
Effect of Hydrocortisone on Angiotensinogen (AGT) Mutation–Causing Autosomal Recessive Renal Tubular Dysgenesis

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
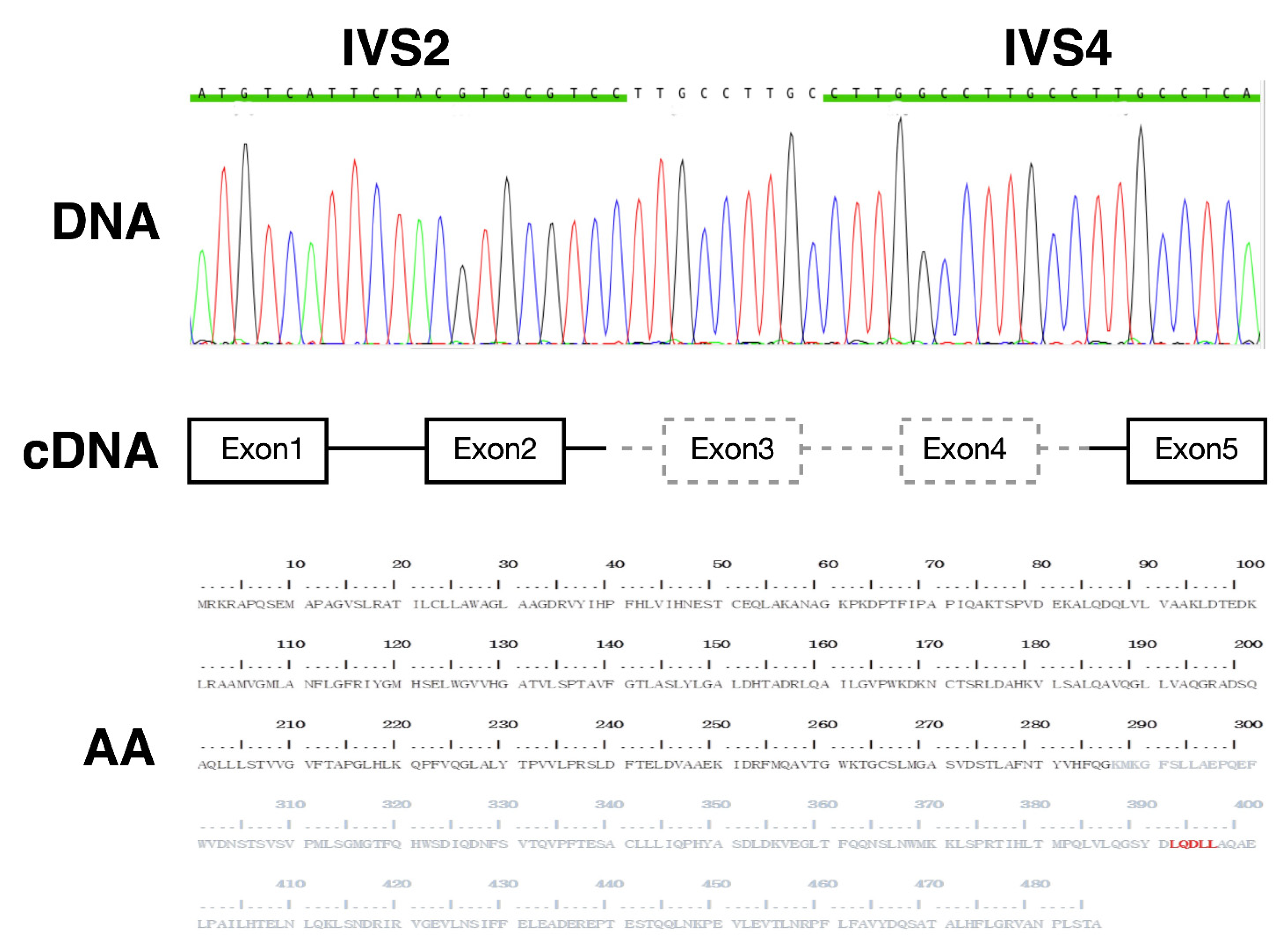
2.1. Index Case
2.2. Construction of Plasmids
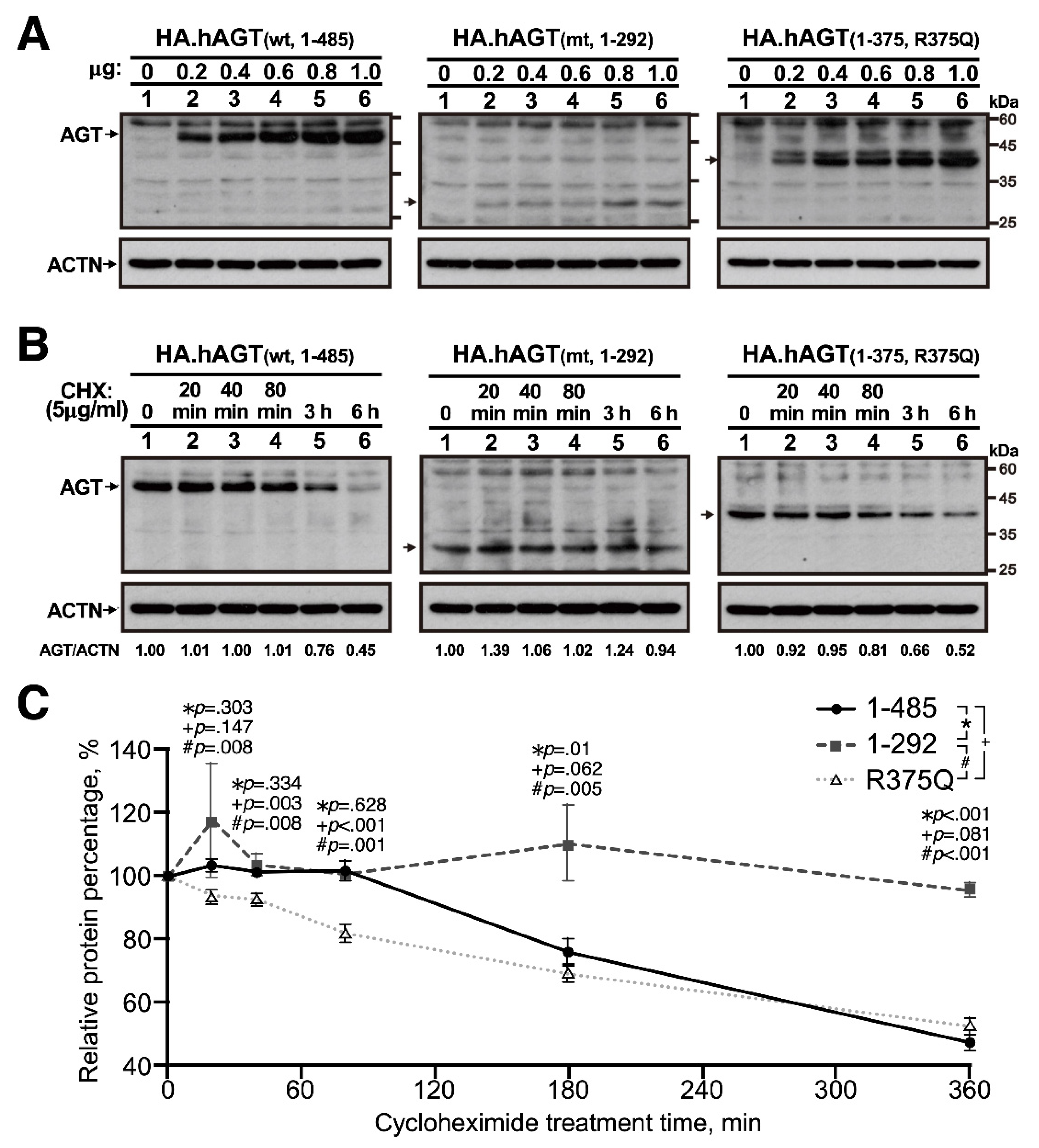
2.3. Cell Culture, Transfection and Protein Stability Assay
2.4. Western Blotting
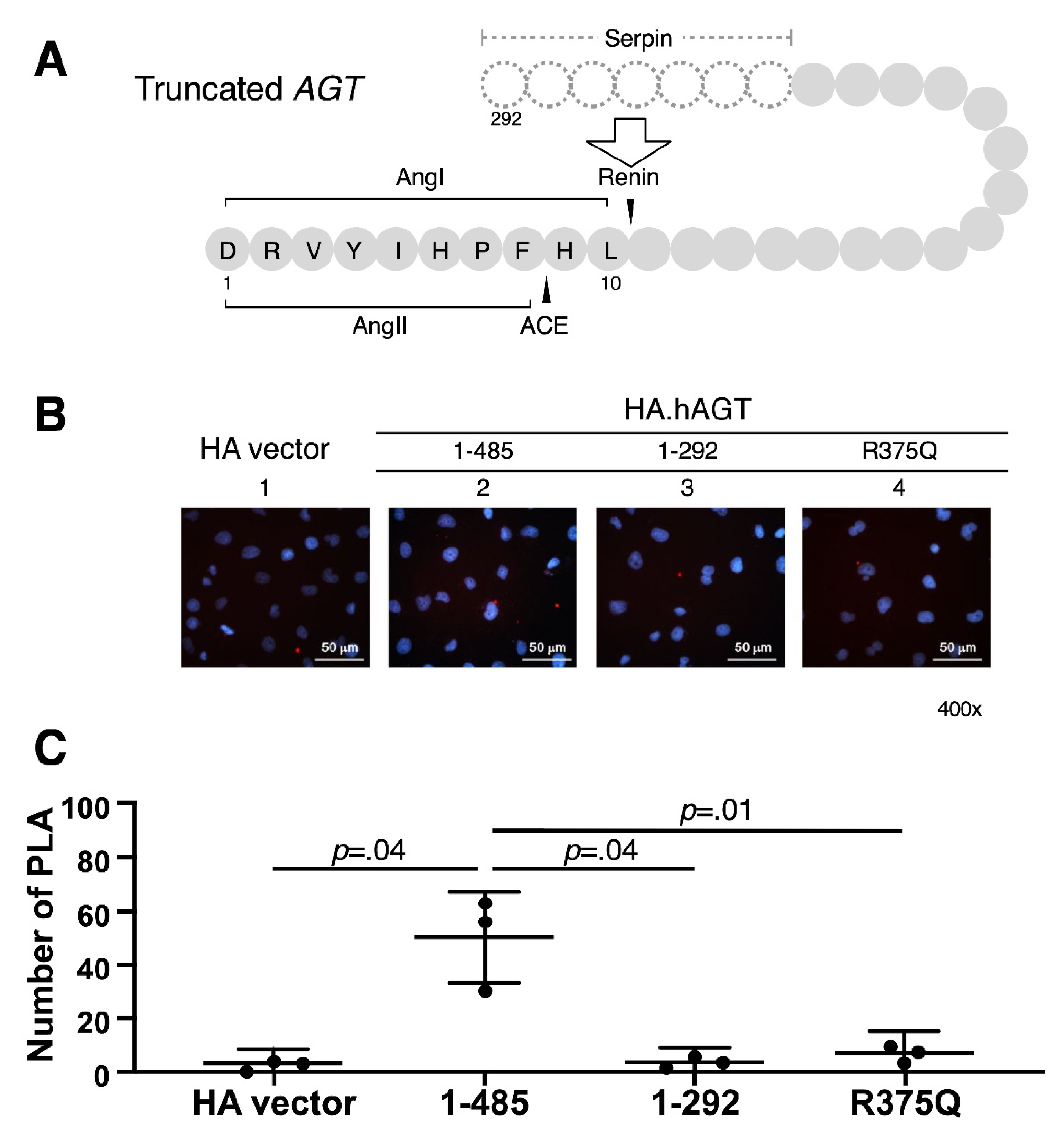
2.5. Proximity Ligation Assay (PLA)
2.6. Luciferase Reporter Analysis of Glucocorticoid Receptor (GR)-Dependent Transactivation
2.7. Statistical Analysis
3. Results
3.1. In Vitro Expression of Truncated AGT
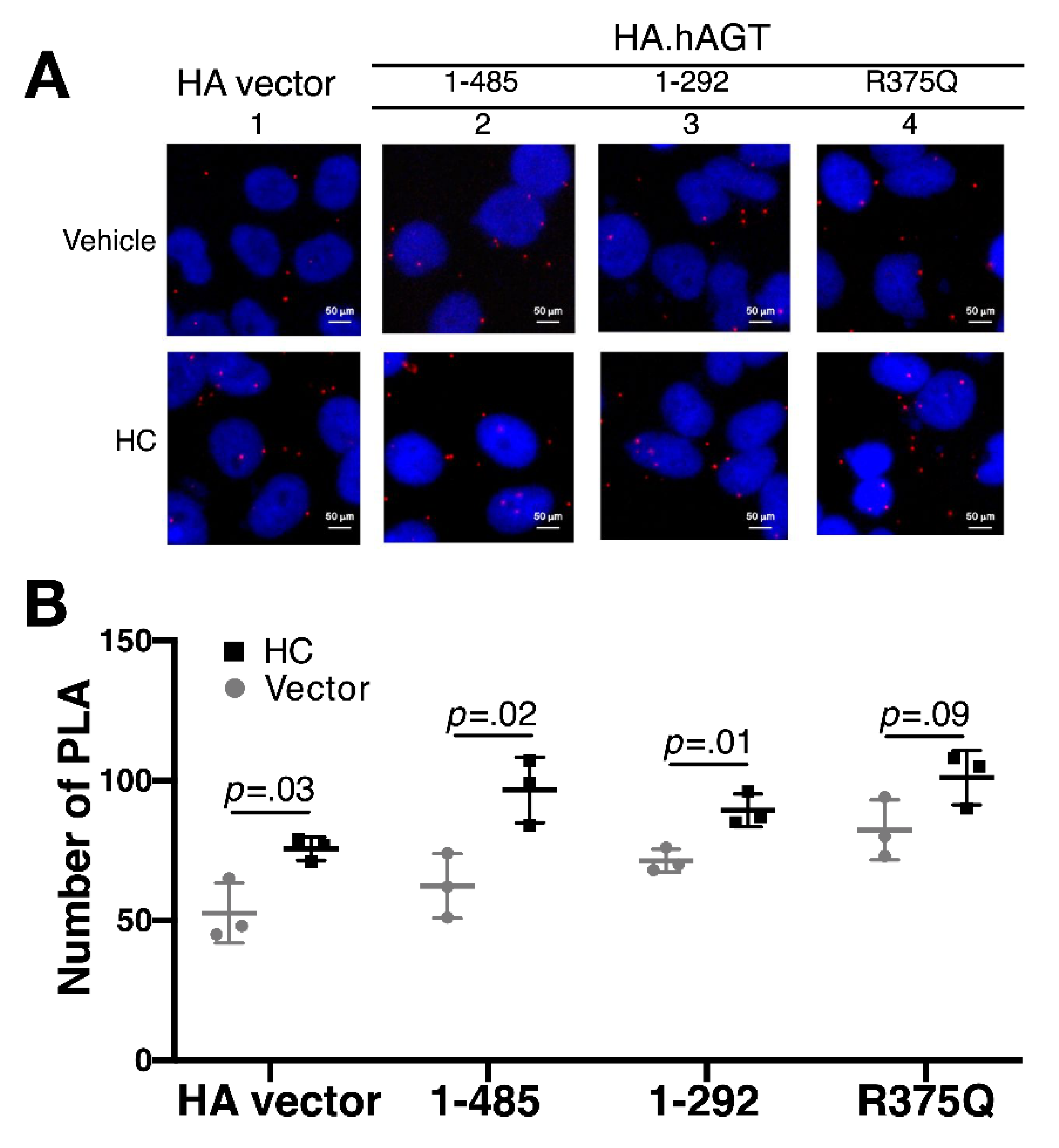
3.2. Measurement of Renin-AGT Interaction in Kidney
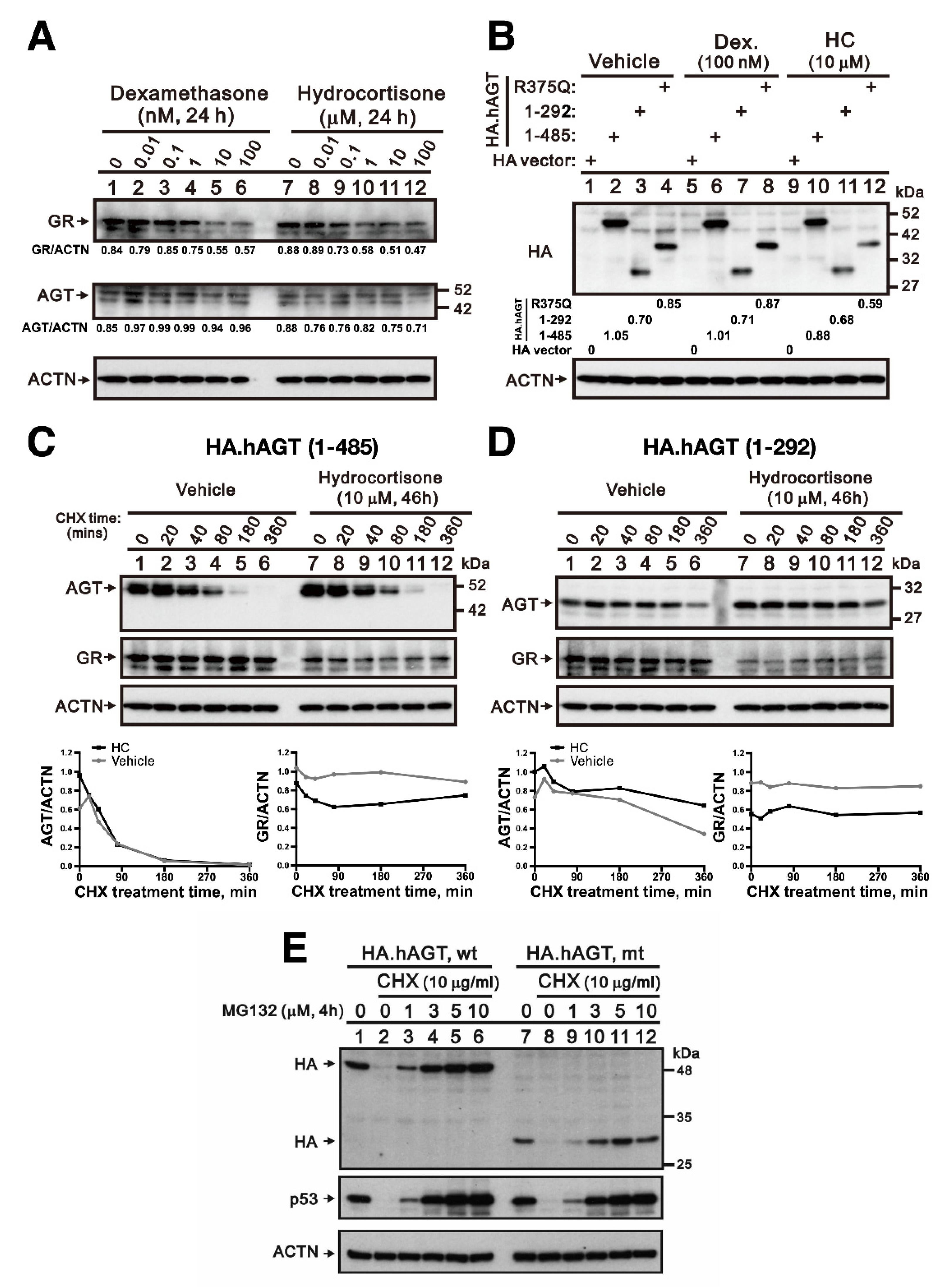
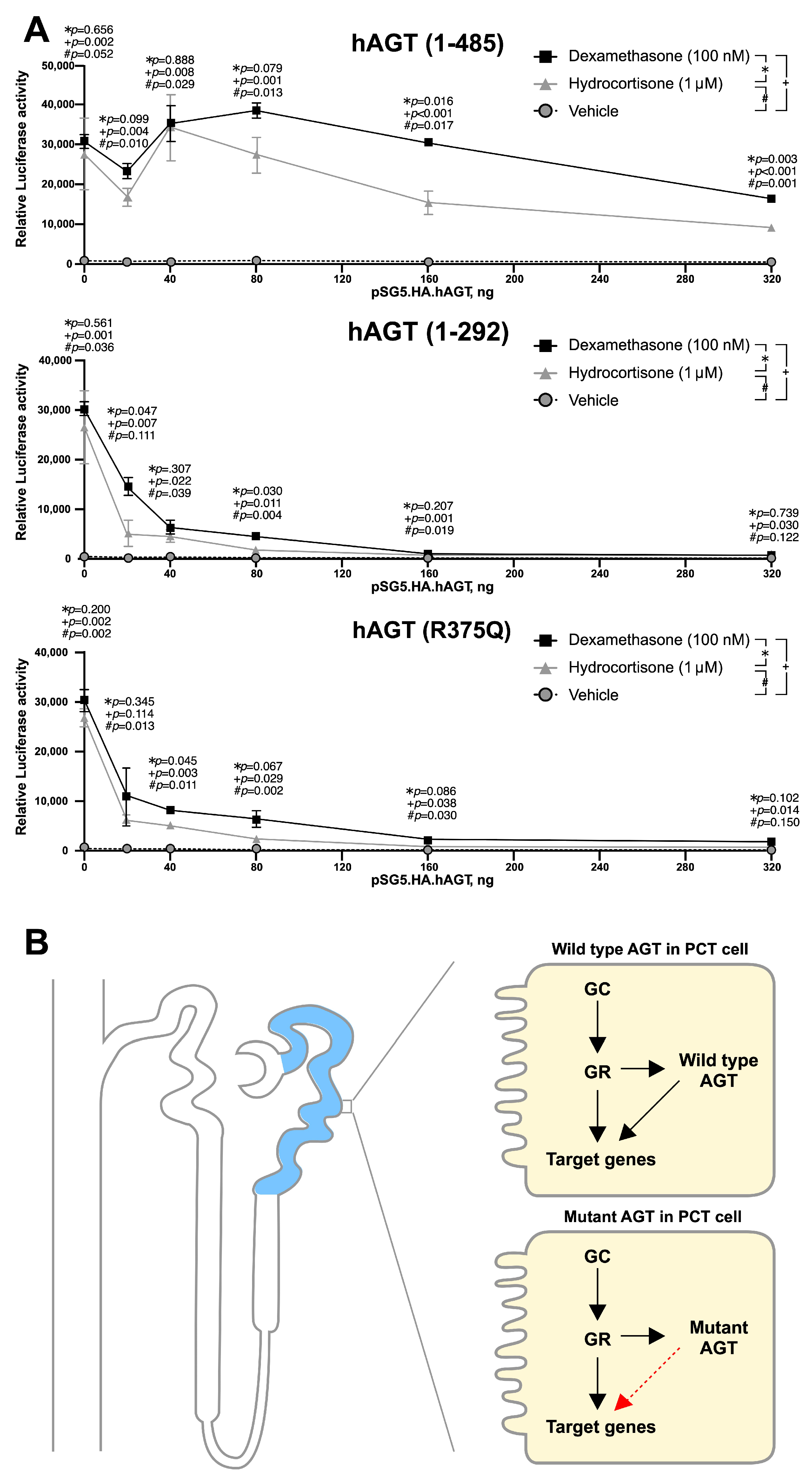
3.3. Effect of Hydrocortisone on the Expression of AGT in Liver
3.4. Effect of Hydrocortisone on the Interaction between Renin and Mutant AGT in Liver
3.5. Mutant AGT Impaired the Glucocorticoid Receptor (GR)-Dependent Transactivation
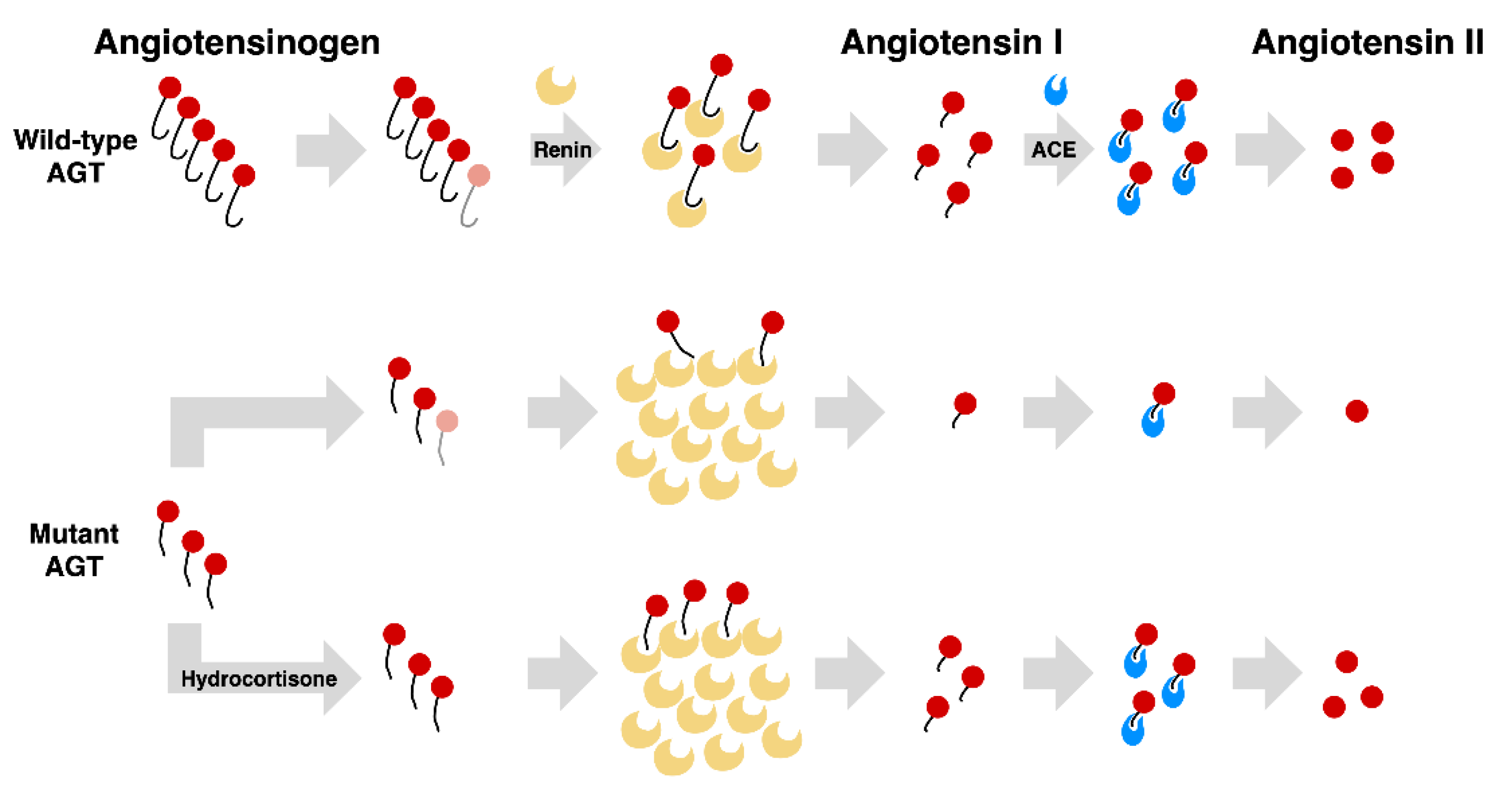
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allanson, J.E.; Pantzar, J.T.; Macleod, P.M. Possible new autosomal recessive syndrome with unusual renal histopathological changes. Am. J. Med. Genet. 1983, 16, 57–60. [Google Scholar] [CrossRef]
