
FLYWCH1, a Multi-Functional Zinc Finger Protein Contributes to the DNA Repair Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture and Treatment
2.3. Generation of ATM Knock-Out Cell Lines
2.4. Generation of Cell Lines Transiently Overexpressing FLYWCH1
2.5. Immunofluorescence
2.6. Western Blotting
2.7. RNA Isolation and Quantitative PCR (qPCR)
2.8. Statistical Analysis
3. Results
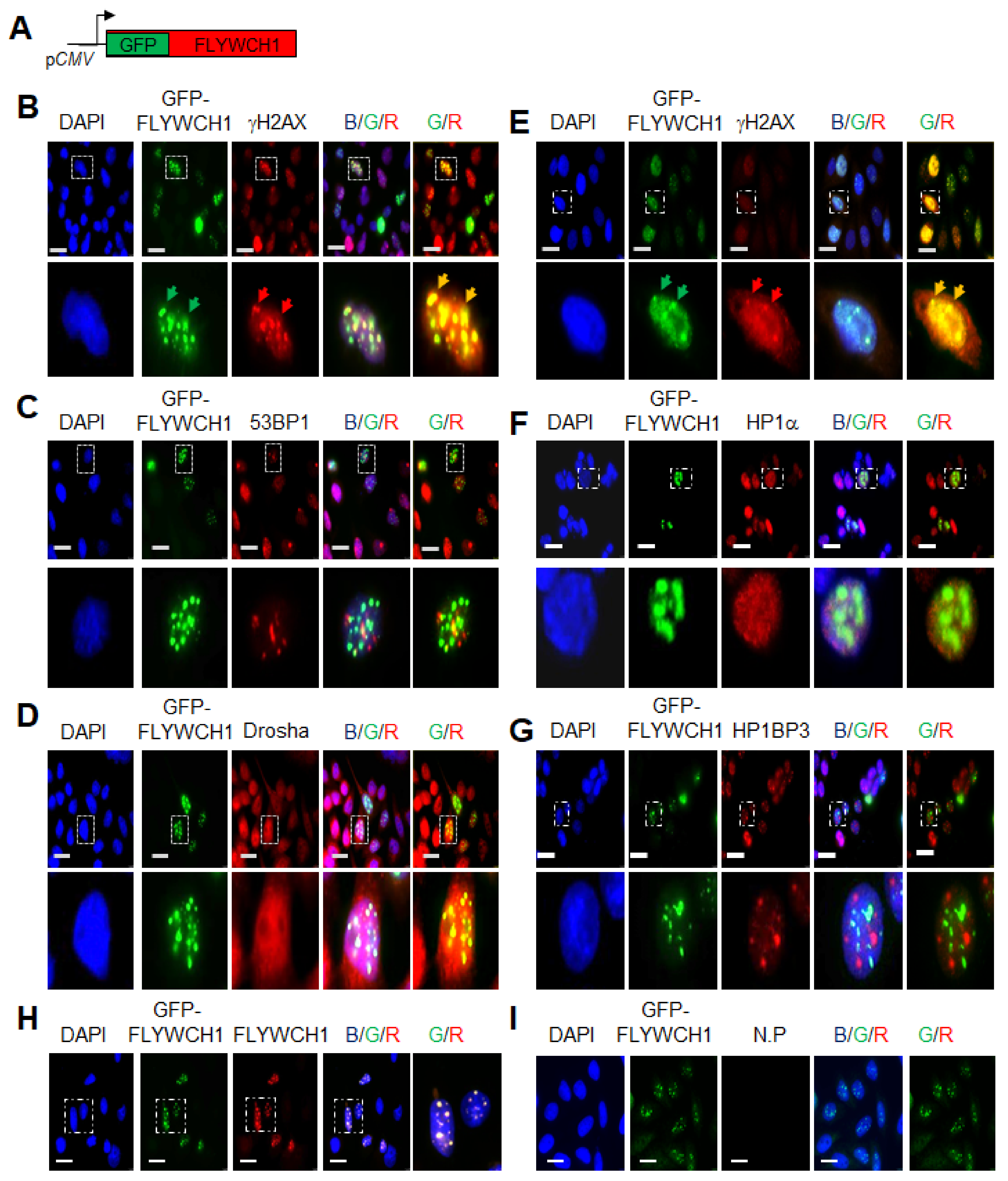
3.1. FLYWCH1 Is Localised in Nuclear Speckles and Colocalised with γH2AX
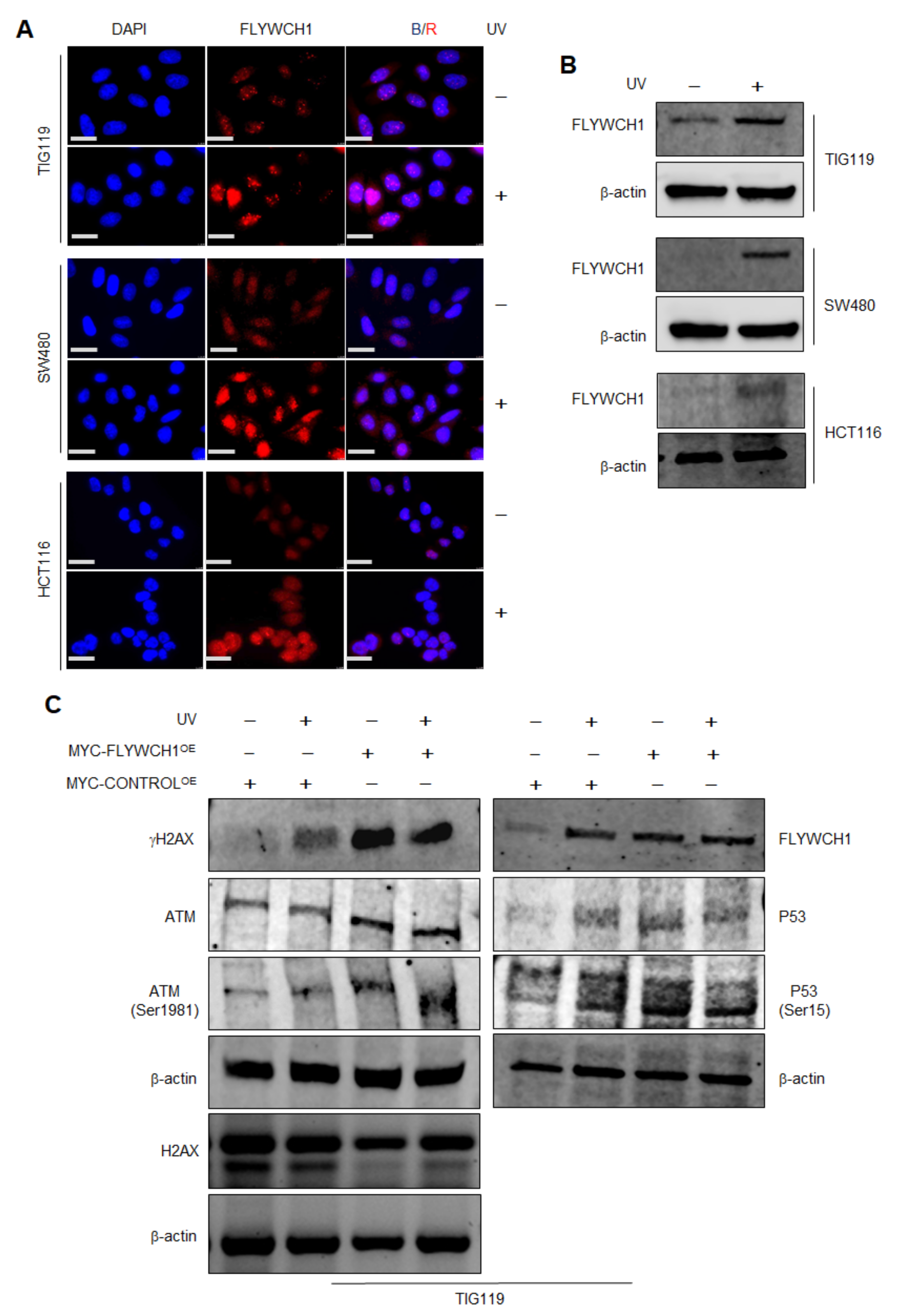
3.2. The Effects of UV-Radiation on FLYWCH1 Expression in Normal vs. CRC Cells
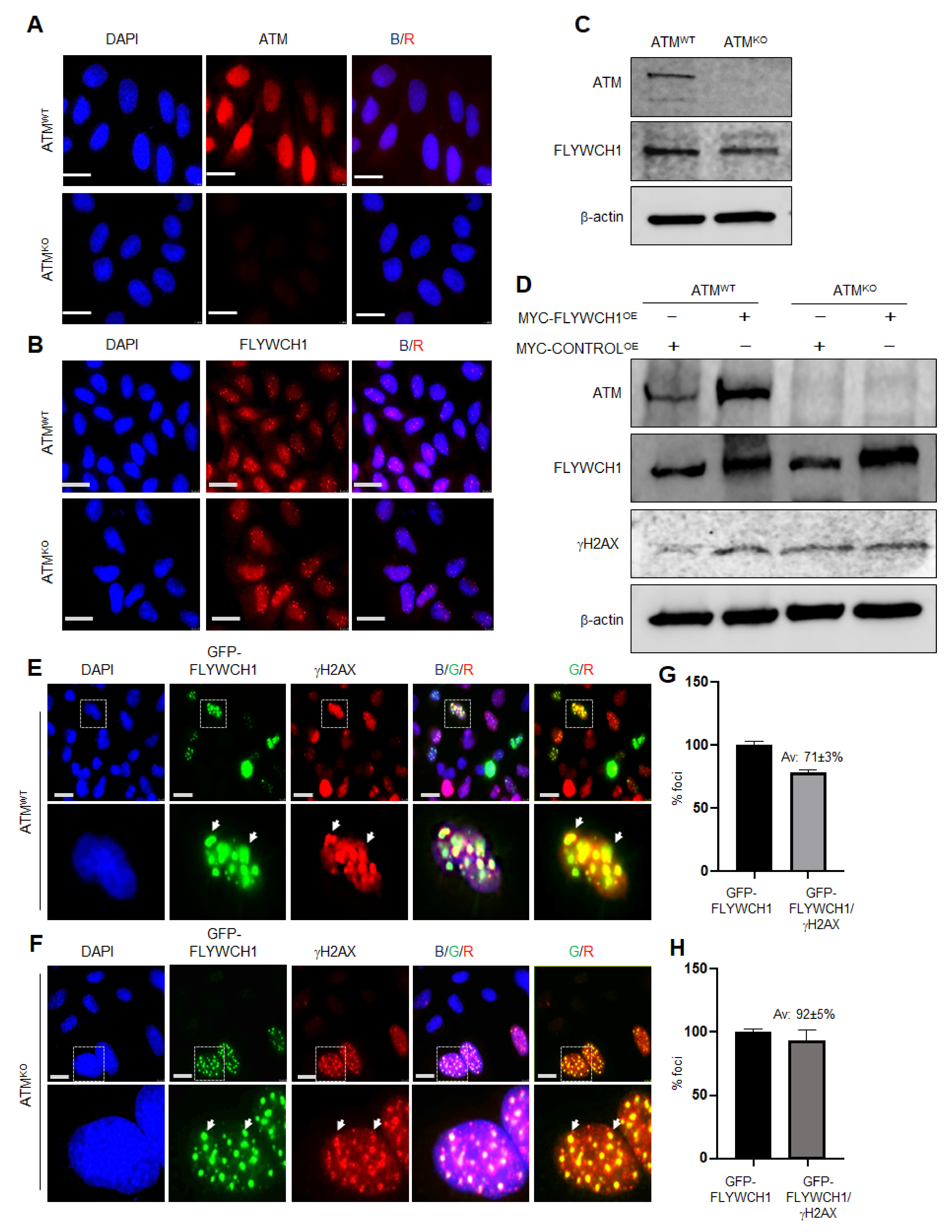
3.3. FLYWCH1-Mediated Induction of Phosphorylated H2AX Level Is Independent of ATM Protein
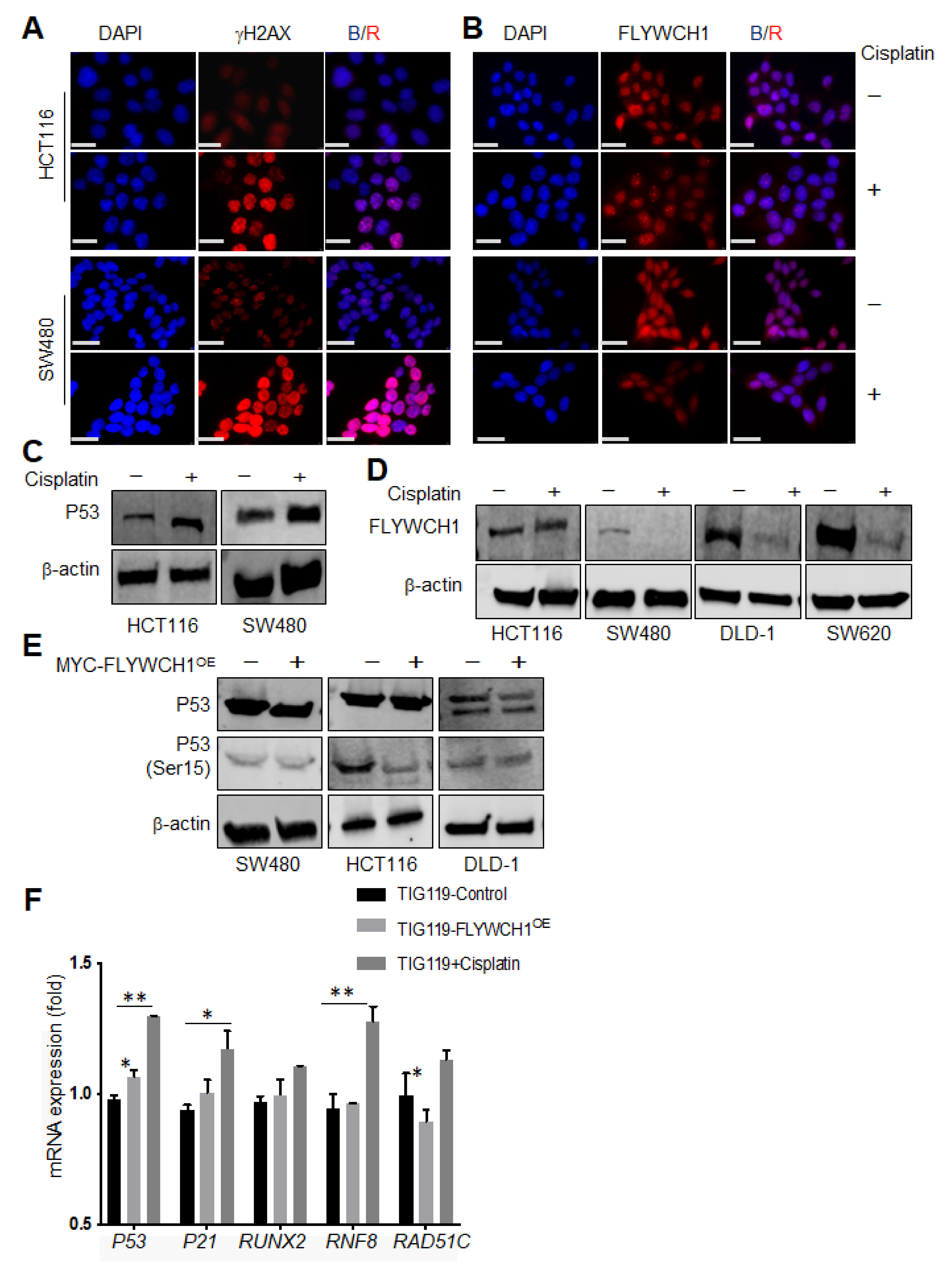
3.4. Cisplatin Treatment Reduced FLYWCH1 Protein Level in CRC Cells but Not in HCT116
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BRCA1 | Breast cancer type 1 susceptibility protein |
| BSA | Bovine Serum Albumin |
| β-TRCP | Beta-Transducin Repeat-Containing Protein |
| C2H2 | Cystein2-Histidine2 |
| CRC | Colorectal Cancer |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DDR | DNA Damage Repair |
| DNA | Deoxyribonucleic acid |
| dH2O | Distilled Water |
| dsRNA | Double stranded RNA |
| DSB | Double-strand breaks |
| Drosha | Double-stranded RNA-specific ribonuclease (RNase) III |
| FLYWCH1 | FLYWCH-type zinc finger 1 |
