
Mechanism of Na-K-ATPase Inhibition by PGE2 in Intestinal Epithelial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Viability Assays
2.4. Crude Plasma Membrane Preparation
2.5. Na-K-ATPase Activity Assay
2.6. 86Rb+ Uptake for Na-K-ATPase Activity
2.7. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.8. cAMP Measurement
2.9. Western Blot Analysis
2.10. Immunocytochemistry (ICC) Staining
2.11. Protein Determination
2.12. Statistical Analysis
3. Results
3.1. Effect of PGE2 on Na-K-ATPase in IEC-18 Cells
3.2. Effect of PGE2 on Cell Viability
3.3. Prostaglandin Receptor Antagonist Blocked PGE2 Effect on Na-K-ATPase
3.4. Effect of PGE2 on Intracellular cAMP
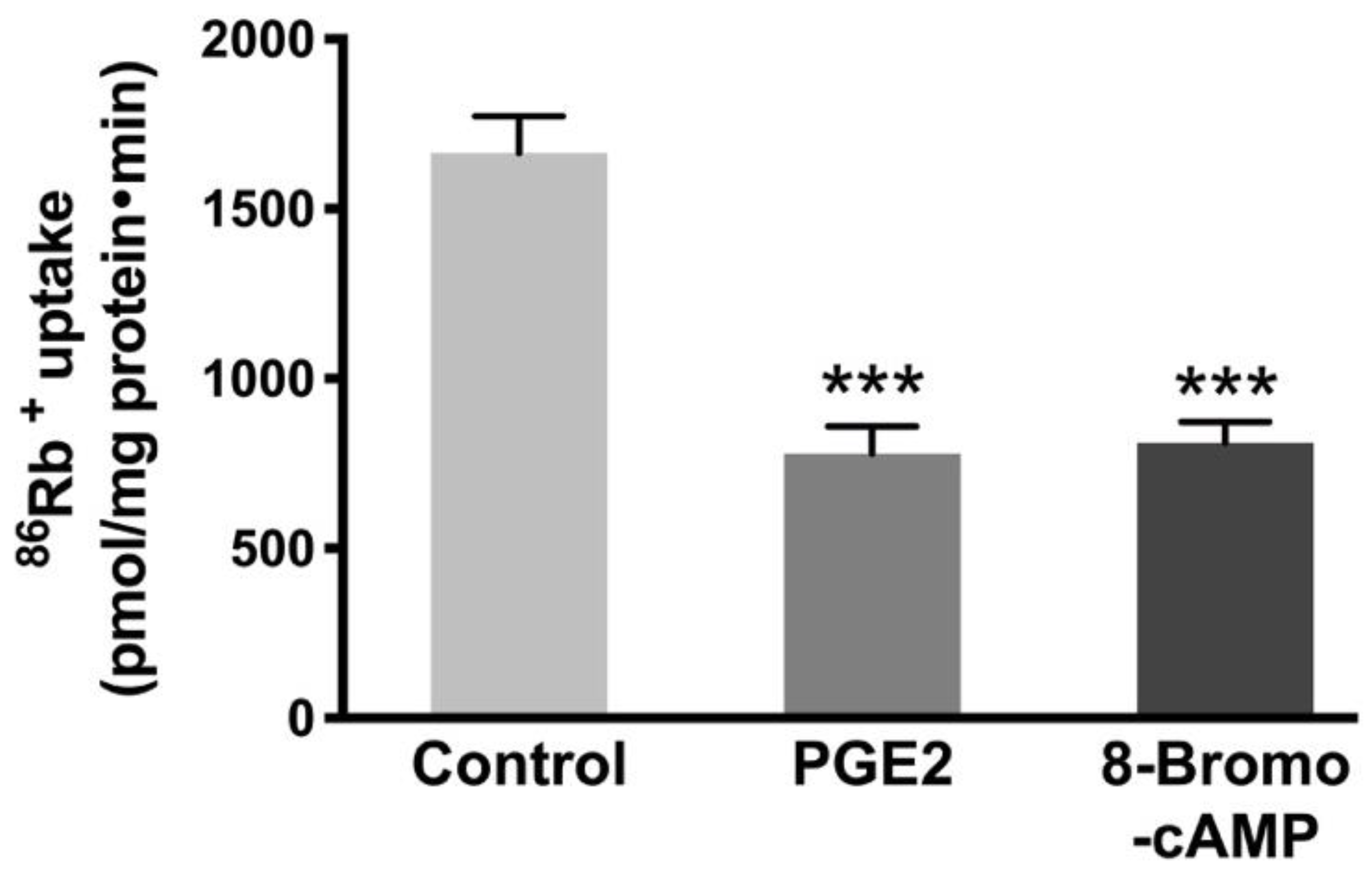
3.5. Effect of an Analog of cAMP on Na-K-ATPase Activity in IEC-18 Cells
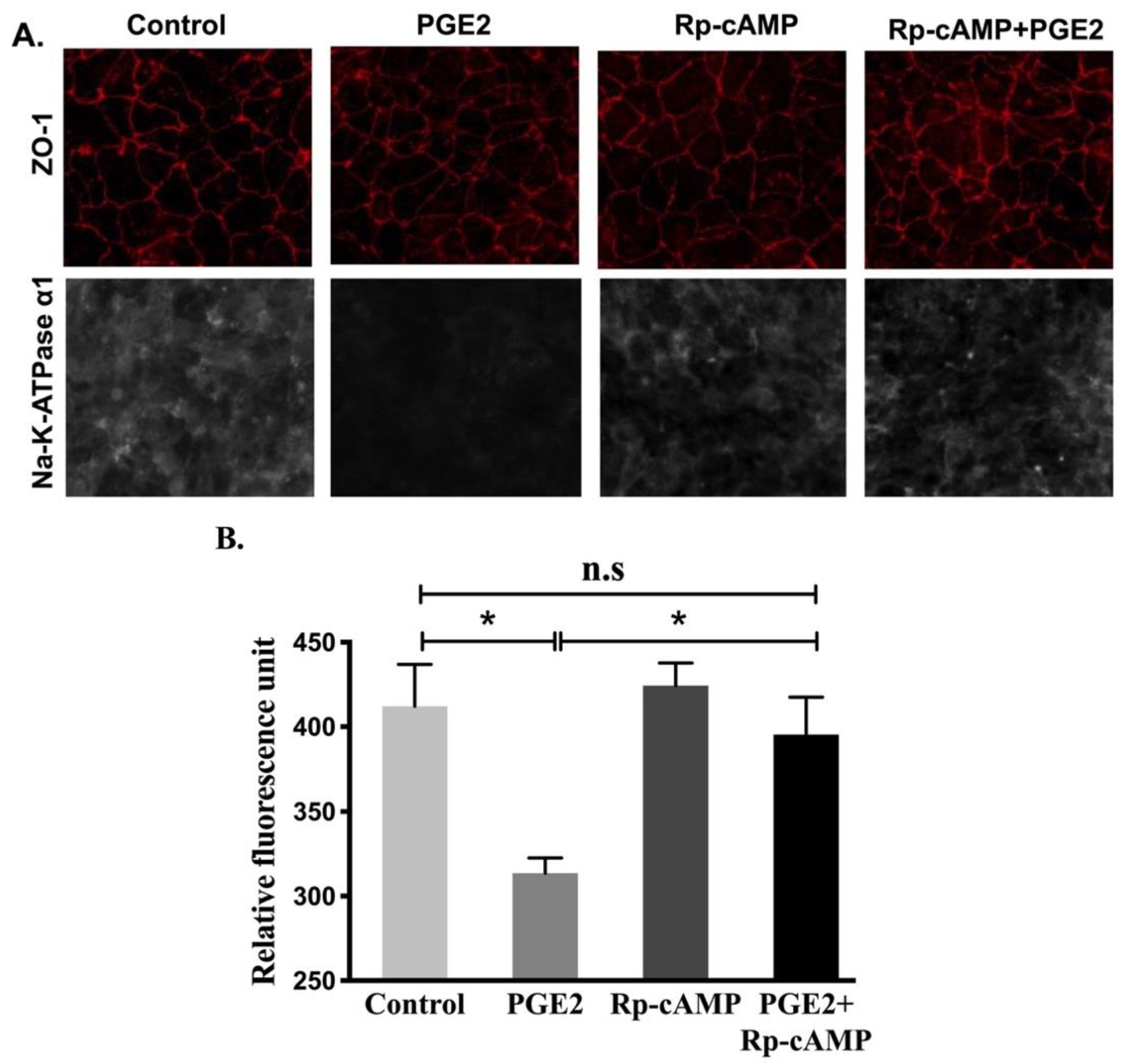
3.6. Effect of PKA Pathway Inhibition on Na-K-ATPase Activity in IEC-18 Cells
3.7. Na-K-ATPase α1 and β1 Subunit mRNA Abundance after PGE2 Treatment
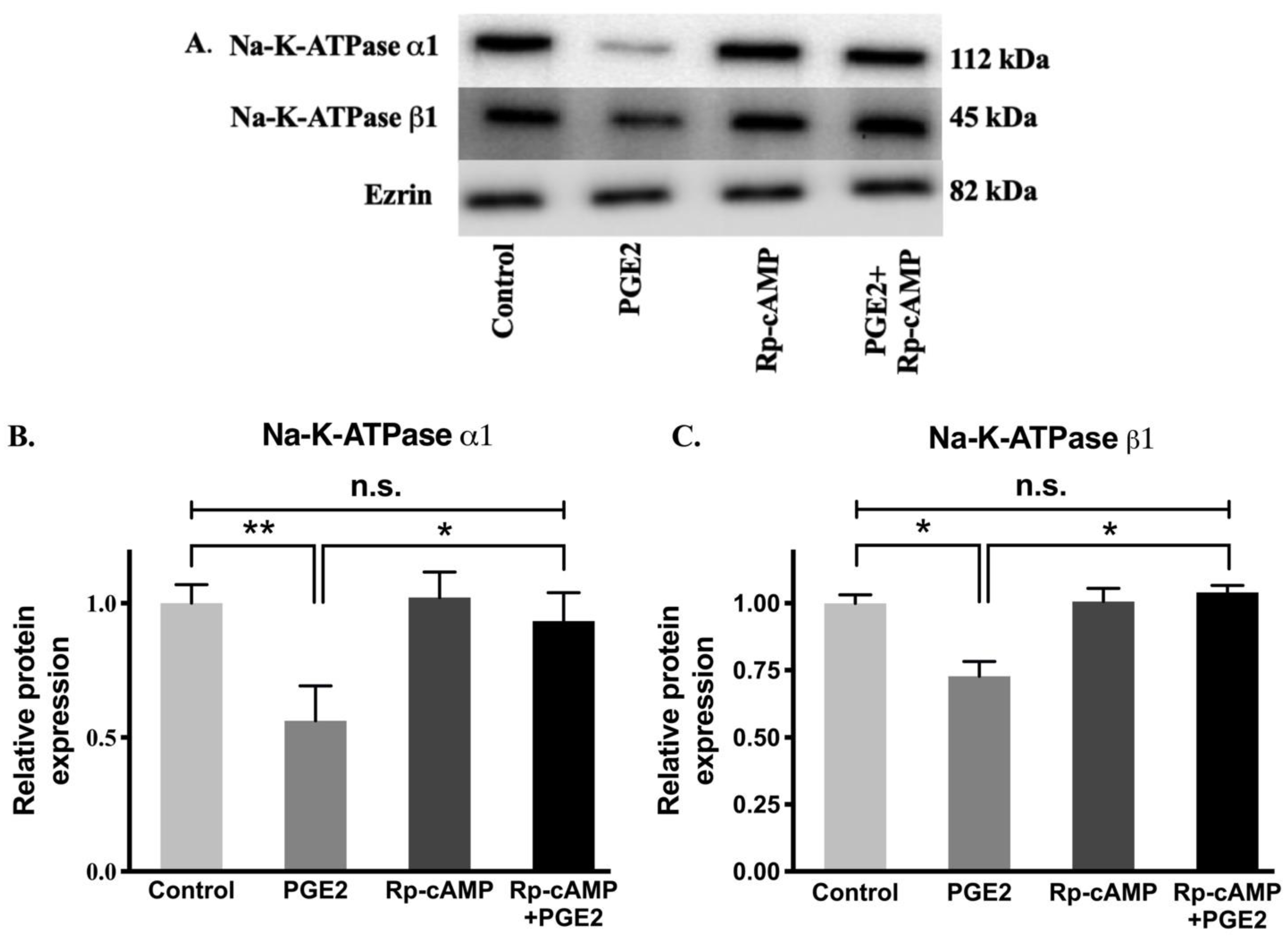
3.8. Na-K-ATPase α1 and β1 Subunit Protein Expression after PGE2 Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blanco, G.; Melton, R.J.; Sanchez, G.; Mercer, R.W. Functional characterization of a testes-specific alpha-subunit isoform of the sodium/potassium adenosinetriphosphatase. Biochemistry 1999, 38, 13661–13669. [Google Scholar] [CrossRef] [PubMed]
- Geering, K. The functional role of the beta-subunit in the maturation and intracellular transport of Na,K-ATPase. FEBS Lett. 1991, 285, 189–193. [Google Scholar] [CrossRef]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef]
- Mercer, R.W. Structure of the Na,K-ATPase. Int. Rev. Cytol. 1993, 137C, 139–168. [Google Scholar]
- Saha, P.; Manoharan, P.; Arthur, S.; Sundaram, S.; Kekuda, R.; Sundaram, U. Molecular mechanism of regulation of villus cell Na-K-ATPase in the chronically inflamed mammalian small intestine. Biochim. Biophys. Acta 2015, 1848, 702–711. [Google Scholar] [CrossRef]
- Barada, K.; Okolo, C.; Field, M.; Cortas, N. Na,K-ATPase in diabetic rat small intestine. Changes at protein and mRNA levels and role of glucagon. J. Clin. Investig. 1994, 93, 2725–2731. [Google Scholar] [CrossRef]
