRef]
- Jury, E.C.; Kabouridis, P.S.; Abba, A.; Mageed, R.A.; Isenberg, D.A. Increased ubiquitination and reduced expression of LCK in T lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 1343–1354. [Google Scholar] [CrossRef]
- Moulton, V.R.; Tsokos, G.C. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J. Clin. Investig. 2015, 125, 2220–2227. [Google Scholar] [CrossRef]
- Xavier, R.; Brennan, T.; Li, Q.; McCormack, C.; Seed, B. Membrane Compartmentation Is Required for Efficient T Cell Activation. Immunity 1998, 8, 723–732. [Google Scholar] [CrossRef]
- Packard, T.A.; Cambier, J.C. B lymphocyte antigen receptor signaling: Initiation, amplification, and regulation. F1000prime Rep. 2013, 5, 40. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, W.; Xiong, S. Blockade of Notch1 signaling alleviates murine lupus via blunting macrophage activation and M2b polarization. J. Immunol. 2010, 184, 6465–6478. [Google Scholar] [CrossRef]
- Kang, J.-A.; Kim, W.-S.; Park, S.-G. Notch1 is an important mediator for enhancing of B-cell activation and antibody secretion by Notch ligand. Immunology 2014, 143, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Calamito, M.; Srivastava, B.; Maillard, I.; Pear, W.S.; Allman, D. Notch activity synergizes with B-cell-receptor and CD40 signaling to enhance B-cell activation. Blood 2007, 109, 3342–3350. [Google Scholar] [CrossRef] [PubMed]
- Karrar, S.; Cunninghame Graham, D.S. Abnormal B Cell Development in Systemic Lupus Erythematosus: What the Genetics Tell Us. Arthritis Rheumatol. 2018, 70, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Sadras, T.; Cutler, J.; Aguade-Gorgorio, J.; Chen, Z.; Cosgun, K.N.; Pandey, A.; Muschen, M. Cooperation between SYK and ZAP70 Kinases As a Driver of Oncogenic BCR-Signaling in B-Cell Malignancies. Blood 2018, 132, 3922. [Google Scholar] [CrossRef]
- Chen, J.; Moore, A.; Ringshausen, I. ZAP-70 Shapes the Immune Microenvironment in B Cell Malignancies. Front. Oncol. 2020, 10, 2188. [Google Scholar] [CrossRef]
- Pollyea, D.; Gore, L.; Gutman, J.; Eckhardt, S.G.; Hagelstrom, N.; Coutre, S.; Thirman, M.; Byrd, J. A Dose Escalation Study of Ibrutinib with Lenalidomide for Relapsed and Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. Ann. Oncol. 2013, 24, i33. [Google Scholar] [CrossRef]
- Robert, G.; Jacquel, A.; Auberger, P. Chaperone-Mediated Autophagy and Its Emerging Role in Hematological Malignancies. Cells 2019, 8, 1260. [Google Scholar] [CrossRef]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef]
- Visperas, P.R.; Wilson, C.G.; Winger, J.A.; Yan, Q.; Lin, K.; Arkin, M.R.; Weiss, A.; Kuriyan, J. Identification of Inhibitors of the Association of ZAP-70 with the T Cell Receptor by High-Throughput Screen. SLAS Discov. Adv. Life Sci. R D 2017, 22, 324–331. [Google Scholar] [CrossRef]