- Schwartz, B.R.; Lage, J.M.; Pober, B.R.; Driscoll, S.G. Isolated congenital renal tubular immaturity in siblings. Hum. Pathol. 1986, 17, 1259–1263. [Google Scholar] [CrossRef]
- Swinford, A.E.; Bernstein, J.; Toriello, H.V.; Higgins, J.V.; Opitz, J.M.; Reynolds, J.F. Renal tubular dysgenesis: Delayed onset of oligohydramnios. Am. J. Med. Genet. 1989, 32, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Allanson, J.E.; Hunter, A.G.W.; Mettler, G.S.; Jimenez, C. Renal tubular dysgenesis: A not not uncommon autosomal recessive syndrome: A review. Am. J. Med. Genet. 1992, 43, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Ariel, I.; Wells, T.R.; Landing, B.H.; Sagi, M.; Bar-Oz, B.; Ron, N.; Rosenmann, E. Familial renal tubular dysgenesis: A disorder not isolated to proximal convoluted tubules. Pediatr. Pathol. Lab. Med. 1995, 15, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Gribouval, O.; Gonzales, M.; Neuhaus, T.; Aziza, J.; Bieth, E.; Laurent, N.; Bouton, J.M.; Feuillet, F.; Makni, S.; Ben Amar, H.; et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat. Genet. 2005, 37, 964–968. [Google Scholar] [CrossRef]