- Blanco, G.; Sanchez, G.; Mercer, R.W. Differential regulation of Na,K-ATPase isozymes by protein kinases and arachidonic acid. Arch. Biochem. Biophys. 1998, 359, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Musch, M.W.; Miller, R.J.; Field, M.; Siegel, M.I. Stimulation of colonic secretion by lipoxygenase metabolites of arachidonic acid. Science 1982, 217, 1255–1256. [Google Scholar] [CrossRef] [PubMed]
- Serhan, M.F.; Kreydiyyeh, S.I. Insulin targets the Na+/K+ ATPase in enterocytes via PI3K, PKC, and MAPKS. J. Recept. Signal Transduct. Res. 2011, 31, 299–306. [Google Scholar] [CrossRef]
- Bertorello, A.M.; Katz, A.I. Short-term regulation of renal Na-K-ATPase activity: Physiological relevance and cellular mechanisms. Am. J. Physiol. 1993, 265 Pt 2, F743–F755. [Google Scholar] [CrossRef]
- Ewart, H.S.; Klip, A. Hormonal regulation of the Na(+)-K(+)-ATPase: Mechanisms underlying rapid and sustained changes in pump activity. Am. J. Physiol. 1995, 269 Pt 1, C295–C311. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.A.; Farley, R.A. Regulation of Na,K-ATPase activity. Curr. Opin. Nephrol. Hypertens. 1993, 2, 725–734. [Google Scholar] [CrossRef]
- Ejderhamn, J.; Finkel, Y.; Strandvik, B. Na,K-ATPase activity in rectal mucosa of children with ulcerative colitis and Crohn’s disease. Scand. J. Gastroenterol. 1989, 24, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Rachmilewitz, D.; Karmeli, F.; Sharon, P. Decreased colonic Na-K-ATPase activity in active ulcerative colitis. Isr. J. Med. Sci. 1984, 20, 681–684. [Google Scholar]
- Sundaram, U.; Wisel, S.; Fromkes, J.J. Unique mechanism of inhibition of Na+-amino acid cotransport during chronic ileal inflammation. Am. J. Physiol. 1998, 275 Pt 1, G483–G489. [Google Scholar]
- Sundaram, U.; Wisel, S.; Rajendren, V.M.; West, A.B. Mechanism of inhibition of Na+-glucose cotransport in the chronically inflamed rabbit ileum. Am. J. Physiol. 1997, 273 Pt 1, G913–G919. [Google Scholar] [CrossRef]
- Stadnyk, A.W. Intestinal epithelial cells as a source of inflammatory cytokines and chemokines. Can. J. Gastroenterol. 2002, 16, 241–246. [Google Scholar] [CrossRef]
