
Leukocyte TNFR1 and TNFR2 Expression Contributes to the Peripheral Immune Response in Cases with Ischemic Stroke

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval and Consent to Participate
2.2. Participants
2.3. Patient Demographics
2.4. Sample Collection and Handling
2.5. Chemiluminescence Analysis
2.6. Simoa Analysis
2.7. ELISA Analysis
2.8. Leukocyte Isolation
2.9. Flow Cytometry
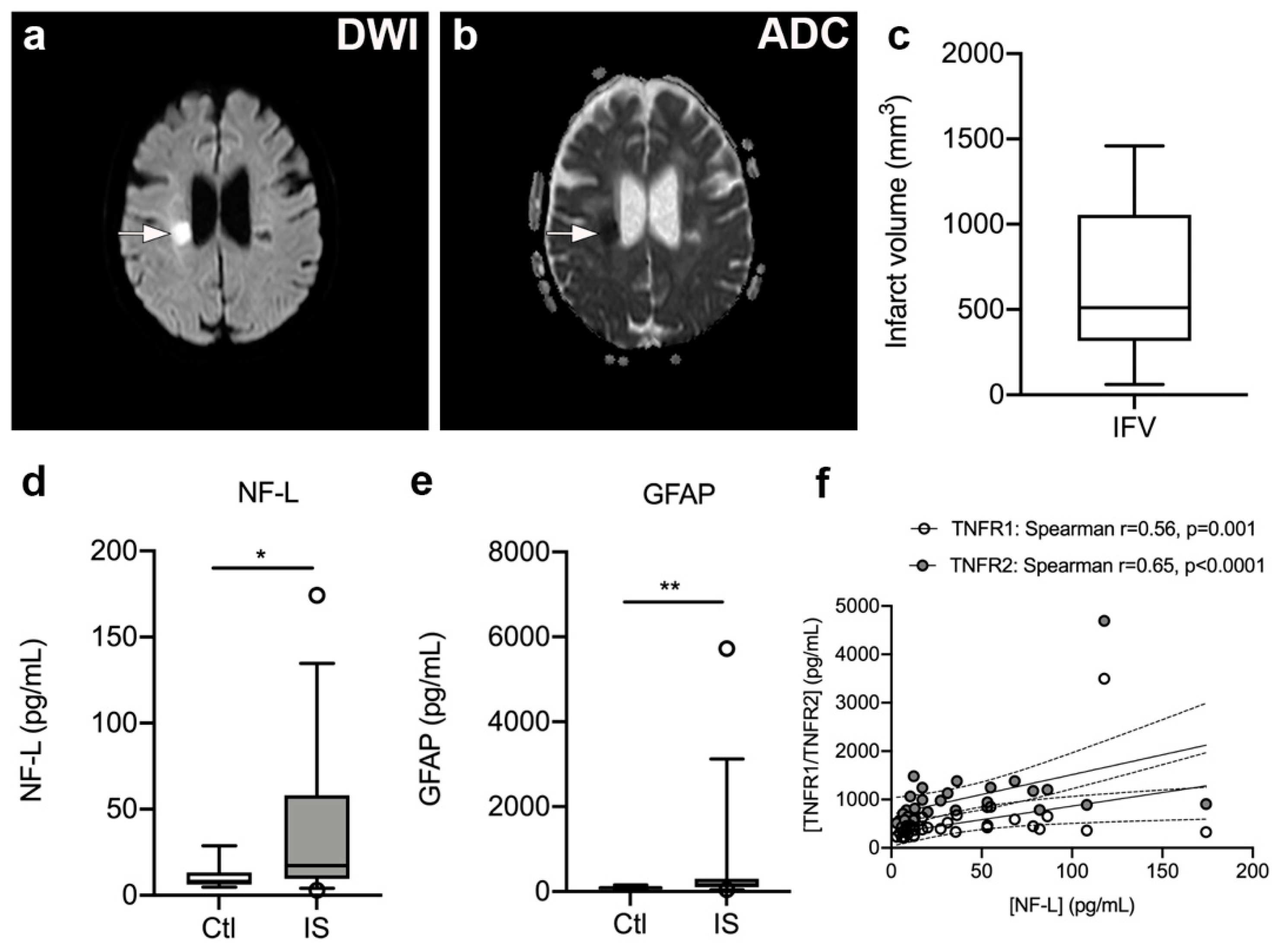
2.10. Infarct Volume Estimation
2.11. Statistical Analysis
3. Results
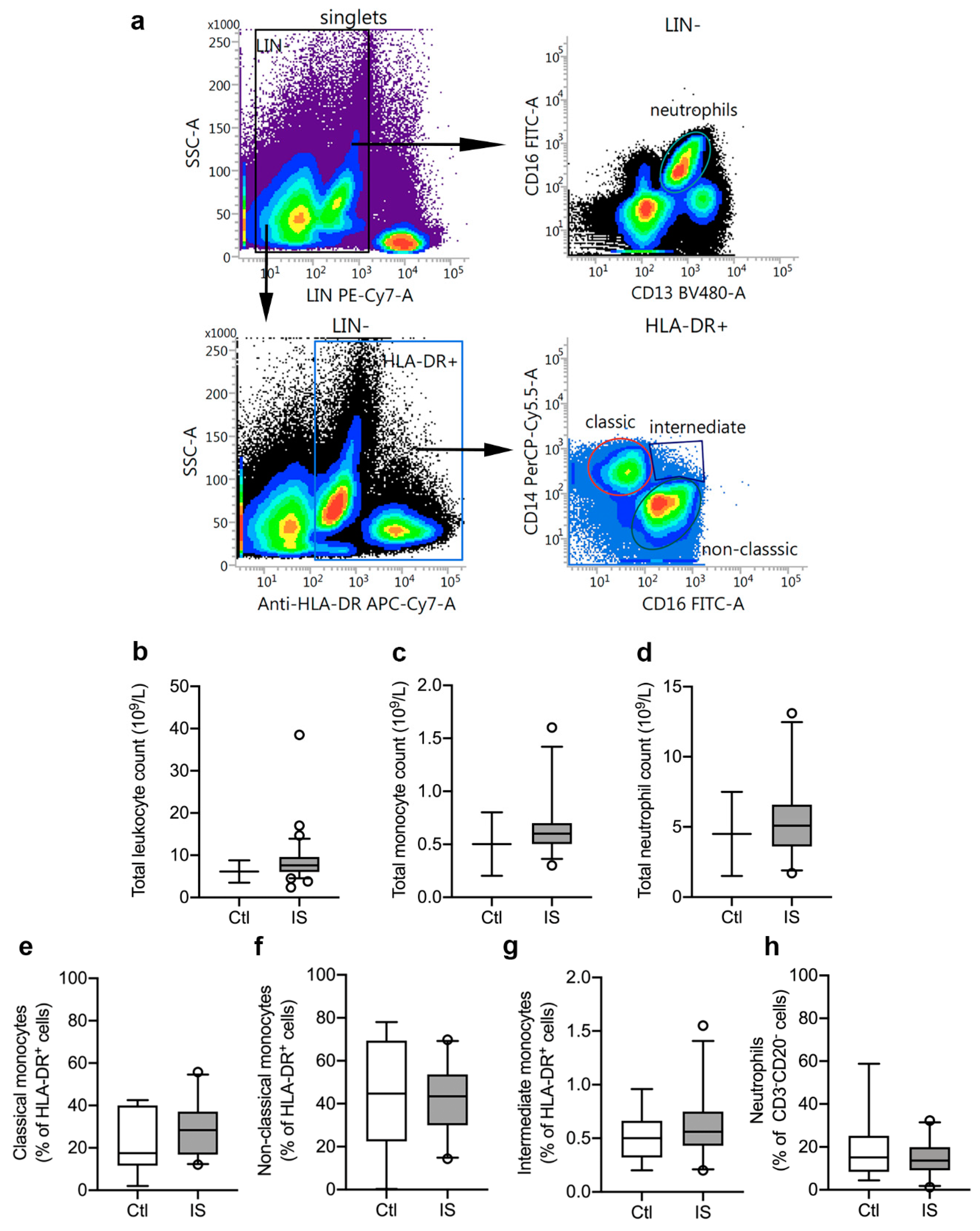
3.1. Distribution of Peripheral Immune Cells
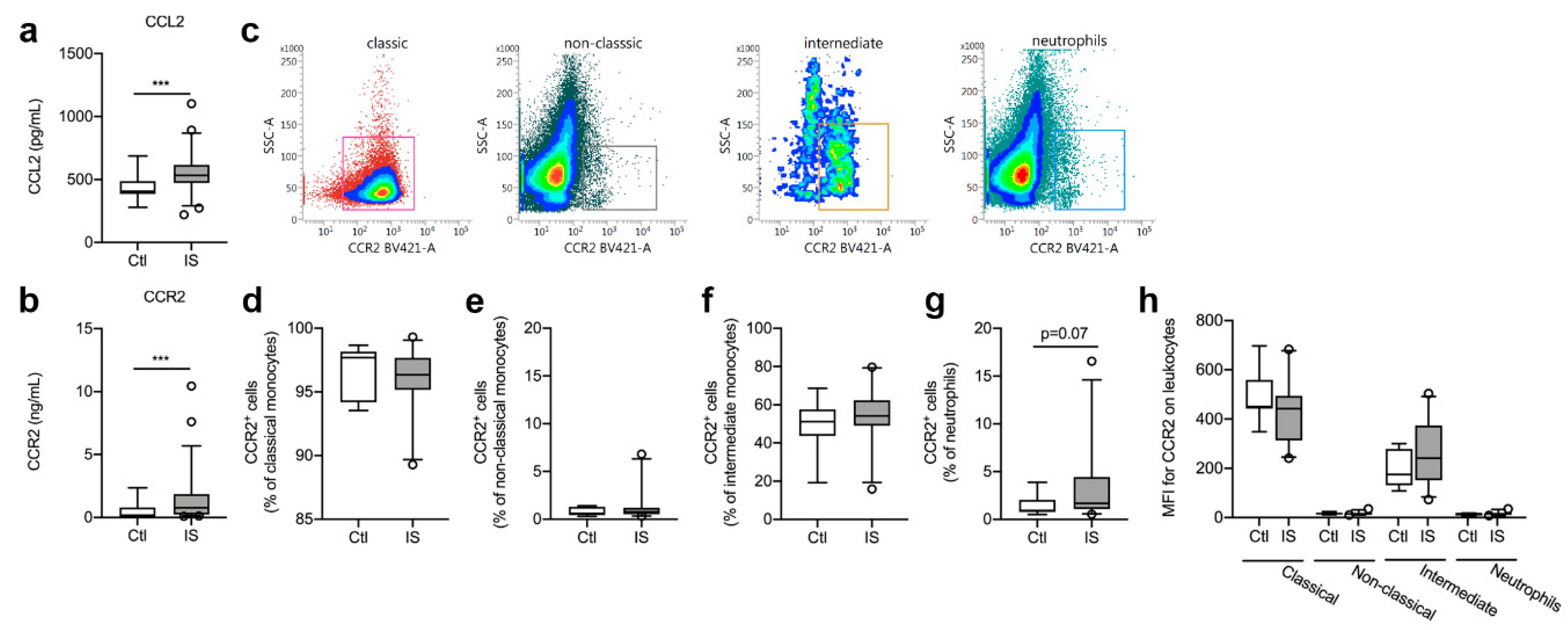
3.2. CCR2 and CCL2 Blood Levels
3.3. CCR2+ Cell Populations were Comparable in Healthy Controls and Ischemic Stroke Patients
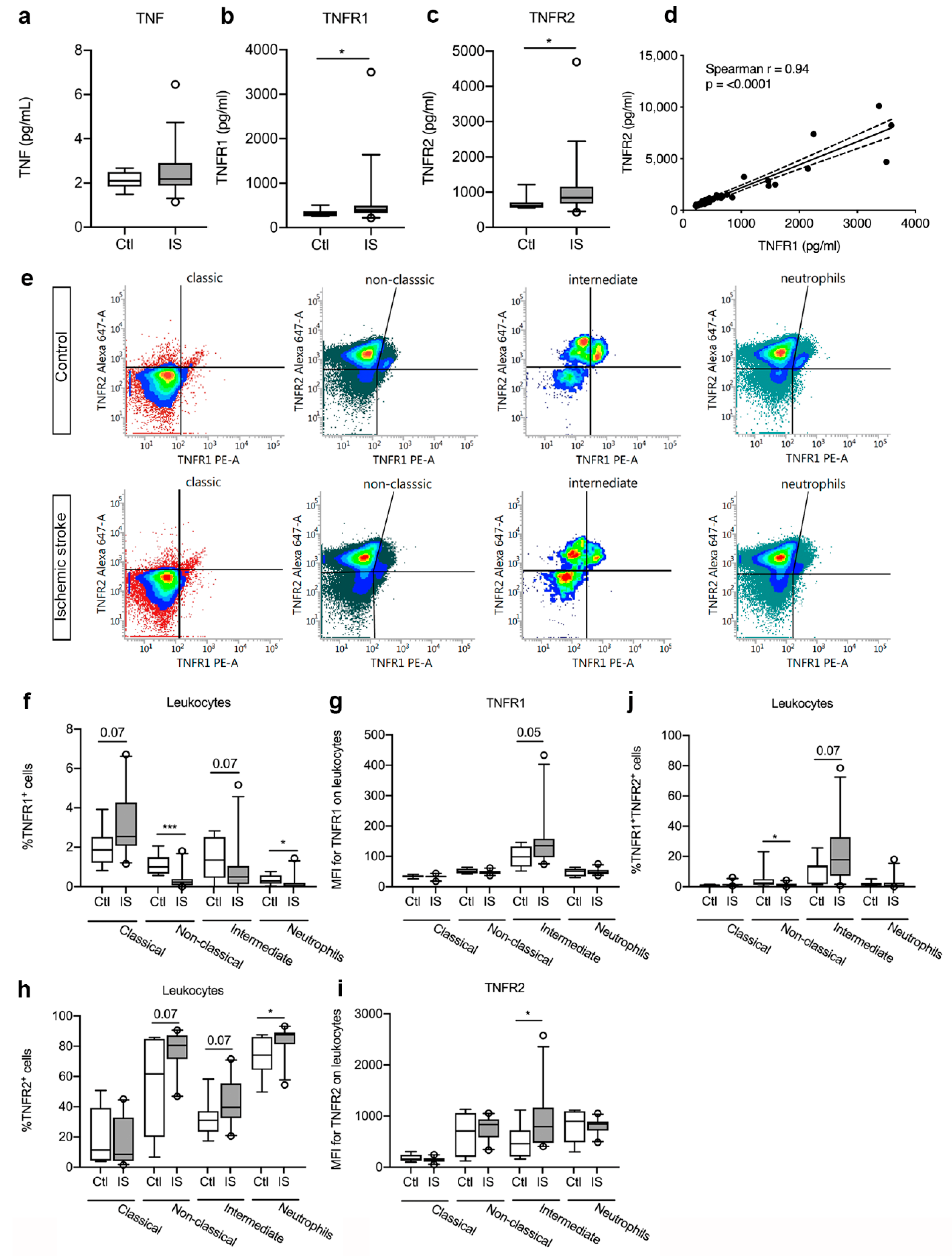
3.4. Plasma Levels of TNFR1 and TNFR2 were Significantly Increased in Ischemic Stroke Patients
3.5. TNFR Expression on Peripheral Immune Cells
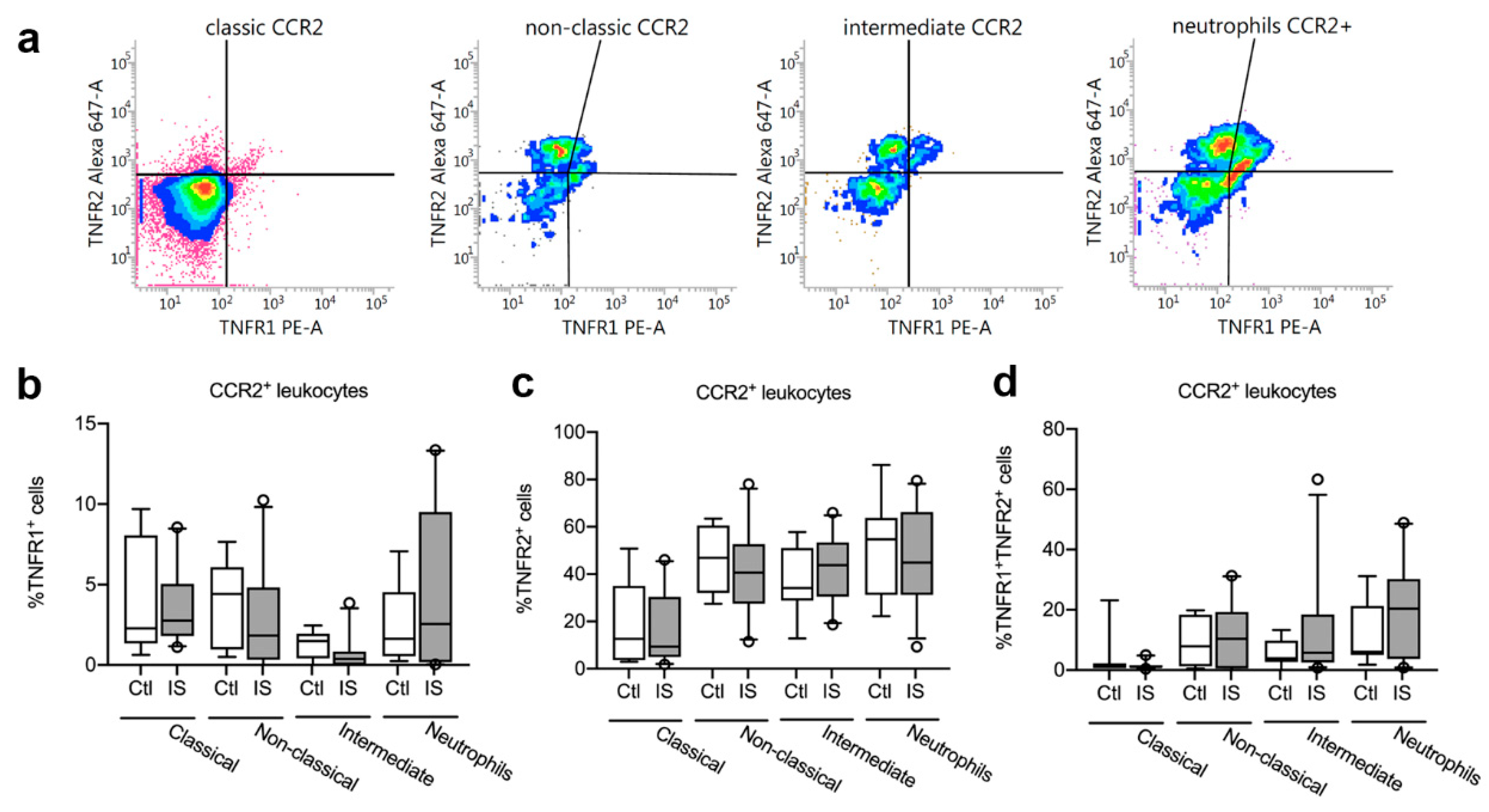
3.6. TNFR1 and TNFR2 Expression on CCR2+ Leukocytes was Unchanged in Acute Ischemic Stroke in Humans
3.7. Serum NF-L and GFAP Levels Are Significantly Increased in Ischemic Stroke Patients
3.8. Plasma TNFR1 and TNFR2 Levels Correlate with Serum NF-L Levels but Not with Infarct Volume, Stroke Severity, or Functional Outcome
3.9. Plasma IL-6 Levels Were Increased in Ischemic Stroke Patients and Correlated with Plasma TNFR1 and TNFR2 Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lambertsen, K.L.; Finsen, B.; Clausen, B.H. Post-stroke inflammation-target or tool for therapy? Acta Neuropathol. 2018, 137, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chen, C.H.; Yang, Y.H.; Lin, R.T.; Lin, F.C.; Liu, C.K. Eligibility for recombinant tissue plasminogen activator in acute ischemic stroke: Way to endeavor. Cereb. Dis. 2006, 22, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.H.; Wirenfeldt, M.; Hogedal, S.S.; Frich, L.H.; Nielsen, H.H.; Schroder, H.D.; Ostergaard, K.; Finsen, B.; Kristensen, B.W.; Lambertsen, K.L. Characterization of the TNF and IL-1 systems in human brain and blood after ischemic stroke. Acta Neuropathol. Commun. 2020, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.H.; Degn, M.; Sivasaravanaparan, M.; Fogtmann, T.; Andersen, M.G.; Trojanowsky, M.D.; Gao, H.; Hvidsten, S.; Baun, C.; Deierborg, T.; et al. Conditional ablation of myeloid TNF increases lesion volume after experimental stroke in mice, possibly via altered ERK1/2 signaling. Sci. Rep. 2016, 6, 29291. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, J.; Losy, J. Early TNF-alpha levels correlate with ischaemic stroke severity. Acta Neurol. Scand 2001, 104, 288–295. [Google Scholar] [CrossRef]
- Intiso, D.; Zarrelli, M.M.; Lagioia, G.; Di Rienzo, F.; Checchia De Ambrosio, C.; Simone, P.; Tonali, P.; Cioffi, R.P. Tumor necrosis factor alpha serum levels and inflammatory response in acute ischemic stroke patinets. Neurol. Sci. 2003, 24, 390–396. [Google Scholar] [CrossRef]
- Sotgiu, S.; Zanda, B.; Marchetti, B.; Fois, M.L.; Arru, G.; Pes, G.M.; Salaris, F.S.; Arru, A.; Pirisi, A.; Rosati, G. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur. J. Neurol. 2006, 13, 505–513. [Google Scholar] [CrossRef]
