
Crk and CrkL as Therapeutic Targets for Cancer Treatment


Abstract
1. Introduction
2. Regulation of the Cytoskeletal Network by Crk and CrkL
3. Regulation of In Vitro Cell Proliferation by Crk and CrkL
4. Regulation of Cell Transformation by Crk and CrkL
5. Regulation of In Vivo Tumor Growth and Metastasis by Crk and CrkL
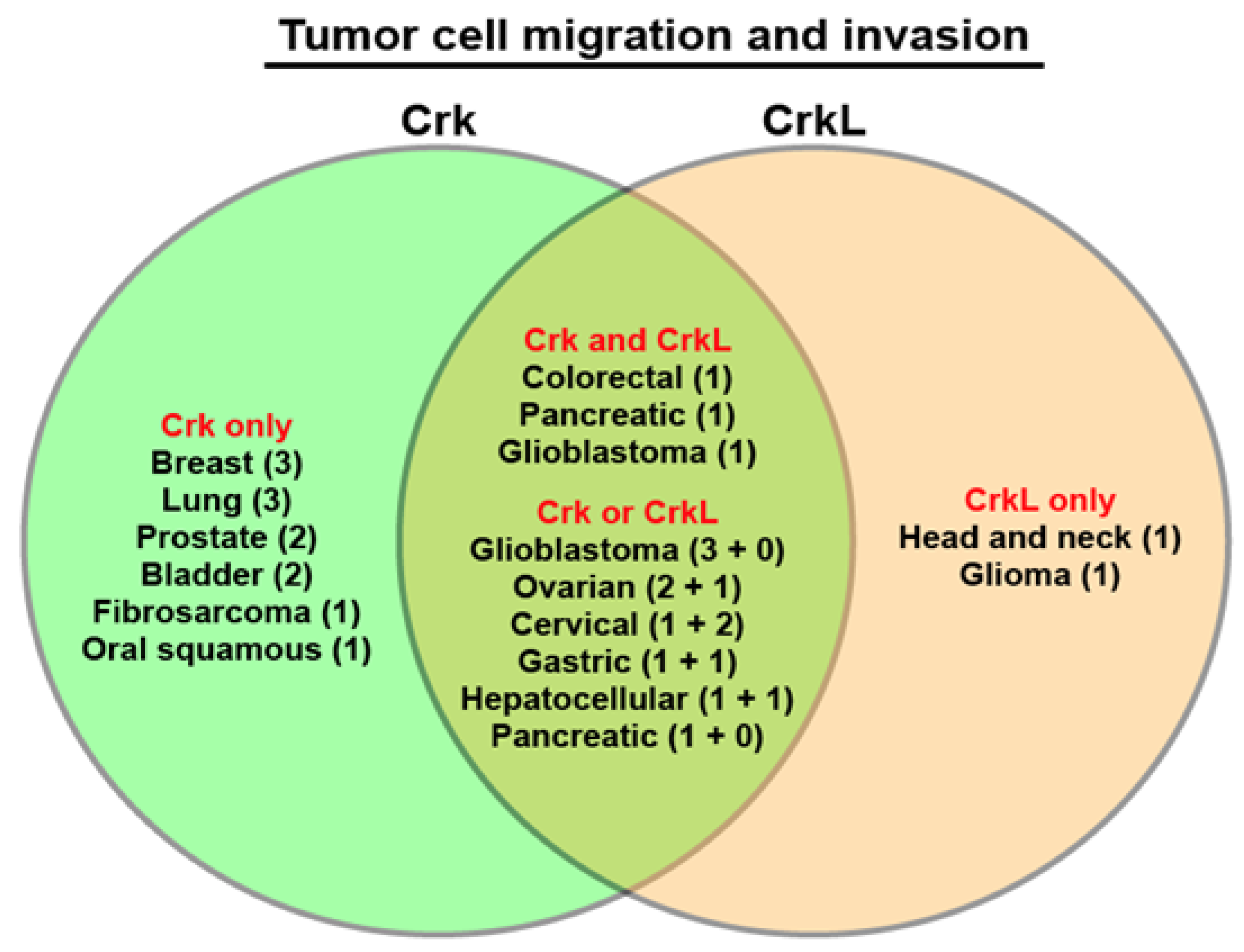
6. Regulation of Cell Migration and Invasion by Crk and CrkL
7. Regulation of EMT and Chemoresistance by Crk and CrkL
8. Overexpression of Crk and CrkL in Human Cancers and Lower Survival
9. Fibroblast Growth Factor Signaling and Tumor Cell Functions
10. Summary
11. Future Directions for Therapeutic Intervention
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Crk | CT10 regulator of kinase |
| CrkL | Crk-like |
| GBM | Glioblastoma |
| EMT | Epithelial-mesenchymal transition |
| TCGA | The Cancer Genome Atlas |
| OQL | Onco Query Language |
| NSCLC | Non-small cell lung cancer |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
References
- Mayer, B.J.; Hamaguchi, M.; Hanafusa, H. A novel viral oncogene with structural similarity to phospholipase C. Nat. Cell Biol. 1988, 332, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Tsuchie, H.; Chang, C.H.; Yoshida, M.; Vogt, P.K. A newly isolated avian sarcoma virus, ASV-1, carries the crk oncogene. Oncogene 1989, 4, 1281–1284. [Google Scholar]
- Reichman, C.T.; Mayer, B.J.; Keshav, S.; Hanafusa, H. The product of the cellular crk gene consists primarily of SH2 and SH3 regions. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 1992, 3, 451–460. [Google Scholar]
- Hoeve, J.T.; Morris, C.; Heisterkamp, N.; Groffen, J. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene 1993, 8, 2469–2474. [Google Scholar]
- Prosser, S.; Sorokina, E.; Pratt, P.; Sorokin, A. CrkIII: A novel and biologically distinct member of the Crk family of adaptor proteins. Oncogene 2003, 22, 4799–4806. [Google Scholar] [CrossRef]
- Feller, S.M. Crk family adaptors–signalling complex formation and biological roles. Oncogene 2001, 20, 6348–6371. [Google Scholar] [CrossRef]
- Birge, R.B.; Knudsen, B.S.; Besser, D.; Hanafusa, H. SH2 and SH3-containing adaptor proteins: Redundant or independent mediators of intracellular signal transduction. Genes Cells 1996, 1, 595–613. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Kalodimos, C.; Inagaki, F.; Tanaka, S. Crk and CrkL adaptor proteins: Networks for physiological and pathological signaling. Cell Commun. Signal. 2009, 7, 13. [Google Scholar] [CrossRef]
- Park, T.-J.; Curran, T. Crk and Crk-Like Play Essential Overlapping Roles Downstream of Disabled-1 in the Reelin Pathway. J. Neurosci. 2008, 28, 13551–13562. [Google Scholar] [CrossRef] [PubMed]
- Hallock, P.T.; Xu, C.-F.; Park, T.-J.; Neubert, T.A.; Curran, T.; Burden, S.J. Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes Dev. 2010, 24, 2451–2461. [Google Scholar] [CrossRef]
