
Distinct Classes of Flavonoids and Epigallocatechin Gallate, Polyphenol Affects an Oncogenic Mutant p53 Protein, Cell Growth and Invasion in a TNBC Breast Cancer Cell Line

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Plant-Derived Compounds
2.2. Cell Proliferation Assay
2.3. Lentiviral-Mediated Expression of R249S Mutant
2.4. Western Blotting
2.5. qRT-PCR
2.6. Cell Count and Doubling Time Determination
2.7. Cell Cycle Analysis
2.8. Invasion Assay
2.9. Statistical Analysis
3. Results
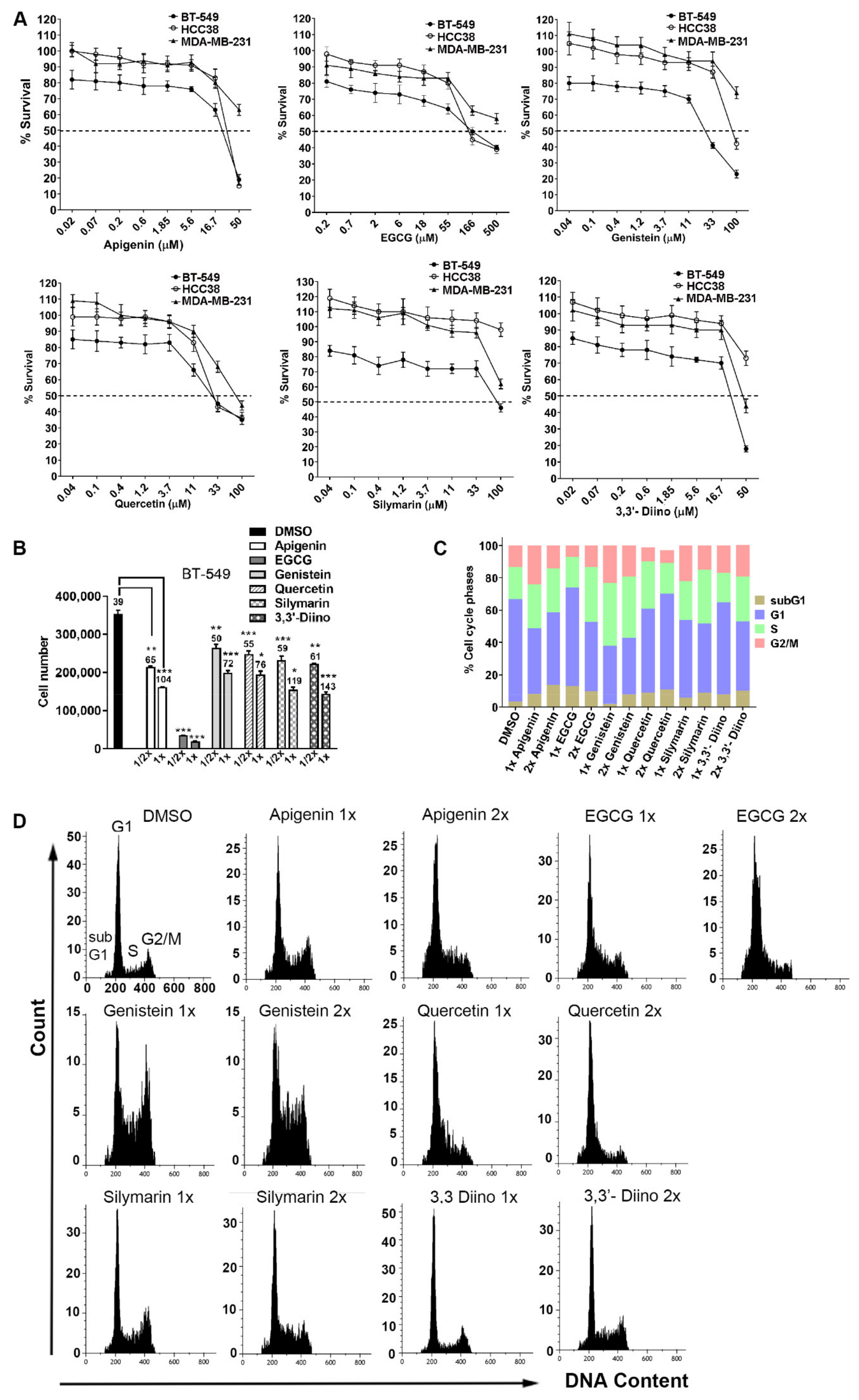
3.1. mtp53 Cell Lines Displayed Differential Sensitivities, Compounds Reduced Cell Growth and Affected Cell Cycle Distribution
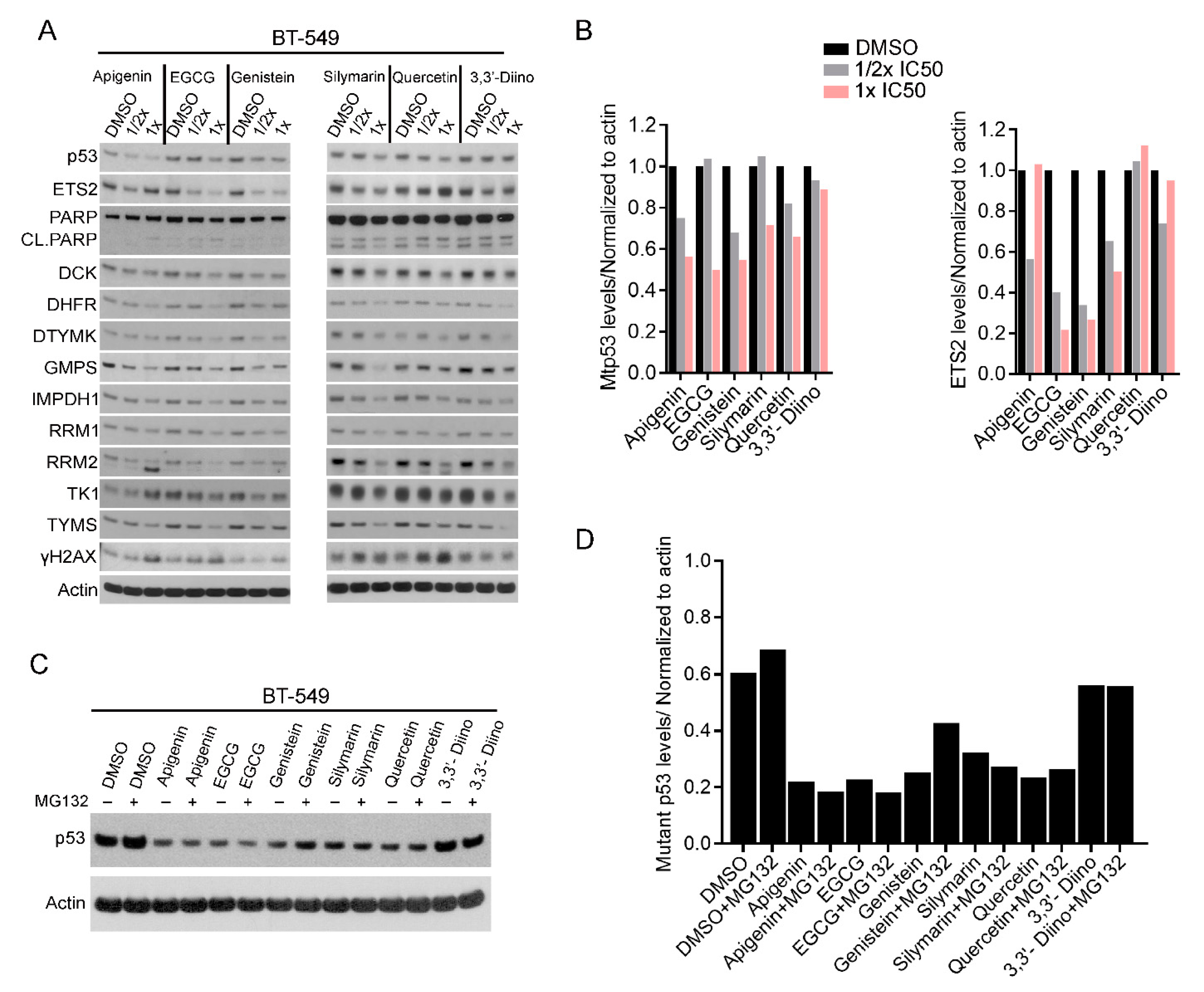
3.2. Plant-Derived Compounds Reduced mtp53 Protein Levels
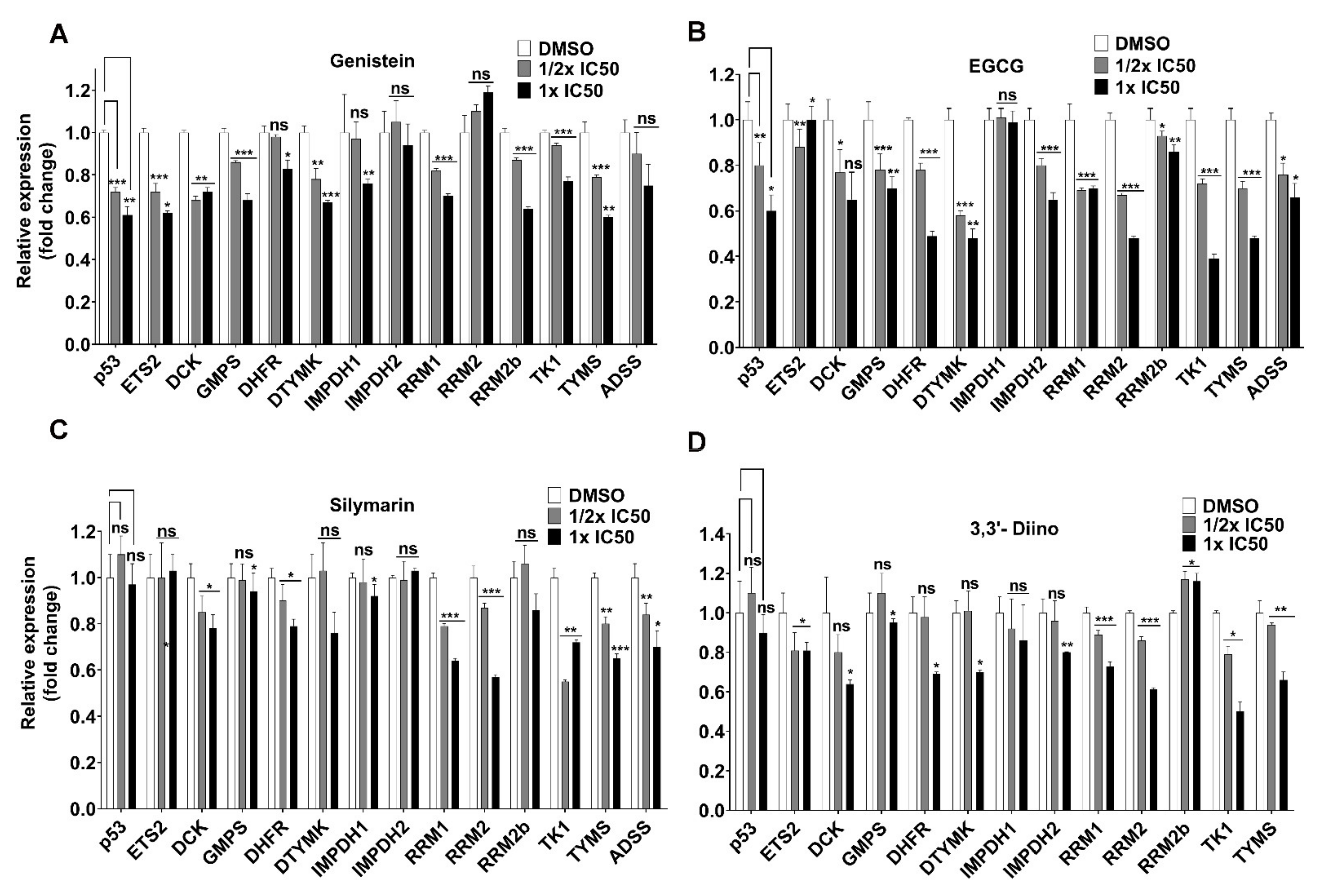
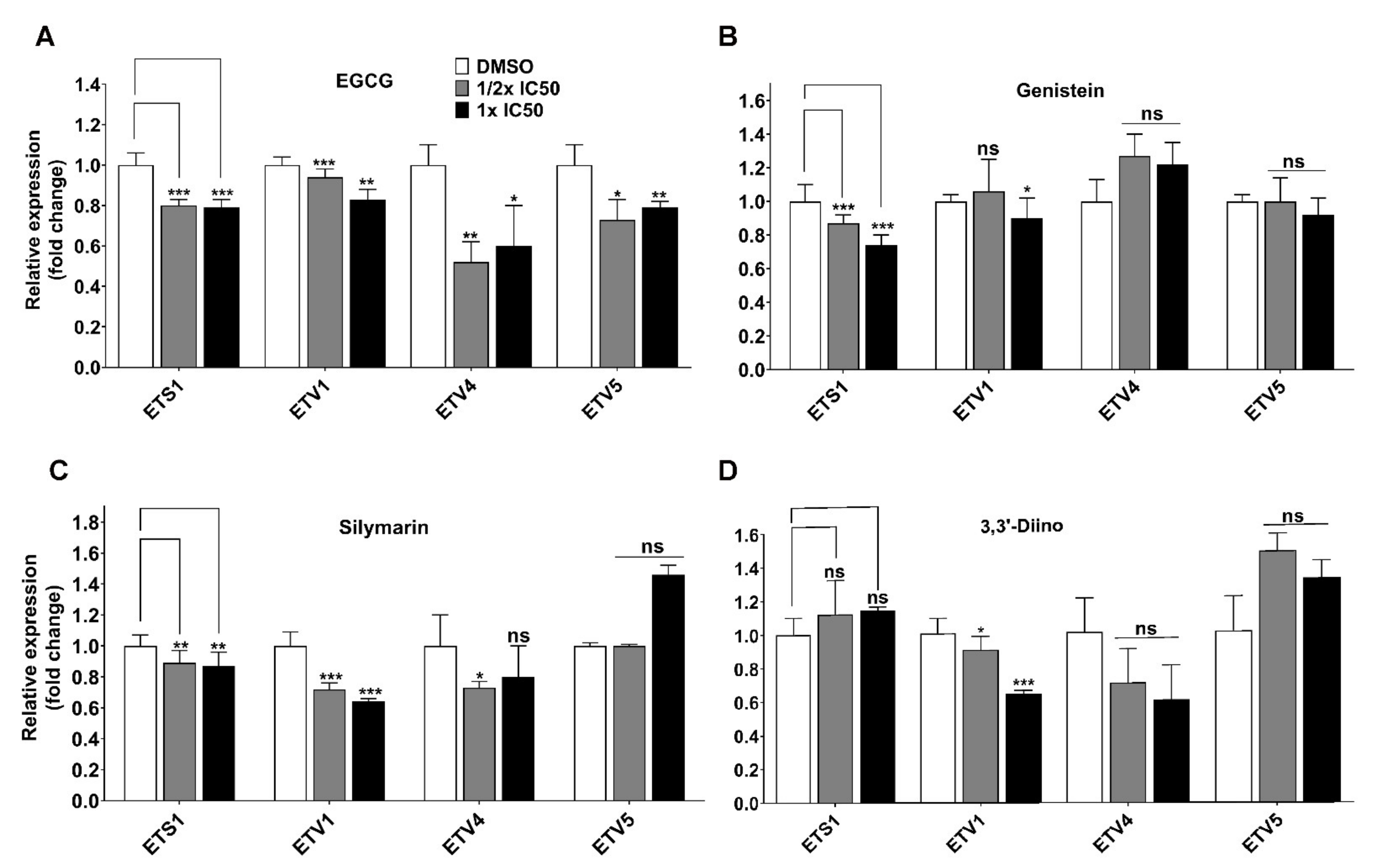
3.3. Direct Transcriptional Targets of Mutant p53 Were Affected
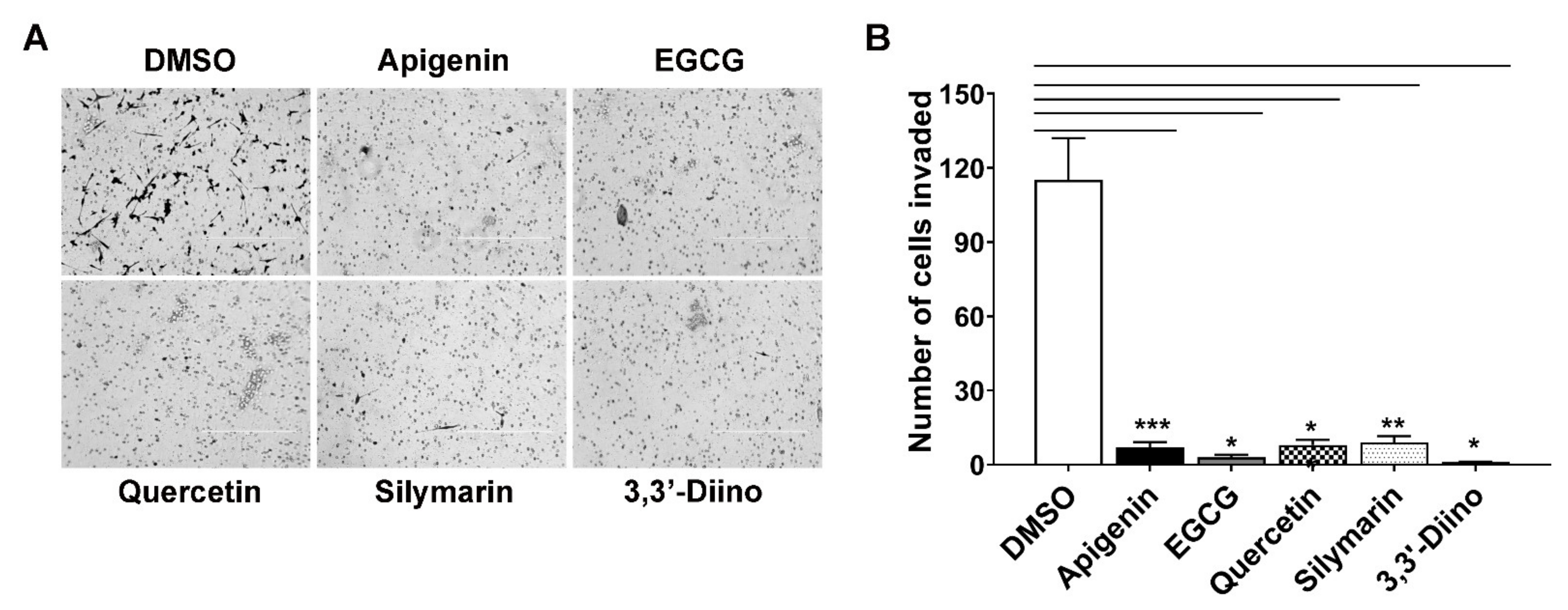
3.4. Treatment with Natural Compounds Perturbed Invasion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Tu, H.; Kollareddy, M.; Pant, V.; Li, Q.; Zhang, Y.; Jackson, J.G.; Suh, Y.-A.; Elizondo-Fraire, A.C.; Yang, P.; et al. Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc. Natl. Acad. Sci. USA 2014, 111, 11145–11150. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, Y. Targeting p53 for Novel Anticancer Therapy. Transl. Oncol. 