- Licata, G.; Tuttolomondo, A.; Di Raimondo, D.; Corrao, S.; Di Sciacca, R.; Pinto, A. Immuno-inflammatory activation in acute cardio-embolic strokes in comparison with other subtypes of ischaemic stroke. Thromb. Haemost. 2009, 101, 929–937. [Google Scholar]
- Mazzotta, G.; Sarchielli, P.; Caso, V.; Paciaroni, M.; Floridi, A.; Floridi, A.; Gallai, V. Different cytokine levels in thrombolysis patients as predictors for clinical outcome. Eur. J. Neurol. 2004, 11, 377–381. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Sciacca, R.; Di Raimondo, D.; Serio, A.; D’Aguanno, G.; La Placa, S.; Pecoraro, R.; Arnao, V.; Marino, L.; Monaco, S.; et al. Plasma levels of inflammatory and thrombotic/fibrinolytic markers in acute ischemic strokes: Relationship with TOAST subtype, outcome and infarct site. J. Neuroimmunol. 2009, 215, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Wang, Y.; Liu, J.; Wang, L.; Weng, S.; Chen, K.; Domino, E.F.; Yang, G.Y. Pro-inflammatory cytokine network in peripheral inflammation response to cerebral ischemia. Neurosci. Lett. 2013, 548, 4–9. [Google Scholar] [CrossRef]
- Vila, N.; Castillo, J.; Davalos, A.; Chamorro, A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Koh, C.S.; Yahikozawa, H.; Yanagisawa, N.; Yagita, H.; Ishihara, Y.; Kim, B.S. The level of tumor necrosis factor-alpha producing cells in the spinal cord correlates with the degree of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. Int. Immunol. 1996, 8, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Emsley, H.C.; Smith, C.J.; Gavin, C.M.; Georgiou, R.F.; Vail, A.; Barberan, E.M.; Illingworth, K.; Scarth, S.; Wickramasinghe, V.; Hoadley, M.E.; et al. Clinical outcome following acute ischaemic stroke relates to both activation and autoregulatory inhibition of cytokine production. BMC Neurol. 2007, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Boehme, A.K.; McClure, L.A.; Zhang, Y.; Luna, J.M.; Del Brutto, O.H.; Benavente, O.R.; Elkind, M.S. Inflammatory Markers and Outcomes After Lacunar Stroke: Levels of Inflammatory Markers in Treatment of Stroke Study. Stroke 2016, 47, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Wang, H.; Li, R.; Zhang, L.; Li, Z.; Wang, Y.; Hui, R.; Ding, H.; Wang, D.W. Polymorphism of tumor necrosis factor alpha (TNF-alpha) gene promoter, circulating TNF-alpha level, and cardiovascular risk factor for ischemic stroke. J. Neuroinflamm. 2012, 9, 235. [Google Scholar] [CrossRef]
- Svensson, E.H.; Soderholm, M.; Abul-Kasim, K.; Engstrom, G. Tumor Necrosis Factor Receptor 1 and 2 are Associated with Risk of Intracerebral Hemorrhage. Stroke 2017, 48, 2710–2715. [Google Scholar] [CrossRef]
- Ruuls, S.R.; Hoek, R.M.; Ngo, V.N.; McNeil, T.; Lucian, L.A.; Janatpour, M.J.; Korner, H.; Scheerens, H.; Hessel, E.M.; Cyster, J.G.; et al. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity 2001, 15, 533–543. [Google Scholar] [CrossRef]
- Holtmann, M.H.; Neurath, M.F. Differential TNF-signaling in chronic inflammatory disorders. Curr. Mol. Med. 2004, 4, 439–444. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef]