- Gribouval, O.; Morinière, V.; Pawtowski, A.; Arrondel, C.; Sallinen, S.-L.; Saloranta, C.; Clericuzio, C.; Viot, G.; Tantau, J.; Blesson, S.; et al. Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum. Mutat. 2012, 33, 316–326. [Google Scholar] [CrossRef]
- Schreiber, R.; Gubler, M.-C.C.; Gribouval, O.; Shalev, H.; Landau, D. Inherited renal tubular dysgenesis may not be universally fatal. Pediatr. Nephrol. 2010, 25, 2531–2534. [Google Scholar] [CrossRef]
- Bernstein, J.; Barajas, L. Renal tubular dysgenesis: Evidence of abnormality in the renin-angiotensin system. J. Am. Soc. Nephrol. 1994, 5, 224–227. [Google Scholar]
- Bacchetta, J.; Dijoud, F.; Bouvier, R.; Putet, G.; Gubler, M.-C.; Cochat, P. Dysgénésie tubulaire proximale et mutation du gène de la rénine. Arch. Pediatr. 2007, 14, 1084–1087. [Google Scholar] [CrossRef]
- Gubler, M.C.; Antignac, C. Renin-angiotensin system in kidney development: Renal tubular dysgenesis. Kidney Int. 2010, 77, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Corvol, P.; Michaud, A.; Gribouval, O.; Gasc, J.M.; Gubler, M.C. Can we live without a functional renin-angiotensin system? Clin. Exp. Pharmacol. Physiol. 2008, 35, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Niimura, F.; Labosky, P.A.; Kakuchi, J.; Okubo, S.; Yoshida, H.; Oikawa, T.; Ichiki, T.; Naftilan, A.J.; Fogo, A.; Inagami, T. Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J. Clin. Investig. 1995, 96, 2947–2954. [Google Scholar] [CrossRef] [PubMed]