- Huang, Y.; Clarke, F.; Karimi, M.; Roy, N.H.; Williamson, E.K.; Okumura, M.; Mochizuki, K.; Chen, E.J.; Park, T.-J.; Debes, G.F.; et al. CRK proteins selectively regulate T cell migration into inflamed tissues. J. Clin. Investig. 2015, 125, 1019–1032. [Google Scholar] [CrossRef]
- Collins, T.N.; Mao, Y.; Li, H.; Bouaziz, M.; Hong, A.; Feng, G.-S.; Wang, F.; Quilliam, L.A.; Chen, L.; Park, T.; et al. Crk proteins transduce FGF signaling to promote lens fiber cell elongation. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Nabekura, T.; Chen, Z.; Schroeder, C.; Park, T.; Vivier, E.; Lanier, L.L.; Liu, N. Crk Adaptor Proteins Regulate NK Cell Expansion and Differentiation during Mouse Cytomegalovirus Infection. J. Immunol. 2018, 200, 3420–3428. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Fan, Q.; Dlugos, C.P.; Soofi, A.A.; Zhang, J.; Verma, R.; Park, T.-J.; Wong, H.; Curran, T.; Nihalani, D.; et al. Crk1/2 and CrkL form a hetero-oligomer and functionally complement each other during podocyte morphogenesis. Kidney Int. 2014, 85, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Curran, T. Requirement for Crk and CrkL during postnatal lens development. Biochem. Biophys. Res. Commun. 2020, 529, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Park, T.-J.; Curran, T. Essential roles of Crk and CrkL in fibroblast structure and motility. Oncogene 2013, 33, 5121–5132. [Google Scholar] [CrossRef]
- Park, T.; Koptyra, M.; Curran, T. Fibroblast Growth Requires CT10 Regulator of Kinase (Crk) and Crk-like (CrkL). J. Biol. Chem. 2016, 291, 26273–26290. [Google Scholar] [CrossRef]
- Imamoto, A.; Ki, S.; Li, L.; Iwamoto, K.; Maruthamuthu, V.; Devany, J.; Lu, O.; Kanazawa, T.; Zhang, S.; Yamada, T.; et al. Essential role of the Crk family-dosage in DiGeorge-like anomaly and metabolic homeostasis. Life Sci. Alliance 2020, 3, e201900635. [Google Scholar] [CrossRef]
- Guris, D.L.; A Fantes, J.; Tara, D.; Druker, B.J.; Imamoto, A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 2001, 27, 293–298. [Google Scholar] [CrossRef]
- Park, T.-J.; Boyd, K.; Curran, T. Cardiovascular and Craniofacial Defects in Crk-Null Mice. Mol. Cell. Biol. 2006, 26, 6272–6282. [Google Scholar] [CrossRef]
- Moon, A.M.; Guris, D.L.; Seo, J.-H.; Li, L.; Hammond, J.; Talbot, A.; Imamoto, A. Crkl Deficiency Disrupts Fgf8 Signaling in a Mouse Model of 22q11 Deletion Syndromes. Dev. Cell 2006, 10, 71–80. [Google Scholar] [CrossRef]
- Lopez-Rivera, E.; Liu, Y.P.; Verbitsky, M.; Anderson, B.R.; Capone, V.P.; Otto, E.A.; Yan, Z.; Mitrotti, A.; Martino, J.; Steers, N.J.; et al. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N. Engl. J. Med. 2017, 376, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Austgen, K.; Johnson, E.T.; Park, T.-J.; Curran, T.; Oakes, S.A. The adaptor protein CRK is a pro-apoptotic transducer of endoplasmic reticulum stress. Nat. Cell Biol. 2011, 14, 87–92. [Google Scholar] [CrossRef][Green Version]
- George, B.; Verma, R.; Soofi, A.A.; Garg, P.; Zhang, J.; Park, T.-J.; Giardino, L.; Ryzhova, L.; Johnstone, D.B.; Wong, H.; et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J. Clin. Investig. 2012, 122, 674–692. [Google Scholar] [CrossRef]