- Fresegna, D.; Bullitta, S.; Musella, A.; Rizzo, F.R.; De Vito, F.; Guadalupi, L.; Caioli, S.; Balletta, S.; Sanna, K.; Dolcetti, E.; et al. Re-Examining the Role of TNF in MS Pathogenesis and Therapy. Cells 2020, 9, 2290. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Pecoraro, R.; Pinto, A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: A review of the evidence to date. Drug Des. Devel. Ther. 2014, 8, 2221–2238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Li, Y.; Deng, Y.; Alzheimer’s Disease Neuroimaging Initiative. TNF receptors are associated with tau pathology and conversion to Alzheimer’s dementia in subjects with mild cognitive impairment. Neurosci. Lett. 2020, 738, 135392. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.K.; Harrison, J.L.; Zhang, H.; Bachstetter, A.D.; Hesson, D.P.; O’Hara, B.F.; Greene, M.I.; Lifshitz, J. Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J. Neuroinflamm. 2018, 15, 154. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.J.; Boling, W.; Kindy, M.S.; Peschon, J.; Kraemer, P.J.; Carpenter, M.K.; Holtsberg, F.W.; Mattson, M.P. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med. 1996, 2, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Gary, D.S.; Bruce-Keller, A.J.; Kindy, M.S.; Mattson, M.P. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J. Cereb. Blood Flow Metab. 1998, 18, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Petit, E.; Divoux, D.; Tseveleki, V.; Mengozzi, M.; Roberts, M.L.; Valable, S.; Ghezzi, P.; Quackenbush, J.; Brines, M.; et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc. Natl. Acad. Sci. USA 2008, 105, 6185–6190. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef]
- Madsen, P.M.; Clausen, B.H.; Degn, M.; Thyssen, S.; Kristensen, L.K.; Svensson, M.; Ditzel, N.; Finsen, B.; Deierborg, T.; Brambilla, R.; et al. Genetic ablation of soluble tumor necrosis factor with preservation of membrane tumor necrosis factor is associated with neuroprotection after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2016, 36, 1553–1569. [Google Scholar] [CrossRef]
- Hughes, P.M.; Allegrini, P.R.; Rudin, M.; Perry, V.H.; Mir, A.K.; Wiessner, C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J. Cereb. Blood Flow Metab. 2002, 22, 308–317. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Saederup, N.; Cardona, A.E.; Croft, K.; Mizutani, M.; Cotleur, A.C.; Tsou, C.L.; Ransohoff, R.M.; Charo, I.F. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE 2010, 5, e13693. [Google Scholar] [CrossRef]
- Miro-Mur, F.; Perez-de-Puig, I.; Ferrer-Ferrer, M.; Urra, X.; Justicia, C.; Chamorro, A.; Planas, A.M. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav. Immun. 2016, 53, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 2007, 38, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Urra, X.; Villamor, N.; Amaro, S.; Gomez-Choco, M.; Obach, V.; Oleaga, L.; Planas, A.M.; Chamorro, A. Monocyte subtypes predict clinical course and prognosis in human stroke. J. Cereb. Blood Flow Metab. 