- Castro, G.A. Immunological regulation of electrolyte transport. In Text-Book of Secretory Diarrhea; Labenthal, E., Duffey, M.E., Eds.; Raven Press: New York, NY, USA, 1991; p. 456. [Google Scholar]
- Hinterleitner, T.A.; Powell, D.W. Immune system control of intestinal ion transport. Proc. Soc. Exp. Biol. Med. 1991, 197, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Pathogenetic and clinical relevance of cytokines in inflammatory bowel disease. Immunol. Res. 1991, 10, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Hamberg, M.; Svensson, J.; Samuelsson, B. Prostaglandin endoperoxides. A new concept concerning the mode of action and release of prostaglandins. Proc. Natl. Acad. Sci. USA 1974, 71, 3824–3828. [Google Scholar] [CrossRef]
- Mohajer, B.; Ma, T.Y. Eicosanoids and the small intestine. Prostaglandins Other Lipid Mediat. 2000, 61, 125–143. [Google Scholar] [CrossRef]
- Arthur, S.; Singh, S.; Sundaram, U. Cyclooxygenase pathway mediates the inhibition of Na-glutamine co-transporter B0AT1 in rabbit villus cells during chronic intestinal inflammation. PLoS ONE 2018, 13, e0203552. [Google Scholar] [CrossRef]
- Singh, S.; Arthur, S.; Sundaram, U. Mechanism of inhibition of Na-Glutamine Co-Transport by Prostaglandin E2 in Intestinal Epithelial Cells. Gastroenterology 2014, 146, S493. [Google Scholar] [CrossRef]
- Nepal, N.; Arthur, S.; Sundaram, U. Unique Regulation of Na-K-ATPase during Growth and Maturation of Intestinal Epithelial Cells. Cells 2019, 8, 593. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Forbush, B., 3rd. Assay of Na,K-ATPase in plasma membrane preparations: Increasing the permeability of membrane vesicles using sodium dodecyl sulfate buffered with bovine serum albumin. Anal. Biochem. 1983, 128, 159–163. [Google Scholar] [CrossRef]
- Beck, F.X.; Dörge, A.; Blümner, E.; Giebisch, G.; Thurau, K. Cell rubidium uptake: A method for studying functional heterogeneity in the nephron. Kidney Int. 1988, 33, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Dietz, T.H.; Byrne, R.A. Potassium and Rubidium Uptake in Freshwater Bivalves. J. Exp. Biol. 1990, 150, 395–405. [Google Scholar]