| GFP | Green Fluorescent Protein |
| gRNA | Guide RNA |
| H2AX | Histone H2A variant |
| HDAC | Histone Deacetylases |
| HP1a | Heterochromatin protein 1 isoform a |
| HP1BP3 | Heterochromatin protein 1 binding protein 3 |
| HR | Homologous recombination |
| MMR | Mismatch repair |
| MSI | Microsatellite Instability |
| MSS | Microsatellite Stability |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous end-joining |
| NLS | Nuclear localization signals |
| ROS | Reactive Oxygen Species |
| UV | Ultraviolet |
References
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Foroughi Asl, H.; Talukdar, H.A.; Kindt, A.S.; Jain, R.K.; Ermel, R.; Ruusalepp, A.; Nguyen, K.D.; Dobrin, R.; Reilly, D.F.; Schunkert, H.; et al. Expression quantitative trait Loci acting across multiple tissues are enriched in inherited risk for coronary artery disease. Circ. Cardiovasc. Genet. 2015, 8, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Haskell, G.T.; Jensen, B.C.; Skrzynia, C.; Pulikkotil, T.; Tilley, C.R.; Lu, Y.; Marchuk, D.S.; Ann Samsa, L.; Wilhelmsen, K.C.; Lange, E.; et al. Genetic complexity of mitral valve prolapse revealed by clinical and genetic evaluation of a large family. J. Heart Valve Dis. 2017, 26, 569–580. [Google Scholar]