2009, 29, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Merali, Z.; Huang, K.; Mikulis, D.; Silver, F.; Kassner, A. Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS ONE 2017, 12, e0171558. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Zhang, L.; Tsang, W.; Soltanian-Zadeh, H.; Morris, D.; Zhang, R.; Goussev, A.; Powers, C.; Yeich, T.; Chopp, M. Correlation of VEGF and angiopoietin expression with disruption of blood-brain barrier and angiogenesis after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2002, 22, 379–392. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.H.; Soares, C.B.; Hogedal, S.S.; Madsen, J.S.; Hansen, R.B.; Christensen, A.A.; Madsen, C.; Clausen, B.H.; Frich, L.H.; Degn, M.; et al. Acute Neurofilament Light Chain Plasma Levels Correlate with Stroke Severity and Clinical Outcome in Ischemic Stroke Patients. Front. Neurol. 2020, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- De Marchis, G.M.; Katan, M.; Barro, C.; Fladt, J.; Traenka, C.; Seiffge, D.J.; Hert, L.; Gensicke, H.; Disanto, G.; Sutter, R.; et al. Serum neurofilament light chain in patients with acute cerebrovascular events. Eur. J. Neurol. 2018, 25, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Tiedt, S.; Duering, M.; Barro, C.; Kaya, A.G.; Boeck, J.; Bode, F.J.; Klein, M.; Dorn, F.; Gesierich, B.; Kellert, L.; et al. Serum neurofilament light: A biomarker of neuroaxonal injury after ischemic stroke. Neurology 2018, 91, e1338–e1347. [Google Scholar] [CrossRef] [PubMed]
- Hjalmarsson, C.; Bjerke, M.; Andersson, B.; Blennow, K.; Zetterberg, H.; Aberg, N.D.; Olsson, B.; Eckerstrom, C.; Bokemark, L.; Wallin, A. Neuronal and glia-related biomarkers in cerebrospinal fluid of patients with acute ischemic stroke. J. Cent. Nerv. Syst. Dis. 2014, 6, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Cheng, Y.W.; Chen, Y.F.; Tang, S.C.; Jeng, J.S. Plasma neurofilament light chain and glial fibrillary acidic protein predict stroke in CADASIL. J. Neuroinflamm. 2020, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- Seder, D.B. Serum Glial Fibrillary Acidic Protein in Acute Stroke. Neurocrit Care 2020, 33, 35–36. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Rosengren, L.; Blomstrand, C.; Wikkelso, C.; Jensen, C.; Ekholm, S.; Tarkowski, A. Early intrathecal production of interleukin-6 predicts the size of brain lesion in stroke. Stroke 1995, 26, 1393–1398. [Google Scholar] [CrossRef]
- Dziewulska, D.; Mossakowski, M.J. Cellular expression of tumor necrosis factor a and its receptors in human ischemic stroke. Clin. Neuropathol. 2003, 22, 35–40. [Google Scholar]
- Group, S.S.S. Multicenter trial of hemodilution in ischemic stroke--background and study protocol. Scandinavian Stroke Study Group. Stroke 1985, 16, 885–890. [Google Scholar]
- Broderick, J.P.; Adeoye, O.; Elm, J. Evolution of the Modified Rankin Scale and Its Use in Future Stroke Trials. Stroke 2017, 48, 2007–2012. [Google Scholar] [CrossRef]
- Clausen, B.H.; Lambertsen, K.L.; Finsen, B. Glyceraldehyde-3-phosphate dehydrogenase versus toluidine blue as a marker for infarct volume estimation following permanent middle cerebral artery occlusion in mice. Exp. Brain Res. 2006, 175, 60–67. [Google Scholar] [CrossRef]