- Rodrigues, S.P.; Fathers, K.E.; Chan, G.; Zuo, D.; Halwani, F.; Meterissian, S.; Park, M. CrkI and CrkII Function as Key Signaling Integrators for Migration and Invasion of Cancer Cells. Mol. Cancer Res. 2005, 3, 183–194. [Google Scholar] [CrossRef] [PubMed]
- E Fathers, K.; Bell, E.S.; Rajadurai, C.V.; Cory, S.; Zhao, H.; Mourskaia, A.; Zuo, D.; Madore, J.; Monast, A.; Mes-Masson, A.-M.; et al. Crk adaptor proteins act as key signaling integrators for breast tumorigenesis. Breast Cancer Res. 2012, 14, R74. [Google Scholar] [CrossRef]
- Park, T.; Large, N.; Curran, T. Quantitative assessment of glioblastoma phenotypes in vitro establishes cell migration as a robust readout of Crk and CrkL activity. J. Biol. Chem. 2021, 296, 100390. [Google Scholar] [CrossRef] [PubMed]
- Linghu, H.; Tsuda, M.; Makino, Y.; Sakai, M.; Watanabe, T.; Ichihara, S.; Sawa, H.; Nagashima, K.; Mochizuki, N.; Tanaka, S. Involvement of adaptor protein Crk in malignant feature of human ovarian cancer cell line MCAS. Oncogene 2006, 25, 3547–3556. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Che, Y.-L.; Li, G.; Liu, B.; Shen, T.-M.; Wang, H.; Linghu, H. Crk and CrkL present with different expression and significance in epithelial ovarian carcinoma. Mol. Carcinog. 2011, 50, 506–515. [Google Scholar] [CrossRef]
- Watanabe, T.; Tsuda, M.; Tanaka, S.; Ohba, Y.; Kawaguchi, H.; Majima, T.; Sawa, H.; Minami, A. Adaptor Protein Crk Induces Src-Dependent Activation of p38 MAPK in Regulation of Synovial Sarcoma Cell Proliferation. Mol. Cancer Res. 2009, 7, 1582–1592. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, X.; Guo, C.; Sun, M.; Liu, S. CRKII overexpression promotes the in vitro proliferation, migration and invasion potential of murine hepatocarcinoma Hca‑P cells. Oncol. Lett. 2019, 17, 5169–5174. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, Q.-Z.; Fu, L.; Stoecker, M.; Wang, E.; Wang, E.-H. Overexpression of crkl correlates with poor prognosis and cell proliferation in non-small cell lung cancer. Mol. Carcinog. 2012, 52, 890–899. [Google Scholar] [CrossRef]
- Yeung, C.L.; Ngo, V.N.; Grohar, P.J.; I Arnaldez, F.; Asante, A.; Wan, X.; Khan, J.; Hewitt, S.M.; Khanna, C.; Staudt, L.M.; et al. Loss-of-function screen in rhabdomyosarcoma identifies CRKL-YES as a critical signal for tumor growth. Oncogene 2013, 32, 5429–5438. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Miao, Z.; Wang, Z.; Xu, Y.; Wu, J.; Liu, X.; You, Y.; Li, J. Overexpression of CRKL correlates with malignant cell proliferation in breast cancer. Tumor Biol. 2013, 34, 2891–2897. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, X.; Li, P.; Su, L.; Yu, B.; Cai, Q.; Li, J.; Yu, Y.; Liu, B.; Zhu, Z. CRKL promotes cell proliferation in gastric cancer and is negatively regulated by miR-126. Chem. Interact. 2013, 206, 230–238. [Google Scholar] [CrossRef]