- Boohaker, R.J.; Lee, M.W.; Vishnubhotla, P.; Perez, J.M.; Khaled, A.R. The use of therapeutic peptides to target and to kill cancer cells. Curr. Med. Chem. 2012, 19, 3794–3804. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.M.; Iegre, J.; O’ Donovan, D.H.; Ölwegård Halvarsson, M.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide–drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rho BTB1 | Rho BTB2 | RhoH | Rnd1 | Rnd2 | Rnd3 | RhoD | RhoF | RhoA | RhoC | RhoB | Wrch2 | Wrch1 | TC10 | TCL | Cdc42 | RhoG | Rac2 | Rac1 | Rac3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RhoBTB1 | 70 | 34 | 33 | 31 | 30 | 34 | 28 | 38 | 37 | 38 | 34 | 32 | 38 | 35 | 40 | 40 | 41 | 42 | 41 | |
| RhoBTB2 | 70 | 32 | 32 | 31 | 29 | 34 | 28 | 39 | 37 | 38 | 33 | 32 | 39 | 37 | 40 | 40 | 42 | 42 | 41 | |
| RhoH | 34 | 32 | 29 | 32 | 36 | 38 | 33 | 40 | 40 | 41 | 41 | 38 | 40 | 39 | 42 | 40 | 40 | 41 | 40 | |
| Rnd1 | 32 | 32 | 29 | 53 | 61 | 37 | 39 | 41 | 42 | 41 | 31 | 32 | 36 | 34 | 37 | 37 | 39 | 39 | 38 | |
| Rnd2 | 31 | 31 | 32 | 53 | 63 | 39 | 41 | 46 | 47 | 43 | 28 | 31 | 36 | 35 | 37 | 41 | 40 | 41 | 39 | |
| Rnd3 | 29 | 29 | 36 | 61 | 63 | 37 | 40 | 48 | 48 | 47 | 31 | 32 | 39 | 35 | 38 | 41 | 39 | 42 | 40 | |
| RhoD | 34 | 35 | 38 | 37 | 39 | 37 | 49 | 49 | 49 | 49 | 39 | 36 | 42 | 38 | 43 | 44 | 46 | 49 | 49 | |
| RhoF | 28 | 28 | 33 | 39 | 41 | 40 | 49 | 47 | 48 | 47 | 36 | 37 | 46 | 43 | 43 | 46 | 50 | 59 | 47 | |
| RhoA | 38 | 39 | 40 | 41 | 46 | 48 | 49 | 47 | 92 | 85 | 40 | 44 | 51 | 48 | 53 | 55 | 53 | 57 | 55 | |
| RhoC | 37 | 37 | 40 | 42 | 47 | 48 | 49 | 48 | 92 | 85 | 40 | 44 | 50 | 49 | 51 | 55 | 53 | 57 | 54 | |
| RhoB | 38 | 38 | 41 | 41 | 43 | 47 | 49 | 47 | 85 | 85 | 42 | 45 | 51 | 48 | 50 | 53 | 54 | 55 | 54 | |
| Wrch2 | 34 | 33 | 41 | 31 | 28 | 31 | 39 | 36 | 40 | 40 | 42 | 59 | 51 | 48 | 53 | 46 | 51 | 52 | 53 | |
| Wrch1 | 32 | 32 | 37 | 32 | 31 | 32 | 36 | 37 | 44 | 44 | 45 | 59 | 50 | 46 | 56 | 48 | 54 | 54 | 54 | |
| TC10 | 38 | 39 | 40 | 36 | 36 | 39 | 42 | 46 | 51 | 50 | 51 | 51 | 50 | 76 | 66 | 54 | 60 | 62 | 61 | |
| TCL | 35 | 37 | 39 | 34 | 35 | 35 | 38 | 44 | 48 | 49 | 48 | 48 | 46 | 76 | 63 | 53 | 58 | 60 | 59 | |
| Cdc42 | 40 | 40 | 42 | 37 | 37 | 38 | 43 | 43 | 53 | 51 | 50 | 53 | 56 | 66 | 63 | 61 | 69 | 71 | 70 | |
| RhoG | 39 | 40 | 40 | 37 | 41 | 41 | 44 | 46 | 55 | 55 | 53 | 46 | 48 | 54 | 53 | 60 | 72 | 72 | 70 | |
| Rac2 | 41 | 42 | 40 | 39 | 40 | 39 | 46 | 50 | 53 | 53 | 54 | 50 | 54 | 60 | 58 | 69 | 72 | 92 | 89 | |
| Rac1 | 42 | 42 | 41 | 39 | 41 | 42 | 49 | 49 | 57 | 57 | 55 | 52 | 54 | 62 | 60 | 71 | 72 | 92 | 93 | |
| Rac3 | 41 | 41 | 40 | 38 | 39 | 40 | 49 | 47 | 55 | 54 | 54 | 53 | 54 | 61 | 59 | 70 | 70 | 89 | 93 |
| Group | Rho Protein | C-Terminal Sequence | Lipid Modification | Ref |
|---|---|---|---|---|