- Abramovitz, M.; Adam, M.; Boie, Y.; Carriere, M.; Denis, D.; Godbout, C.; Lamontagne, S.; Rochette, C.; Sawyer, N.; Tremblay, N.M.; et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim. Biophys. Acta 2000, 1483, 285–293. [Google Scholar] [CrossRef]
- Casibang, M.; Moody, T.W. AH6809 antagonizes non-small cell lung cancer prostaglandin receptors. Lung Cancer 2002, 36, 33–42. [Google Scholar] [CrossRef]
- Rahman, N.; Buck, J.; Levin, L.R. pH sensing via bicarbonate-regulated “soluble” adenylyl cyclase (sAC). Front. Physiol. 2013, 4, 343. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Poole, J.A.; Nordgren, T.M.; DeVasure, J.M.; Heires, A.J.; Bailey, K.L.; Romberger, D.J. cAMP-dependent protein kinase activation decreases cytokine release in bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L643–L651. [Google Scholar] [CrossRef]
- Walsh, D.A.; van Patten, S.M. Multiple pathway signal transduction by the cAMP-dependent protein kinase. FASEB J. 1994, 8, 1227–1236. [Google Scholar] [CrossRef]
- De Wit, R.J.; Hekstra, D.; Jastorff, B.; Stec, W.J.; Baraniak, J.; van Driel, R.; van Haastert, P.J. Inhibitory action of certain cyclophosphate derivatives of cAMP on cAMP-dependent protein kinases. Eur. J. Biochem. 1984, 142, 255–260. [Google Scholar] [CrossRef]
- Van Haastert, P.J.; van Driel, R.; Jastorff, B.; Baraniak, J.; Stec, W.J.; de Wit, R.J. Competitive cAMP antagonists for cAMP-receptor proteins. J. Biol. Chem. 1984, 259, 10020–10024. [Google Scholar] [CrossRef]
- Ligumsky, M.; Karmeli, F.; Sharon, P.; Zor, U.; Cohen, F.; Rachmilewitz, D. Enhanced thromboxane A2 and prostacyclin production by cultured rectal mucosa in ulcerative colitis and its inhibition by steroids and sulfasalazine. Gastroenterology 1981, 81, 444–449. [Google Scholar] [CrossRef]
- Sharon, P.; Ligumsky, M.; Rachmilewitz, D.; Zor, U. Role of prostaglandins in ulcerative colitis. Enhanced production during active disease and inhibition by sulfasalazine. Gastroenterology 1978, 75, 638–640. [Google Scholar] [CrossRef]