- Song, Q.; Yi, F.; Zhang, Y.; Li, D.K.J.; Wei, Y.; Yu, H.; Zhang, Y. CRKL regulates alternative splicing of cancer-related genes in cervical cancer samples and HeLa cell. BMC Cancer 2019, 19, 499. [Google Scholar] [CrossRef]
- Ji, H.; Li, B.; Zhang, S.; He, Z.; Zhou, Y.; Ouyang, L. Crk-like adapter protein is overexpressed in cervical carcinoma, facilitates proliferation, invasion and chemoresistance, and regulates Src and Akt signaling. Oncol. Lett. 2016, 12, 3811–3817. [Google Scholar] [CrossRef]
- Cai, L.; Wang, H.; Yang, Q. CRKL overexpression promotes cell proliferation and inhibits apoptosis in endometrial carcinoma. Oncol. Lett. 2017, 13, 51–56. [Google Scholar] [CrossRef][Green Version]
- Ren, Y.; Shang, J.; Li, J.; Liu, W.; Zhang, Z.; Yuan, J.; Yang, M. The long noncoding RNA PCAT-1 links the microRNA miR-215 to oncogene CRKL-mediated signaling in hepatocellular carcinoma. J. Biol. Chem. 2017, 292, 17939–17949. [Google Scholar] [CrossRef] [PubMed]
- Franke, F.C.; Slusarenko, B.O.; Engleitner, T.; Johannes, W.; Laschinger, M.; Rad, R.; Nitsche, U.; Janssen, K. Novel role for CRK adaptor proteins as essential components of SRC/FAK signaling for epithelial–mesenchymal transition and colorectal cancer aggressiveness. Int. J. Cancer 2020, 147, 1715–1731. [Google Scholar] [CrossRef]
- Wang, L.; Tabu, K.; Kimura, T.; Tsuda, M.; Linghu, H.; Tanino, M.; Kaneko, S.; Nishihara, H.; Tanaka, S. Signaling adaptor protein Crk is indispensable for malignant feature of glioblastoma cell line KMG4. Biochem. Biophys. Res. Commun. 2007, 362, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Qin, J.; Yi, R.; Coreman, M.; Shi, R.; Kang, H.; Yao, C. Crkl Efficiently Mediates Cell Proliferation, Migration, and Invasion Induced by TGF-β Pathway in Glioblastoma. J. Mol. Neurosci. 2013, 51, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, X.; Hu, B.; Wang, X.-J.; Wang, Q.; Wang, W.-L. The effects of Micro-429 on inhibition of cervical cancer cells through targeting ZEB1 and CRKL. Biomed. Pharmacother. 2016, 80, 311–321. [Google Scholar] [CrossRef]
- Kumar, S.; Davra, V.; Obr, A.E.; Geng, K.; Wood, T.L.; De Lorenzo, M.S.; Birge, R.B. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. OncoImmunology 2017, 7, e1376155. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; Tsuda, M.; Wang, L.; Maishi, N.; Abe, T.; Kimura, T.; Tanino, M.; Nishihara, H.; Hida, K.; Ohba, Y.; et al. Adaptor protein CRK induces epithelial–mesenchymal transition and metastasis of bladder cancer cells through HGF /c-Met feedback loop. Cancer Sci. 2015, 106, 709–717. [Google Scholar] [CrossRef]
- Yanagi, H.; Wang, L.; Nishihara, H.; Kimura, T.; Tanino, M.; Yanagi, T.; Fukuda, S.; Tanaka, S. CRKL plays a pivotal role in tumorigenesis of head and neck squamous cell carcinoma through the regulation of cell adhesion. Biochem. Biophys. Res. Commun. 2012, 418, 104–109. [Google Scholar] [CrossRef]