- Tseng, M.-H.; Huang, S.-M.; Huang, J.-L.; Fan, W.-L.; Konrad, M.; Shaw, S.W.; Lien, R.; Chien, H.-P.; Ding, J.-J.; Wu, T.-W.; et al. Autosomal recessive renal tubular dysgenesis caused by a founder mutation of AGT (Angiotensinogen). Kidney Int. Rep. 2020, 5, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ma, H.; Hong, H.; Koh, S.S.; Huang, S.-M.; Schurter, B.T.; Aswad, D.W.; Stallcup, M. Regulation of transcription by a protein methyltransferase. Science 1999, 284, 2174–2177. [Google Scholar] [CrossRef]
- Huang, S.-M.; Stallcup, M.R. Mouse Zac1, a Transcriptional Coactivator and Repressor for Nuclear Receptors. Mol. Cell. Biol. 2000, 20, 1855–1867. [Google Scholar] [CrossRef]
- Wallace, A.D.; Cidlowski, J.A. Proteasome-mediated Glucocorticoid Receptor Degradation Restricts Transcriptional Signaling by Glucocorticoids. J. Biol. Chem. 2001, 276, 42714–42721. [Google Scholar] [CrossRef]
- Zingg-Schenk, A.; Bacchetta, J.; Corvol, P.; Michaud, A.; Stallmach, T.; Cochat, P.; Gribouval, O.; Gubler, M.-C.; Neuhaus, T.J. Inherited renal tubular dysgenesis: The first patients surviving the neonatal period. Eur. J. Pediatr. 2008, 167, 311–316. [Google Scholar] [CrossRef]