2010, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Tye, L.; Bresnick, E.; Birt, D.F. Inhibitory effect of apigenin, a plant flavonoid, on epidermal ornithine decarboxylase and skin tumor promotion in mice. Cancer Res. 1990, 50, 499–502. [Google Scholar] [PubMed]
- King, J.C.; Lu, Q.-Y.; Li, G.; Moro, A.; Takahashi, H.; Chen, M.; Go, V.L.W.; Reber, H.A.; Eibl, G.; Hines, O.J. Evidence for activation of mutated p53 by apigenin in human pancreatic cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1823, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Elias, R.J. The antioxidant and pro-oxidant activities of green tea polyphenols: A role in cancer prevention. Arch. Biochem. Biophys. 2010, 501, 65–72. [Google Scholar] [CrossRef]
- Braicu, C.; Pileczki, V.; Pop, L.; Petric, R.C.; Chira, S.; Pointiere, E.; Achimaş-Cadariu, P.; Berindan-Neagoe, I. Dual Targeted Therapy with p53 siRNA and Epigallocatechingallate in a Triple Negative Breast Cancer Cell Model. PLoS ONE 2015, 10, e0120936. [Google Scholar] [CrossRef]
- Chen, Z.P.; Schell, J.B.; Ho, C.-T.; Chen, K.Y. Green tea epigallocatechin gallate shows a pronounced growth inhibitory effect on cancerous cells but not on their normal counterparts. Cancer Lett. 1998, 129, 173–179. [Google Scholar] [CrossRef]