- Franke, F.C.; Müller, J.; Abal, M.; Medina, E.D.; Nitsche, U.; Weidmann, H.; Chardonnet, S.; Ninio, E.; Janssen, K.-P. The Tumor Suppressor SASH1 Interacts With the Signal Adaptor CRKL to Inhibit Epithelial–Mesenchymal Transition and Metastasis in Colorectal Cancer. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 33–53. [Google Scholar] [CrossRef]
- Petit, V.; Boyer, B.; Lentz, D.; Turner, C.E.; Thiery, J.P.; Vallés, A.M. Phosphorylation of Tyrosine Residues 31 and 118 on Paxillin Regulates Cell Migration through an Association with Crk in Nbt-II Cells. J. Cell Biol. 2000, 148, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Takino, T.; Nakada, M.; Miyamori, H.; Yamashita, J.; Yamada, K.M.; Sato, H. CrkI adapter protein modulates cell migration and invasion in glioblastoma. Cancer Res. 2003, 63, 2335–2337. [Google Scholar]
- Kumar, S.; Lu, B.; Dixit, U.; Hossain, S.; Liu, Y.; Li, J.; Hornbeck, P.; Zheng, W.; Sowalsky, A.G.; Kotula, L.; et al. Reciprocal regulation of Abl kinase by Crk Y251 and Abi1 controls invasive phenotypes in glioblastoma. Oncotarget 2015, 6, 37792–37807. [Google Scholar] [CrossRef] [PubMed]
- Takino, T.; Tamura, M.; Miyamori, H.; Araki, M.; Matsumoto, K.; Sato, H.; Yamada, K.M. Tyrosine phosphorylation of the CrkII adaptor protein modulates cell migration. J. Cell Sci. 2003, 116, 3145–3155. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Watanabe, T.; Tsuda, M.; Makino, Y.; Ichihara, S.; Sawa, H.; Minami, A.; Mochizuki, N.; Nagashima, K.; Tanaka, S. Adaptor Molecule Crk Is Required for Sustained Phosphorylation of Grb2-Associated Binder 1 and Hepatocyte Growth Factor–Induced Cell Motility of Human Synovial Sarcoma Cell Lines. Mol. Cancer Res. 2006, 4, 499–510. [Google Scholar] [CrossRef]
- Yamada, S.-I.; Yanamoto, S.; Kawasaki, G.; Rokutanda, S.; Yonezawa, H.; Kawakita, A.; Nemoto, T.K. Overexpression of CRKII increases migration and invasive potential in oral squamous cell carcinoma. Cancer Lett. 2011, 303, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Qi, L.; Zhang, X.; Li, Y.; Chen, M.; Zu, X. CrkI and p130Cas complex regulates the migration and invasion of prostate cancer cells. Cell Biochem. Funct. 2011, 29, 625–629. [Google Scholar] [CrossRef]
- Dhupkar, P.; Zhao, H.; Mujoo, K.; An, Z.; Zhang, N. Crk II silencing down-regulates IGF-IR and inhibits migration and invasion of prostate cancer cells. Biochem. Biophys. Rep. 2016, 8, 382–388. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kumar, S.; Lu, B.; Davra, V.; Hornbeck, P.; Machida, K.; Birge, R.B. Crk Tyrosine Phosphorylation Regulates PDGF-BB–inducible Src Activation and Breast Tumorigenicity and Metastasis. Mol. Cancer Res. 2017, 16, 173–183. [Google Scholar] [CrossRef]
- Pezeshkpour, G.H.; Moatamed, F.; Lewis, M.; Hoang, B.; Rettig, M.; Mortazavi, F. CRK SH3N Domain Diminishes Cell Invasiveness of Non-Small Cell Lung Cancer. Genes Cancer 2013, 4, 315–324. [Google Scholar] [CrossRef][Green Version]
- Rettig, M.; Trinidad, K.; Pezeshkpour, G.; Frost, P.; Sharma, S.; Moatamed, F.; Tamanoi, F.; Mortazavi, F. PAK1 Kinase Promotes Cell Motility and Invasiveness through CRK-II Serine Phosphorylation in Non-Small Cell Lung Cancer Cells. PLoS ONE 2012, 7, e42012. [Google Scholar] [CrossRef]