| Typical | RhoA | KDGVREVFEMATRAALQARRGKKKSGCLVL | GG | [11] |
| RhoB | VREVFETATRAALQKRYGSQNGCINCCKVL | GG, F, P | ||
| RhoC | KEGVREVFEMATRAGLQVRKNKRRRGCPIL | GG | ||
| Rac1 | RGLKTVFDEAIRAVLCPPPVKKRKRKCLLL | GG, P | ||
| Rac2 | RGLKTVFDEAIRAVLCPQPTRQQKRACSLL | GG | ||
| Rac3 | RGLKTVFDEAIRAVLCPPPVKKPGKKCTVF | GG | ||
| RhoG | QDGVKEVFAEAVRAVLNPTPIKRGRSCILL | GG | ||
| Cdc42 | QKGLKNVFDEAILAALEPPEPKKSRRCVLL | GG | ||
| TCL | AVFDEAILTIFHPKKKKKRCSEGHSCCSII | F | ||
| TC10 | DEAIIAILTPKKHTVKKRIGSRCINCCLIT | F, P | ||
| Atypical | RhoU | QQQPKKSKSRTPDKMKNLSKSWWKKYCCFV | P | [11] |
| RhoV | EHKARLEKKLNAKGVRTLSRCRWKKFFCFV | P | ||
| RhoD | AVFQEAAEVALSSRGRNFWRRITQGFCVVT | F, GG | [12] | |
| RhoF | EDVFREAAKVALSALKKAQRQKKRRLCLLL | F, GG | ||
| Rnd1/ RhoS | LSKRLLHLPSRSELISSTFKKEKAKSCSIM | F | [11] | |
| Rnd2/ RhoN | MQRSAQLSGRPDRGNEGEIHKDRAKSCNLM | F | ||
| Rnd3/ RhoE | KRISHMPSRPELSAVATDLRKDKAKSCTVM | F | ||
| RhoH/ TTF | VFECAVRTAVNQARRRNRRRLFSINECKIF | GG, F | [11,12] | |
| RhoBTB1 | KREREKEDIALNKHRSRRKWCFWNSSPAVA | Unknown | N/A | |
| RhoBTB2 | KRRWLFWNSPSSPSSSAASSSSPSSSSAVV | Unknown |
| Amino Acids 12, 59 and 61 | |||
|---|---|---|---|
| Group | Subfamily | Member | Sequence |
| Classic | Cdc42 | Cdc42 | 12 59 61

 |
| GDGAV---AGQED | |||
| Fast-cycling | RhoU/RhoV | RhoU | GDGAV---AGQED |
| RhoV | GDGAV---AGQDE | ||
| RhoD/RhoF | RhoD | GDGGC---AGQDD | |
| RhoF | GDGGC---AGQED | ||
| GTPase defective | RhoBTB | RhoBTB−1 | GDNAV---FGDHH |
| RhoBTB−2 | GDNAV---FGDHH | ||
| Rnd | Rnd1 | GDVQC---SGSPY | |
| Rnd2 | GDAEC---SGSSY | ||
| Rnd3 | GDVQC---SGSPY | ||
| RhoH | GDSAV---AGNDA | ||
| Amino acids 28 | |||
| Classic | Rac | Rac1 | 28 |
| SYTTNAFPGEYIP | |||
| Fast-cycling | RhoU/RhoV | RhoU | SYTTNGYPTEYIP |
| RhoV | SYTCNGYPARYRP | ||
| RhoD/RhoF | RhoD | VFADGAFPESYTP | |
| RhoF | VYSQGSFPEHYAP | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad Mokhtar, A.M.; Hashim, I.F.; Mohd Zaini Makhtar, M.; Salikin, N.H.; Amin-Nordin, S. The Role of RhoH in TCR Signalling and Its Involvement in Diseases. Cells 2021, 10, 950. https://doi.org/10.3390/cells10040950
Ahmad Mokhtar AM, Hashim IF, Mohd Zaini Makhtar M, Salikin NH, Amin-Nordin S. The Role of RhoH in TCR Signalling and Its Involvement in Diseases. Cells. 2021; 10(4):950. https://doi.org/10.3390/cells10040950
Chicago/Turabian StyleAhmad Mokhtar, Ana Masara, Ilie Fadzilah Hashim, Muaz Mohd Zaini Makhtar, Nor Hawani Salikin, and Syafinaz Amin-Nordin. 2021. "The Role of RhoH in TCR Signalling and Its Involvement in Diseases" Cells 10, no. 4: 950. https://doi.org/10.3390/cells10040950
APA StyleAhmad Mokhtar, A. M., Hashim, I. F., Mohd Zaini Makhtar, M., Salikin, N. H., & Amin-Nordin, S. (2021). The Role of RhoH in TCR Signalling and Its Involvement in Diseases. Cells, 10(4), 950. https://doi.org/10.3390/cells10040950