- Pedersen, A.; Stanne, T.M.; Nilsson, S.; Klasson, S.; Rosengren, L.; Holmegaard, L.; Jood, K.; Blennow, K.; Zetterberg, H.; Jern, C. Circulating neurofilament light in ischemic stroke: Temporal profile and outcome prediction. J. Neurol. 2019, 266, 2796–2806. [Google Scholar] [CrossRef]
- Worthmann, H.; Tryc, A.B.; Goldbecker, A.; Ma, Y.T.; Tountopoulou, A.; Hahn, A.; Dengler, R.; Lichtinghagen, R.; Weissenborn, K. The temporal profile of inflammatory markers and mediators in blood after acute ischemic stroke differs depending on stroke outcome. Cereb. Dis. 2010, 30, 85–92. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Soares, C.B.; Gaist, D.; Nielsen, H.H. Neurofilaments: The C-Reactive Protein of Neurology. Brain Sci. 2020, 10, 56. [Google Scholar] [CrossRef]
- Perini, F.; Morra, M.; Alecci, M.; Galloni, E.; Marchi, M.; Toso, V. Temporal profile of serum anti-inflammatory and pro-inflammatory interleukins in acute ischemic stroke patients. Neurol. Sci 2001, 22, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Emsley, H.C.; Gavin, C.M.; Georgiou, R.F.; Vail, A.; Barberan, E.M.; del Zoppo, G.J.; Hallenbeck, J.M.; Rothwell, N.J.; Hopkins, S.J.; et al. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Wytrykowska, A.; Prosba-Mackiewicz, M.; Nyka, W.M. IL-1beta, TNF-alpha, and IL-6 levels in gingival fluid and serum of patients with ischemic stroke. J. Oral. Sci. 2016, 58, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Faustino, J.; Chip, S.; Derugin, N.; Jullienne, A.; Hamer, M.; Haddad, E.; Butovsky, O.; Obenaus, A.; Vexler, Z.S. CX3CR1-CCR2-dependent monocyte-microglial signaling modulates neurovascular leakage and acute injury in a mouse model of childhood stroke. J. Cereb. Blood Flow Metab. 2019, 39, 1919–1935. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Yang, J.; Beltran, C.D.; Cho, S. Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J. Cereb. Blood Flow Metab. 2014, 34, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Kaito, M.; Araya, S.; Gondo, Y.; Fujita, M.; Minato, N.; Nakanishi, M.; Matsui, M. Relevance of distinct monocyte subsets to clinical course of ischemic stroke patients. PLoS ONE 2013, 8, e69409. [Google Scholar] [CrossRef]
- Hijdra, D.; Vorselaars, A.D.; Grutters, J.C.; Claessen, A.M.; Rijkers, G.T. Differential expression of TNFR1 (CD120a) and TNFR2 (CD120b) on subpopulations of human monocytes. J. Inflamm. 2012, 9, 38. [Google Scholar] [CrossRef]
- Wein, T.; Wanner, K.T.; Rappengluck, S.; Sichler, S.; Niessen, K.V.; Seeger, T.; Worek, F.; Thiermann, H. New Resensitizers for the Nicotinic Acetylcholine Receptor by Ligand-Based Pharmacophore Modeling. Curr Comput Aided Drug Des. 2019, 15, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Hijdra, D.; Vorselaars, A.D.; Grutters, J.C.; Claessen, A.M.; Rijkers, G.T. Phenotypic characterization of human intermediate monocytes. Front. Immunol. 2013, 4, 339. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; Deighton, J.; Walmsley, S.R.; Chilvers, E.R. The survival effect of TNF-alpha in human neutrophils is mediated via NF-kappa B-dependent IL-8 release. Eur. J. Immunol. 2004, 34, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Huang, Q.; Wang, F.; Milner, R.; Li, L. Cerebral ischemia-induced angiogenesis is dependent on tumor necrosis factor receptor 1-mediated upregulation of alpha5beta1 and alphaVbeta3 integrins. J. Neuroinflamm. 2016, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Lopatnikova, J.A.; Vasilyev, F.F.; Alshevskaya, A.A.; Sennikov, S.V. Quantitative flow cytometric analysis of expression of tumor necrosis factor receptor types I and II on mononuclear cells. J. Recept. Signal Transduct Res. 2013, 33, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.X.; Arumugam, T.V.; Gelderblom, M.; Magnus, T.; Drummond, G.R.; Sobey, C.G. Role of CCR2 in inflammatory conditions of the central nervous system. J. Cereb. Blood Flow Metab. 2014, 34, 1425–1429. [Google Scholar] [CrossRef]
- Rayasam, A.; Faustino, J.; Lecuyer, M.; Vexler, Z.S. Neonatal Stroke and TLR1/2 Ligand Recruit Myeloid Cells through the Choroid Plexus in a CX3CR1-CCR2- and Context-Specific Manner. J. Neurosci. 2020, 40, 3849–3861. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Strecker, J.K.; Ringelstein, E.B.; Schabitz, W.R.; Kiefer, R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain Res. 2009, 1289, 79–84. [Google Scholar] [CrossRef]
- Garcia-Bonilla, L.; Brea, D.; Benakis, C.; Lane, D.A.; Murphy, M.; Moore, J.; Racchumi, G.; Jiang, X.; Iadecola, C.; Anrather, J. Endogenous Protection from Ischemic Brain Injury by Preconditioned Monocytes. J. Neurosci. 2018, 38, 6722–6736. [Google Scholar] [CrossRef] [PubMed]
- Verweij, S.L.; Duivenvoorden, R.; Stiekema, L.C.A.; Nurmohamed, N.S.; van der Valk, F.M.; Versloot, M.; Verberne, H.J.; Stroes, E.S.G.; Nahrendorf, M.; Bekkering, S.; et al. CCR2 expression on circulating monocytes is associated with arterial wall inflammation assessed by 18F-FDG PET/CT in patients at risk for cardiovascular disease. Cardiovasc. Res. 2018, 114, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.N.; Campbell, J.J.; Salanga, C.L.; Ertl, L.S.; Wang, Y.; Yau, S.; Dang, T.; Zeng, Y.; McMahon, J.P.; Krasinski, A.; et al. CCR2-Mediated Uptake of Constitutively Produced CCL2: A Mechanism for Regulating Chemokine Levels in the Blood. J. Immunol. 2019, 203, 3157–3165. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.; Alvarez, R.; Batalla, A.; Reguero, J.R.; Alvarez, V.; Astudillo, A.; Cubero, G.I.; Cortina, A.; Coto, E. Genetic variation at the chemokine receptors CCR5/CCR2 in myocardial infarction. Genes Immun. 2001, 2, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cao, Y.; Zhang, X.; Peng, C.; Tian, X.; Yan, C.; Liu, Y.; Liu, M.; Han, Y. Chemokine CC-motif ligand 2 participates in platelet function and arterial thrombosis by regulating PKCalpha-P38MAPK-HSP27 pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2901–2912. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.; Bianchini, F.J.; Nascimento, D.C.; Oliveira, R.D.; Souto, F.O.; Pinto, L.G.; Peres, R.S.; Silva, J.R.; Almeida, S.C.; Louzada-Junior, P.; et al. CCR2 Expression in Neutrophils Plays a Critical Role in Their Migration Into the Joints in Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Makinde, H.M.; Cuda, C.M.; Just, T.B.; Perlman, H.R.; Schwulst, S.J. Nonclassical Monocytes Mediate Secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration after Traumatic Brain Injury. J. Immunol. 2017, 199, 3583–3591. [Google Scholar] [CrossRef] [PubMed]