- Li, X.; Wang, F.; Qi, Y. MiR-126 inhibits the invasion of gastric cancer cell in part by targeting Crk. Eur. Rev. Med Pharmacol. Sci. 2014, 18, 2031–2037. [Google Scholar]
- Uemura, S.; Wang, L.; Tsuda, M.; Suzuka, J.; Tanikawa, S.; Sugino, H.; Nakamura, T.; Mitsuhashi, T.; Hirano, S.; Tanaka, S. Signaling adaptor protein Crk is involved in malignant feature of pancreatic cancer associated with phosphorylation of c-Met. Biochem. Biophys. Res. Commun. 2020, 524, 378–384. [Google Scholar] [CrossRef]
- Cheng, S.; Guo, J.; Yang, Q.; Han, L. Crk-like adapter protein is required for TGF-β-induced AKT and ERK-signaling pathway in epithelial ovarian carcinomas. Tumor Biol. 2014, 36, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Sun, M.-Z.; Guo, C.; Shi, J.; Chen, X.; Liu, S. CRKL overexpression suppresses in vitro proliferation, invasion and migration of murine hepatocarcinoma Hca-P cells. Biomed. Pharmacother. 2015, 69, 11–17. [Google Scholar] [CrossRef]
- Han, G.; Wu, D.; Yang, Y.; Li, Z.; Zhang, J.; Li, C. CrkL meditates CCL20/CCR6-induced EMT in gastric cancer. Cytokine 2015, 76, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Elmansuri, A.Z.; Tanino, M.A.; Mahabir, R.; Wang, L.; Kimura, T.; Nishihara, H.; Kinoshita, I.; Dosaka-Akita, H.; Tsuda, M.; Tanaka, S. Novel signaling collaboration between TGF-β and adaptor protein Crk facilitates EMT in human lung cancer. Oncotarget 2016, 7, 27094–27107. [Google Scholar] [CrossRef][Green Version]
- Cheng, S.; Guo, J.; Yang, Q.; Yang, X. Crk-like adapter protein regulates CCL19/CCR7-mediated epithelial-to-mesenchymal transition via ERK signaling pathway in epithelial ovarian carcinomas. Med Oncol. 2015, 32, 47. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Tanaka, S.; Tsuda, M.; Oikawa, S.; Maeda, M.; Shimizu, M.; Shinomiya, H.; Tanigami, A.; Sawa, H.; Nagashima, K. Molecular and immunohistochemical analysis of signaling adaptor protein Crk in human cancers. Cancer Lett. 2002, 180, 55–61. [Google Scholar] [CrossRef]
- Miller, C.T.; Chen, G.; Gharib, T.G.; Wang, H.; Thomas, D.G.; E Misek, D.; Giordano, T.J.; Yee, J.; Orringer, M.B.; Hanash, S.M.; et al. Increased C-CRK proto-oncogene expression is associated with an aggressive phenotype in lung adenocarcinomas. Oncogene 2003, 22, 7950–7957. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yue, S.; Shi, H.; Han, J.; Zhang, T.; Zhu, W.; Zhang, D. Prognostic value of microRNA-126 and CRK expression in gastric cancer. OncoTargets Ther. 2016, 9, 6127–6135. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Sah, B.K.; Beeharry, M.K.; Yuan, F.; Su, L.; Jin, X.; Yan, M.; Liu, B.; Li, C.; Zhu, Z. Dysregulation of miR-126/Crk protein axis predicts poor prognosis in gastric cancer patients. Cancer Biomark. 2018, 21, 335–343. [Google Scholar] [CrossRef]