- Zhao, H.; Xie, P.; Li, X.; Zhu, W.; Sun, X.; Sun, X.; Chen, X.; Xing, L.; Yu, J. A prospective phase II trial of EGCG in treatment of acute radiation-induced esophagitis for stage III lung cancer. Radiother. Oncol. 2015, 114, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Trudel, D.; Labbé, D.P.; Araya-Farias, M.; Doyen, A.; Bazinet, L.; Duchesne, T.; Plante, M.; Gregoire, J.; Renaud, M.-C.; Bachvarov, D.; et al. A two-stage, single-arm, phase II study of EGCG-enriched green tea drink as a maintenance therapy in women with advanced stage ovarian cancer. Gynecol. Oncol. 2013, 131, 357–361. [Google Scholar] [CrossRef]
- Banerjee, S.; Li, Y.; Wang, Z.; Sarkar, F.H. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008, 269, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, Y.; Marui, N.; Sakai, T.; Satomi, Y.; Yoshida, M.; Matsumoto, K.; Nishino, H.; Aoike, A. Genistein arrests cell cycle progression at G2-M. Cancer Res. 1993, 53, 1328–1331. [Google Scholar] [PubMed]
- Tominaga, Y.; Wang, A.; Wang, R.-H.; Wang, X.; Cao, L.; Deng, C.-X. Genistein inhibits Brca1 mutant tumor growth through activation of DNA damage checkpoints, cell cycle arrest, and mitotic catastrophe. Cell Death Differ. 2006, 14, 472–479. [Google Scholar] [CrossRef]
- Miles, S.L.; McFarland, M.; Niles, R.M. Molecular and physiological actions of quercetin: Need for clinical trials to assess its benefits in human disease. Nutr. Rev. 2014, 72, 720–734. [Google Scholar] [CrossRef]