- Boyette, L.B.; Macedo, C.; Hadi, K.; Elinoff, B.D.; Walters, J.T.; Ramaswami, B.; Chalasani, G.; Taboas, J.M.; Lakkis, F.G.; Metes, D.M. Phenotype, function and differentiation potential of human monocyte subsets. PLoS ONE 2017, 12, e0176460. [Google Scholar] [CrossRef]
- Algra, S.O.; Groeneveld, K.M.; Schadenberg, A.W.; Haas, F.; Evens, F.C.; Meerding, J.; Koenderman, L.; Jansen, N.J.; Prakken, B.J. Cerebral ischemia initiates an immediate innate immune response in neonates during cardiac surgery. J. Neuroinflamm. 2013, 10, 24. [Google Scholar] [CrossRef]
- Urra, X.; Cervera, A.; Villamor, N.; Planas, A.M.; Chamorro, A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience 2009, 158, 1174–1183. [Google Scholar] [CrossRef]
- Works, M.G.; Koenig, J.B.; Sapolsky, R.M. Soluble TNF receptor 1-secreting ex vivo-derived dendritic cells reduce injury after stroke. J. Cereb. Blood Flow Metab. 2013, 33, 1376–1385. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls | Ischemic Stroke | p-Value | b Ischemic Stroke | |
|---|---|---|---|---|
| Number of participants | 10 | 33 | 11 | |
| Age, years, median (IQR) | 59.0 (54.8; 72.3) | 73.0 (62.5; 80.5) | 0.02 c | 62.0 (48.0; 76.0) |
| Sex, n (%) males | 2 (20) | 20 (60.6) | 0.03 d | 7 (63.6) |
Anti-inflammatory medication, n (%)
| 1 (10) 9 (90) | 10 (30.3) 23 (69.7) | 0.41 d | 3 (27.3) 8 (72.7) |
| Smoking, n (%) | ||||
| 1 (10) 5 (50) 4 (40) 0 (0) | 8 (24.2) 9 (27.3) 10 (30.3) 6 (18.2) | 0.26 e | 2 (18.2) 4 (36.4) 2 (18.2) 3 (27.2) |
| Alcohol consumption a, n (%) | ||||
| 10 (100) 0 (0) 0 (0) | 23 (69.7) 1 (3) 9 (27.3) | 0.14 e | 3 (27.3) 7 (63.6) 1 (9.1) |
| SSS score (median + IQR) | 52.0 (43.5; 56.0) (5 missing) | 52.0 (49.3; 57.3) (1 missing) | ||
| mRS score (median + IQR) | 0.0 (0.0; 1.0) (14 missing) | 1.0 (0.5; 1.5) (2 missing) | ||
| Treatment, n (%) | ||||
| 12 (36.4) 1 (3) 20 (60.6) | 0 (0) 0 (0) 11 (100) | ||
| Time to blood sample, hours (median + IQR) | 23.2 (15.1; 32.0) | 10.8 (8.4; 15.58) | ||
| Sodium, mmol/L (median + IQR) | 139.0 (136.5; 141.0) | 139.5 (136.8; 141.0) | ||
| Potassium, mmol/L (median + IQR) | 4.0 (3.9; 4.2) | 3.8 (3.7; 4.1) | ||
| CRP, mg/L (median + IQR) | 2.95 (1.23; 6.88) | 2.55 (0.95; 4.05) |
| Differential Leukocyte Count on Admission a | Mean | Median (IQR) | 2.5–97.5% PCTL | Reference Range of Normal Population 2.5–97.5% PCTL b |
|---|---|---|---|---|
| Total leukocyte count (n = 33) | 8.9 | 7.6 (6.1; 9.6) | 2.4–38.5 | 3.5–8.8 |
| Thrombocyte count | ||||
| 222.3 284.7 | 230.0 (158.0; 264.5) 281.0 (205.5; 336.0) | 130.0–336.0 182.0–418.0 | Men: 145–350 Women: 165–400 |
| Neutrophil count (n = 33) | 5.6 | 5.1 (3.6; 6.6) | 1.7–13.1 | 1.5–7.5 |