- Yosypiv, I.V. Renin-angiotensin system in ureteric bud branching morphogenesis: Implications for kidney disease. Pediatr. Nephrol. 2014, 29, 609–620. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhao, J.; Wang, X.; Li, S.; Qin, S.; Chen, X.; Xi, N. Whole exome sequencing identifies c.963T > A and c.492 + 1G > A mutations in REN responsible for autosomal recessive renal tubular dysgenesis. J. Matern. Fetal. Neonatal Med. 2019, 1–6. [Google Scholar] [CrossRef]
- Moldavsky, M. Renal tubular dysgenesis in Israel: Pathologist’s experience and literature review. Isr. Med. Assoc. J. 2009, 11, 6–10. [Google Scholar]
- Lu, H.; Cassis, L.A.; Vander Kooi, C.W.; Daugherty, A. Structure and functions of angiotensinogen. Hypertens. Res. 2016, 39, 492–500. [Google Scholar] [CrossRef]
- Fournier, D.; Luft, F.C.; Bader, M.; Ganten, D.; Andrade-Navarro, M.A. Emergence and evolution of the renin-angiotensin-aldosterone system. J. Mol. Med. 2012, 90, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, M.; Sakamoto, O.; Nishio, T.; Ohura, T.; Matsuda, T.; Inagaki, T.; Abe, T.; Okamura, K.; Kondo, Y.; Tsuchiya, S. A case surviving for over a year of renal tubular dysgenesis with compound heterozygous angiotensinogen gene mutations. Am. J. Med. Genet. Part A 2006, 140, 2355–2360. [Google Scholar] [CrossRef]