- Avila, A.M.; Velasco, A.J.; Cansado, J.; Notario, V. Quercetin mediates the down-regulation of mutant p53 in the human breast cancer cell line MDA-MB468. Cancer Res. 1994, 54, 2424–2428. [Google Scholar]
- Ramasamy, K.; Agarwal, R. Multitargeted therapy of cancer by silymarin. Cancer Lett. 2008, 269, 352–362. [Google Scholar] [CrossRef]
- Le, H.T.; Schaldach, C.M.; Firestone, G.L.; Bjeldanes, L.F. Plant-derived 3,3′-Diindolylmethane Is a Strong Androgen Antagonist in Human Prostate Cancer Cells. J. Biol. Chem. 2003, 278, 21136–21145. [Google Scholar] [CrossRef]
- Hong, C.; Kim, H.-A.; Firestone, G.L.; Bjeldanes, L.F. 3,3′-Diindolylmethane (DIM) induces a G1 cell cycle arrest in human breast cancer cells that is accompanied by Sp1-mediated activation of p21WAF1/CIP1 expression. Carcinogenesis 2002, 23, 1297–1305. [Google Scholar] [CrossRef]
- Chang, X.; Tou, J.C.; Hong, C.; Kim, H.-A.; Riby, J.E.; Firestone, G.L.; Bjeldanes, L.F. 3,3’-Diindolylmethane inhibits angiogenesis and the growth of transplantable human breast carcinoma in athymic mice. Carcinogenesis 2005, 26, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012, 26, 830–845. [Google Scholar] [CrossRef]