- Almars, A.; Chondrou, P.S.; Onyido, E.K.; Almozyan, S.; Seedhouse, C.; Babaei-Jadidi, R.; Nateri, A.S. Increased FLYWCH1 expression is negatively correlated with Wnt/β-catenin target gene expression in acute myeloid leukemia cells. Int. J. Mol. Sci. 2019, 20, 2739. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, B.A.; Almozyan, S.; Babaei-Jadidi, R.; Onyido, E.K.; Saadeddin, A.; Kashfi, S.H.; Spencer-Dene, B.; Ilyas, M.; Lourdusamy, A.; Behrens, A.; et al. FLYWCH1, a novel suppressor of nuclear beta-catenin, regulates migration and morphology in colorectal cancer. Mol. Cancer Res. 2018, 16, 1977–1990. [Google Scholar] [CrossRef]
- Singh, J.K.; Van Attikum, H. DNA double-strand break repair: Putting zinc fingers on the sore spot. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Helfricht, A.; Thijssen, P.E.; Rother, M.B.; Shah, R.G.; Du, L.; Takada, S.; Rogier, M.; Moritz, J.; IJspeert, H.; Stoepker, C.; et al. Loss of ZBTB24 impairs nonhomologous end-joining and class-switch recombination in patients with ICF syndrome. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Nicolai, S.; Mahen, R.; Raschellà, G.; Marini, A.; Pieraccioli, M.; Malewicz, M.; Venkitaraman, A.R.; Melino, G. ZNF281 is recruited on DNA breaks to facilitate DNA repair by non-homologous end joining. Oncogene 2020, 39, 754–766. [Google Scholar] [CrossRef]
- Chen, G.; Chen, J.; Qiao, Y.; Shi, Y.; Liu, W.; Zeng, Q.; Xie, H.; Shi, X.; Sun, Y.; Liu, X.; et al. ZNF830 mediates cancer chemoresistance through promoting homologous-recombination repair. Nucleic Acids Res. 2018, 46, 1266–1279. [Google Scholar] [CrossRef]