- Ishida, J.; Sugiyama, F.; Tanimoto, K.; Taniguchi, K.; Syouji, M.; Takimoto, E.; Horiguchi, H.; Murakami, K.; Yagami, K.-I.; Fukamizu, A. Rescue of angiotensinogen-knockout mice. Biochem. Biophys. Res. Commun. 1998, 252, 610–616. [Google Scholar] [CrossRef]
- Tanimoto, K.; Sugiyama, F.; Goto, Y.; Ishida, J.; Takimoto, E.; Yagami, K.; Fukamizu, A.; Murakami, K. Angiotensinogen-deficient mice with hypotension. J. Biol. Chem. 1994, 269, 31334–31337. [Google Scholar] [CrossRef]
- Savkur, R.S.; Burris, T.P. The coactivator LXXLL nuclear receptor recognition motif. J. Pept. Res. 2004, 63, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Canonica, J.; Frateschi, S.; Boscardin, E.; Ebering, A.; Sergi, C.; Jäger, Y.; Peyrollaz, T.; Mérillat, A.-M.; Maillard, M.; Klusonova, P.; et al. Lack of renal tubular glucocorticoid receptor decreases the thiazide-sensitive Na+/Cl− cotransporter NCC and transiently affects sodium handling. Front. Physiol. 2019, 10, 989. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.A.; Sernia, C. In situ hybridization and immunohistochemistry of renal angiotensinogen in neonatal and adult rat kidneys. Cell Tissue Res. 1995, 281, 197–206. [Google Scholar] [CrossRef]