- Huang, S.K.; White, E.S.; Wettlaufer, S.H.; Grifka, H.; Hogaboam, C.M.; Thannickal, V.J.; Horowitz, J.C.; Peters-Golden, M. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 2009, 23, 4317–4326. [Google Scholar] [CrossRef]
- Hyun, H.S.; Arai, S.; Onaga, T.; Kato, S. PGE2 inhibits glucose uptake in isolated villous epithelial cells of the ovine small intestine. J. Vet. Med. Ser. A 1997, 44, 443–447. [Google Scholar] [CrossRef]
- Talukder, J.R.; Griffin, A.; Jaima, A.; Boyd, B.; Wright, J. Lactoferrin ameliorates prostaglandin E2-mediated inhibition of Na+-glucose cotransport in enterocytes. Can. J. Physiol. Pharmacol. 2014, 92, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Hodeify, R.F.; Kreydiyyeh, S.I. PGE2 reduces net water and chloride absorption from the rat colon by targeting the Na+/H+ exchanger and the Na+ K+ 2Cl− cotransporter. Prostaglandins Leukot. Essent. Fatty Acids 2007, 76, 285–292. [Google Scholar] [CrossRef]
- Moeser, A.J.; Nighot, P.K.; Ryan, K.A.; Wooten, J.G.; Blikslager, A.T. Prostaglandin-mediated inhibition of Na+/H+ exchanger isoform 2 stimulates recovery of barrier function in ischemia-injured intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G885–G894. [Google Scholar] [CrossRef] [PubMed]
- Markossian, S.; Kreydiyyeh, S.I. TNF-α down-regulates the Na+−K+ ATPase and the Na+−K+−2Cl−cotransporter in the rat colon via PGE2. Cytokine 2005, 30, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Suzuki, K.; Kawamoto, A.; Ishibashi, F.; Nakata, T.; Murano, T.; Ito, G.; Shimizu, H.; Mizutani, T.; Oshima, S.; et al. PGE2 is a direct and robust mediator of anion/fluid secretion by human intestinal epithelial cells. Sci. Rep. 2016, 6, 36795. [Google Scholar] [CrossRef] [PubMed]
- Arthur, S.; Saha, P.; Sundaram, S.; Kekuda, R.; Sundaram, U. Regulation of sodium-glutamine cotransport in villus and crypt cells by glucocorticoids during chronic enteritis. Inflamm. Bowel Dis. 2012, 18, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Skayian, Y.; Kreydiyyeh, S.I. Tumor necrosis factor alpha alters Na+−K+ ATPase activity in rat cardiac myocytes: Involvement of NF-κB, AP-1 and PGE2. Life Sci. 2006, 80, 173–180. [Google Scholar] [CrossRef]