- Razin, S.V.; Borunova, V.V.; Maksimenko, O.G.; Kantidze, O.L. Cys2His2 zinc finger protein family: Classification, functions, and major members. Biochemistry 2012, 77, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Fedotova, A.A.; Bonchuk, A.N.; Mogila, V.A.; Georgiev, P.G. C2H2 zinc finger proteins: The largest but poorly explored family of higher eukaryotic transcription factors. Acta Nat. 2017, 9, 47–58. [Google Scholar] [CrossRef]
- Najafabadi, H.S.; Mnaimneh, S.; Schmitges, F.W.; Garton, M.; Lam, K.N.; Yang, A.; Albu, M.; Weirauch, M.T.; Radovani, E.; Kim, P.M.; et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015, 33, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, D.; Moye, C.; Morton, J.; Sykoudis, K.; Lin, Y.; Carroll, D.; Wilson, J.H. Zinc-finger directed double-strand breaks within CAG repeat tracts promote repeat instability in human cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9607–9612. [Google Scholar] [CrossRef]
- Mirman, Z.; Lange, D.T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef]
- Lu, W.-T.; Hawley, B.R.; Skalka, G.L.; Baldock, R.A.; Smith, E.M.; Bader, A.S.; Malewicz, M.; Watts, F.Z.; Wilczynska, A.; Bushell, M. Drosha drives the formation of DNA: RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liang, C.; Kollipara, R.K.; Matsui, M.; Ke, X.; Jeong, B.