| Lymphocyte count (n = 31) | 2.5 | 1.6 (1.2; 1.9) | 0.6–31.5 | 1.0–4.0 |
| Monocyte count (n = 31) | 0.7 | 0.6 (0.5; 0.7) | 0.3–1.6 | 0.2–0.8 |
| Protein (pg/mL) | Controls (n = 10) | Ischemic Stroke (n = 33) | U | p-Value | Mean CV (%) |
|---|---|---|---|---|---|
| TNF | 2.1 (1.8; 2.5) | 2.2 (1.9–2.9) | 141 | 0.50 | 5.9 |
| TNFR1 | 306.5 (257.4; 366.5) | 395.1 (330.4; 494.5) | 93 | 0.04 | 3.8 |
| TNFR2 | 615.4 (561.4; 704.3) | 845.7 (675.3; 1156.0) | 90 | 0.03 | 4.1 |
| IL-6 | 0.76 (0.41; 1.13) | 1.25 (0.65; 2.33) | 95 | 0.04 | 1.4 |
| VEGF-A | 29.1 (22.0; 39.6) | 30.2 (20.4; 38.6) | 162 | 0.84 | 8.9 |
| Cell Population | Controls (n = 8) | Ischemic Stroke (n = 23) | U | p-Value |
|---|---|---|---|---|
| TNFR1+ CCR2+ classical monocytes | 2.29% (1.35–8.06) | 2.77% (1.80–5.05) | 89 | 0.91 |
| TNFR1+ CCR2+ non-classical monocytes | 4.42% (0.97–6.09) | 1.83% (0.33–4.82) | 68 | 0.21 |
| TNFR1+ CCR2+ intermediate monocytes | 1.50% (0.42–1.94) | 0.38% (0.00–0.84) | 55.5 | 0.10 |
| TNFR1+ CCR2+ neutrophils | 1.63% (0.55–4.53) | 2.56% (0.18–9.51) | 78 | 0.55 |
| TNFR2+ CCR2+ classical monocytes | 12.59% (3.65–35.02) | 9.37% (4.92–30.35) | 89.5 | 0.92 |
| TNFR2+ CCR2+ non-classical monocytes | 46.92% (32.01–60.51) | 40.65% (27.48–52.73) | 75 | 0.46 |
| TNFR2+ CCR2+ intermediate monocytes | 34.09% (28.79–51.01) | 43.82% (30.47–53.36) | 67.5 | 0.28 |
| TNFR2+ CCR2+ neutrophils | 54.70% (31.19–63.78) | 44.87% (31.22–66.21) | 82 | 0.67 |
| TNFR1+TNFR2+ CCR2+ classical monocytes | 1.10% (0.67–2.24) | 1.04% (0.77–1.61) | 88 | 0.52 |
| TNFR1+TNFR2+ CCR2+ non-classical monocytes | 7.92% (1.24–18.39) | 10.37% (0.54–19.32) | 88 | 0.88 |
| TNFR1+TNFR2+ CCR2+ intermediate monocytes | 3.88% (2.72–9.66) | 5.76% (2.51–18.41) | 75 | 0.87 |
| TNFR1+TNFR2+ CCR2+ neutrophils | 6.05% (5.00–21.29) | 20.36% (3.68–30.21) | 77 | 0.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, R.B.; Laursen, C.C.H.; Nawaz, N.; Madsen, J.S.; Nielsen, H.H.; Kruuse, C.; Møller, A.; Degn, M.; Lambertsen, K.L. Leukocyte TNFR1 and TNFR2 Expression Contributes to the Peripheral Immune Response in Cases with Ischemic Stroke. Cells 2021, 10, 861. https://doi.org/10.3390/cells10040861
Hansen RB, Laursen CCH, Nawaz N, Madsen JS, Nielsen HH, Kruuse C, Møller A, Degn M, Lambertsen KL. Leukocyte TNFR1 and TNFR2 Expression Contributes to the Peripheral Immune Response in Cases with Ischemic Stroke. Cells. 2021; 10(4):861. https://doi.org/10.3390/cells10040861
Chicago/Turabian StyleHansen, Rikke B., Cathrine C. H. Laursen, Niala Nawaz, Jonna S. Madsen, Helle H. Nielsen, Christina Kruuse, Arne Møller, Matilda Degn, and Kate L. Lambertsen. 2021. "Leukocyte TNFR1 and TNFR2 Expression Contributes to the Peripheral Immune Response in Cases with Ischemic Stroke" Cells 10, no. 4: 861. https://doi.org/10.3390/cells10040861
APA StyleHansen, R. B., Laursen, C. C. H., Nawaz, N., Madsen, J. S., Nielsen, H. H., Kruuse, C., Møller, A., Degn, M., & Lambertsen, K. L. (2021). Leukocyte TNFR1 and TNFR2 Expression Contributes to the Peripheral Immune Response in Cases with Ischemic Stroke. Cells, 10(4), 861. https://doi.org/10.3390/cells10040861