- Kollareddy, M.; Dimitrova, E.; Vallabhaneni, K.C.; Chan, A.; Le, T.; Chauhan, K.M.; Carrero, Z.I.; Ramakrishnan, G.; Watabe, K.; Haupt, Y.; et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat. Commun. 2015, 6, 7389. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanaka, M.; Inagaki, A.; Wanibuchi, H.; Izumi, Y.; Miura, K.; Nagayama, K.; Shiota, M.; Iwao, H. Establishment of a 5-fluorouracil-resistant triple-negative breast cancer cell line. Int. J. Oncol. 2013, 43, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Elstrodt, F.; Nagel, J.H.; Kallemeijn, W.W.; Schutte, M. Phosphatidylinositol-3-OH Kinase or RAS Pathway Mutations in Human Breast Cancer Cell Lines. Mol. Cancer Res. 2007, 5, 195–201. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Hill, D.L.; Chen, X.; Wang, H.; Zhang, R.; Poage, G.M.; Butler, R.A.; Houseman, E.A.; McClean, M.D.; et al. Genistein, a Dietary Isoflavone, Down-Regulates the MDM2 Oncogene at Both Transcriptional and Posttranslational Levels. Cancer Res. 2005, 65, 8200–8208. [Google Scholar] [CrossRef]
- Oikawa, T. ETS transcription factors: Possible targets for cancer therapy. Cancer Sci. 2004, 95, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.L.; Yamamoto, H.; Singh, G. Role of the transcription factor Ets-1 in cisplatin resistance. Mol. Cancer Ther. 2004, 3, 823–832. [Google Scholar]
- Verschoor, M.L.; Singh, G. Ets-1 regulates intracellular glutathione levels: Key target for resistant ovarian cancer. Mol. Cancer 2013, 12, 138. [Google Scholar] [CrossRef]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.-H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef]