-C.; Wang, Z.; Yoo, K.S.; Yadav, G.P.; Kinch, L.N.; et al. HP1BP3, a chromatin retention factor for co-transcriptional microRNA processing. Mol. Cell 2016, 63, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Bartova, E.; Malyskova, B.; Komurkova, D.; Legartova, S.; Suchankova, J.; Krejci, J.; Kozubek, S. Function of heterochromatin protein 1 during DNA repair. Protoplasma 2017, 254, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Richa, K.A.; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids 2010, 2010, 592980. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Alabdullah, M.; Miligy, I.; Normatova, M.; Babaei-Jadidi, R.; Nateri, S.A.; Rakha, E.A.; Madhusudan, S. ATM regulated PTEN degradation is XIAP E3 ubiquitin ligase mediated in p85α deficient cancer cells and influence platinum sensitivity. Cells 2019, 8, 1271. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Heijink, A.M.; Everts, M.; Honeywell, M.E.; Richards, R.; Kok, Y.P.; Vries, D.E.G.E.; Lee, M.J.; Van Vugt, M. Modeling of cisplatin-induced signaling dynamics in triple-negative breast cancer cells reveals mediators of sensitivity. Cell Rep. 2019, 28, 2345–2357.e5. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krishnamurthy, S. Cellular responses to cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.A.; Pestell, K.E.; Stefano, D.F.; Workman, P.; Walton, M.I. Characterisation of molecular events following cisplatin treatment of two curable ovarian cancer models: Contrasting role for p53 induction and apoptosis in vivo. Br. J. Cancer 2004, 91, 1614–1623. [Google Scholar] [CrossRef]