- Kreydiyyeh, S.I.; Riman, S.; Serhan, M.; Kassardjian, A. TNF-alpha modulates hepatic Na+-K+ ATPase activity via PGE2 and EP2 receptors. Prostaglandins Other Lipid Mediat. 2007, 83, 295–303. [Google Scholar] [CrossRef]
- Seven, I.; Turkozkan, N.; Cimen, B. The effects of nitric oxide synthesis on the Na+,K(+)-ATPase activity in guinea pig kidney exposed to lipopolysaccharides. Mol. Cell. Biochem. 2005, 271, 107–112. [Google Scholar] [CrossRef]
- Cimen, B.; Turkozkan, N.; Seven, I.; Unlu, A.; Karasu, C. Impaired Na+,K+-ATPase activity as a mechanism of reactive nitrogen species-induced cytotoxicity in guinea pig liver exposed to lipopolysaccharides. Mol. Cell. Biochem. 2004, 259, 53–57. [Google Scholar] [CrossRef]
- Funk, C.D.; Furci, L.; FitzGerald, G.A.; Grygorczyk, R.; Rochette, C.; Bayne, M.A.; Abramovitz, M.; Adam, M.; Metters, K.M. Cloning and expression of a cDNA for the human prostaglandin E receptor EP1 subtype. J. Biol. Chem. 1993, 268, 26767–26772. [Google Scholar] [CrossRef]
- Woodward, D.F.; Pepperl, D.J.; Burkey, T.H.; Regan, J.W. 6-Isopropoxy-9-oxoxanthene-2-carboxylic acid (AH 6809), a human EP2 receptor antagonist. Biochem. Pharmacol. 1995, 50, 1731–1733. [Google Scholar] [CrossRef]
- Sachs, D.; Villarreal, C.; Cunha, F.; Parada, C.; Ferreira, S. The role of PKA and PKCepsilon pathways in prostaglandin E2-mediated hypernociception. Br. J. Pharmacol. 2009, 156, 826–834. [Google Scholar] [CrossRef]
- Sawasdikosol, S.; Pyarajan, S.; Alzabin, S.; Matejovic, G.; Burakoff, S.J. Prostaglandin E2 activates HPK1 kinase activity via a PKA-dependent pathway. J. Biol. Chem. 2007, 282, 34693–34699. [Google Scholar] [CrossRef]
- Oliveira, M.S.; Furian, A.F.; Rambo, L.M.; Ribeiro, L.R.; Royes, L.F.; Ferreira, J.; Calixto, J.B.; Otalora, L.F.; Garrido-Sanabria, E.R.; Mello, C.F. Prostaglandin E2 modulates Na+,K+-ATPase activity in rat hippocampus: Implications for neurological diseases. J. Neurochem. 2009, 109, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, A.; Denis, C.; Vives, M.F.; Girouard, S.; Masse, C.; Nguyen, T.; Yamagata, T.; Grygorczyk, C.; Kothary, R.; Berthiaume, Y. Modulation of alpha-ENaC and alpha1-Na+-K+-ATPase by cAMP and dexamethasone in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L217–L230. [Google Scholar] [CrossRef] [PubMed]