- Rahim, S.; Beauchamp, E.M.; Kong, Y.; Brown, M.L.; Toretsky, J.A.; Uren, A. YK-4-279 Inhibits ERG and ETV1 Mediated Prostate Cancer Cell Invasion. PLoS ONE 2011, 6, e19343. [Google Scholar] [CrossRef] [PubMed]
- Rahim, S.; Minas, T.; Hong, S.-H.; Justvig, S.; Çelik, H.; Kont, Y.S.; Han, J.; Kallarakal, A.T.; Kong, Y.; Rudek, M.A.; et al. A Small Molecule Inhibitor of ETV1, YK-4-279, Prevents Prostate Cancer Growth and Metastasis in a Mouse Xenograft Model. PLoS ONE 2014, 9, e114260. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Kumar, S.; Hideshima, T.; Roccaro, A.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Chauhan, D.; Munshi, N.C.; Green, S.R.; et al. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood 2005, 106, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.W.; Odedra, R.; Heaton, S.P.; Wedge, S.R.; Keen, N.J.; Crafter, C.; Foster, J.R.; Brady, M.C.; Bigley, A.; Brown, E.; et al. AZD1152, a Selective Inhibitor of Aurora B Kinase, Inhibits Human Tumor Xenograft Growth by Inducing Apoptosis. Clin. Cancer Res. 2007, 13, 3682–3688. [Google Scholar] [CrossRef]