- Englinger, B.; Mair, M.; Miklos, W.; Pirker, C.; Mohr, T.; Van Schoonhoven, S.; Lotsch, D.; Korner, W.; Ferk, F.; Knasmuller, S.; et al. Loss of CUL4A expression is underlying cisplatin hypersensitivity in colorectal carcinoma cells with acquired trabectedin resistance. Br. J. Cancer 2017, 116, 489–500. [Google Scholar] [CrossRef]
- Li, N.; Babaei-Jadidi, R.; Lorenzi, F.; Spencer-Dene, B.; Clarke, P.; Domingo, E.; Tulchinsky, E.; Vries, R.G.J.; Kerr, D.; Pan, Y.; et al. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis 2019, 8, 13. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef]
- Bartocci, C.; Denchi, E.L. Put a RING on it: Regulation and inhibition of RNF8 and RNF168 RING finger E3 ligases at DNA damage sites. Front. Genet. 2013, 4, 128. [Google Scholar] [CrossRef]
- Ozaki, T.; Wu, D.; Sugimoto, H.; Nagase, H.; Nakagawara, A. Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis. 2013, 4, e610. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A.; Nagase, H. RUNX family participates in the regulation of p53-dependent DNA damage response. Int. J. Genom. 2013, 2013, 271347. [Google Scholar] [CrossRef] [PubMed]
- Badie, S.; Liao, C.; Thanasoula, M.; Barber, P.; Hill, M.A.; Tarsounas, M. RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J. Cell Biol. 2009, 185, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and ATR: Networking cellular responses to DNA damage. Curr. Opin. Genet. Dev. 2001, 11, 71–77. [Google Scholar] [CrossRef]