- Matlhagela, K.; Taub, M. Prostaglandins regulate transcription by means of prostaglandin response elements located in the promoters of mammalian Na,K-ATPase beta 1 subunit genes. Ann. N. Y. Acad. Sci. 2006, 1091, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Matlhagela, K.; Taub, M. Regulation of the Na-K-ATPase beta(1)-subunit promoter by multiple prostaglandin-responsive elements. Am. J. Physiol. Ren. Physiol. 2006, 291, F635–F646. [Google Scholar] [CrossRef]
- Matlhagela, K.; Borsick, M.; Rajkhowa, T.; Taub, M. Identification of a prostaglandin-responsive element in the Na,K-ATPase beta 1 promoter that is regulated by cAMP and Ca2+. Evidence for an interactive role of cAMP regulatory element-binding protein and Sp1. J. Biol. Chem. 2005, 280, 334–346. [Google Scholar] [CrossRef]
- Mony, S.; Lee, S.J.; Harper, J.F.; Barwe, S.P.; Langhans, S.A. Regulation of Na,K-ATPase beta1-subunit in TGF-beta2-mediated epithelial-to-mesenchymal transition in human retinal pigmented epithelial cells. Exp. Eye Res. 2013, 115, 113–122. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nepal, N.; Arthur, S.; Haynes, J.; Palaniappan, B.; Sundaram, U. Mechanism of Na-K-ATPase Inhibition by PGE2 in Intestinal Epithelial Cells. Cells 2021, 10, 752. https://doi.org/10.3390/cells10040752
Nepal N, Arthur S, Haynes J, Palaniappan B, Sundaram U. Mechanism of Na-K-ATPase Inhibition by PGE2 in Intestinal Epithelial Cells. Cells. 2021; 10(4):752. https://doi.org/10.3390/cells10040752
Chicago/Turabian StyleNepal, Niraj, Subha Arthur, Jennifer Haynes, Balasubramanian Palaniappan, and Uma Sundaram. 2021. "Mechanism of Na-K-ATPase Inhibition by PGE2 in Intestinal Epithelial Cells" Cells 10, no. 4: 752. https://doi.org/10.3390/cells10040752
APA StyleNepal, N., Arthur, S., Haynes, J., Palaniappan, B., & Sundaram, U. (2021). Mechanism of Na-K-ATPase Inhibition by PGE2 in Intestinal Epithelial Cells. Cells, 10(4), 752. https://doi.org/10.3390/cells10040752