- Niimura, F.; Okubo, S.; Fogo, A.; Ichikawa, I. Temporal and spatial expression pattern of the angiotensinogen gene in mice and rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997, 272, R142–R147. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Simoes, A.C.; Maric, C.; Silva, D.M.R.; Machado, R.P.; De Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.; Dipp, S.; Meleg-Smith, S.; El-Dahr, S.S. Ureteric bud derivatives express angiotensinogen and AT1 receptors. Physiol. Genom. 2001, 6, 29–37. [Google Scholar] [CrossRef]
- Deschepper, C.F.; Dallman, M.F. The stimulation of liver angiotensinogen by glucocorticoids depends on the type of steroid and its mode of administration. Endocrinology 1992, 131, 2371–2377. [Google Scholar] [CrossRef]
- Brasier, A.R.; Li, J. Mechanisms for inducible control of angiotensinogen gene transcription. Hypertension 1996, 27, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Krid, H.; Dorison, A.; Salhi, A.; Cheval, L.; Crambert, G. Expression profile of nuclear receptors along male mouse nephron segments reveals a link between ERRβ and thick ascending limb function. PLoS ONE 2012, 7, e34223. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, M.-H.; Huang, S.-M.; Konrad, M.; Huang, J.-L.; Shaw, S.W.; Tian, Y.-C.; Chueh, H.-Y.; Fan, W.-L.; Wu, T.-W.; Ding, J.-J.; et al. Effect of Hydrocortisone on Angiotensinogen (AGT) Mutation–Causing Autosomal Recessive Renal Tubular Dysgenesis. Cells 2021, 10, 782. https://doi.org/10.3390/cells10040782
Tseng M-H, Huang S-M, Konrad M, Huang J-L, Shaw SW, Tian Y-C, Chueh H-Y, Fan W-L, Wu T-W, Ding J-J, et al. Effect of Hydrocortisone on Angiotensinogen (AGT) Mutation–Causing Autosomal Recessive Renal Tubular Dysgenesis. Cells. 2021; 10(4):782. https://doi.org/10.3390/cells10040782
Chicago/Turabian StyleTseng, Min-Hua, Shih-Ming Huang, Martin Konrad, Jing-Long Huang, Steven W. Shaw, Ya-Chung Tian, Ho-Yen Chueh, Wen-Lang Fan, Tai-Wei Wu, Jhao-Jhuang Ding, and et al. 2021. "Effect of Hydrocortisone on Angiotensinogen (AGT) Mutation–Causing Autosomal Recessive Renal Tubular Dysgenesis" Cells 10, no. 4: 782. https://doi.org/10.3390/cells10040782
APA StyleTseng, M.-H., Huang, S.-M., Konrad, M., Huang, J.-L., Shaw, S. W., Tian, Y.-C., Chueh, H.-Y., Fan, W.-L., Wu, T.-W., Ding, J.-J., Chiang, M.-C., & Lin, S.-H. (2021). Effect of Hydrocortisone on Angiotensinogen (AGT) Mutation–Causing Autosomal Recessive Renal Tubular Dysgenesis. Cells, 10(4), 782. https://doi.org/10.3390/cells10040782