- Yajima, H.; Lee, K.J.; Zhang, S.; Kobayashi, J.; Chen, B.P. DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J. Mol. Biol. 2009, 385, 800–810. [Google Scholar] [CrossRef]
- Lu, C.; Shi, Y.; Wang, Z.; Song, Z.; Zhu, M.; Cai, Q.; Chen, T. Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS Lett. 2008, 582, 2703–2708. [Google Scholar] [CrossRef]
- Tu, W.Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.D.; Guan, H.; Zhang, S.M.; Tang, Y.; Rang, W.Q.; Zhou, P.K. gammaH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almozyan, S.; Coulton, J.; Babaei-Jadidi, R.; Nateri, A.S. FLYWCH1, a Multi-Functional Zinc Finger Protein Contributes to the DNA Repair Pathway. Cells 2021, 10, 889. https://doi.org/10.3390/cells10040889
Almozyan S, Coulton J, Babaei-Jadidi R, Nateri AS. FLYWCH1, a Multi-Functional Zinc Finger Protein Contributes to the DNA Repair Pathway. Cells. 2021; 10(4):889. https://doi.org/10.3390/cells10040889
Chicago/Turabian StyleAlmozyan, Sheema, James Coulton, Roya Babaei-Jadidi, and Abdolrahman S. Nateri. 2021. "FLYWCH1, a Multi-Functional Zinc Finger Protein Contributes to the DNA Repair Pathway" Cells 10, no. 4: 889. https://doi.org/10.3390/cells10040889
APA StyleAlmozyan, S., Coulton, J., Babaei-Jadidi, R., & Nateri, A. S. (2021). FLYWCH1, a Multi-Functional Zinc Finger Protein Contributes to the DNA Repair Pathway. Cells, 10(4), 889. https://doi.org/10.3390/cells10040889