- Yim, H. Current clinical trials with polo-like kinase 1 inhibitors in solid tumors. Anti Cancer Drugs 2013, 24, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Kollareddy, M.; Zheleva, D.; Dzubak, P.; Brahmkshatriya, P.S.; Lepsik, M.; Hajduch, M. Aurora kinase inhibitors: Progress towards the clinic. Investig. New Drugs 2012, 30, 2411–2432. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kollareddy, M.; Martinez, L.A. Distinct Classes of Flavonoids and Epigallocatechin Gallate, Polyphenol Affects an Oncogenic Mutant p53 Protein, Cell Growth and Invasion in a TNBC Breast Cancer Cell Line. Cells 2021, 10, 797. https://doi.org/10.3390/cells10040797
Kollareddy M, Martinez LA. Distinct Classes of Flavonoids and Epigallocatechin Gallate, Polyphenol Affects an Oncogenic Mutant p53 Protein, Cell Growth and Invasion in a TNBC Breast Cancer Cell Line. Cells. 2021; 10(4):797. https://doi.org/10.3390/cells10040797
Chicago/Turabian StyleKollareddy, Madhu, and Luis A. Martinez. 2021. "Distinct Classes of Flavonoids and Epigallocatechin Gallate, Polyphenol Affects an Oncogenic Mutant p53 Protein, Cell Growth and Invasion in a TNBC Breast Cancer Cell Line" Cells 10, no. 4: 797. https://doi.org/10.3390/cells10040797
APA StyleKollareddy, M., & Martinez, L. A. (2021). Distinct Classes of Flavonoids and Epigallocatechin Gallate, Polyphenol Affects an Oncogenic Mutant p53 Protein, Cell Growth and Invasion in a TNBC Breast Cancer Cell Line. Cells, 10(4), 797. https://doi.org/10.3390/cells10040797