- Itakura, M.; Terashima, Y.; Shingyoji, M.; Yokoi, S.; Ohira, M.; Kageyama, H.; Matui, Y.; Yoshida, Y.; Ashinuma, H.; Moriya, Y.; et al. High CC chemokine receptor 7 expression improves postoperative prognosis of lung adenocarcinoma patients. Br. J. Cancer 2013, 109, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lv, W.; Shi, R.; Cheng, S.; Zhang, J.; Xu, Z. The clinical implications of Crk-like adaptor protein expression in papillary thyroid microcarcinoma. Tumor Biol. 2014, 35, 12435–12440. [Google Scholar] [CrossRef]
- Shi, X.; Xiao, X.; Yuan, N.; Zhang, S.; Yuan, F.; Wang, X. MicroRNA-379 Suppresses Cervical Cancer Cell Proliferation and Invasion by Directly Targeting V-crk Avian Sarcoma Virus CT10 Oncogene Homolog-Like (CRKL). Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 987–996. [Google Scholar] [CrossRef]
- Wang, L.; Lu, J.; Wu, H.; Wang, L.; Liang, X.; Liang, Z.; Liu, T. Expression of signaling adaptor proteins predicts poor prognosis in pancreatic ductal adenocarcinoma. Diagn. Pathol. 2017, 12, 42. [Google Scholar] [CrossRef][Green Version]
- Matsuda, M.; Tanaka, S.; Nagata, S.; Kojima, A.; Kurata, T.; Shibuya, M. Two species of human CRK cDNA encode proteins with distinct biological activities. Mol. Cell. Biol. 1992, 12, 3482–3489. [Google Scholar] [CrossRef]
- Lamorte, L.; Royal, I.; Naujokas, M.; Park, M. Crk Adapter Proteins Promote an Epithelial–Mesenchymal-like Transition and Are Required for HGF-mediated Cell Spreading and Breakdown of Epithelial Adherens Junctions. Mol. Biol. Cell 2002, 13, 1449–1461. [Google Scholar] [CrossRef]
- Antoku, S.; Saksela, K.; Rivera, G.M.; Mayer, B.J. A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors. J. Cell Sci. 2008, 121, 3071–3082. [Google Scholar] [CrossRef]
- Mortazavi, F.; Dubinett, S.; Rettig, M. c-Crk proto-oncogene contributes to transcriptional repression of p120-catenin in non-small cell lung cancer cells. Clin. Exp. Metastasis 2011, 28, 391–404. [Google Scholar] [CrossRef][Green Version]
- Antoku, S.; Mayer, B.J. Distinct roles for Crk adaptor isoforms in actin reorganization induced by extracellular signals. J. Cell Sci. 2009, 122, 4228–4238. [Google Scholar] [CrossRef]
- Mayer, B.J.; Hanafusa, H. Association of the v-crk oncogene product with phosphotyrosine-containing proteins and protein kinase activity. Proc. Natl. Acad. Sci. USA 1990, 87, 2638–2642. [Google Scholar] [CrossRef]
- Matsuda, M.; Mayer, B.J.; Hanafusa, H. Identification of domains of the v-crk oncogene product sufficient for association with phosphotyrosine-containing proteins. Mol. Cell. Biol. 1991, 11, 1607–1613. [Google Scholar] [CrossRef]
- Matsuda, M.; Reichman, C.T.; Hanafusa, H. Biological and biochemical activity of v-Crk chimeras containing the SH2/SH3 regions of phosphatidylinositol-specific phospholipase C-gamma and Src. J. Virol. 1992, 66, 115–121. [Google Scholar] [CrossRef]
- Senechal, K.; Heaney, C.; Druker, B.; Sawyers, C.L. Structural Requirements for Function of the Crkl Adapter Protein in Fibroblasts and Hematopoietic Cells. Mol. Cell. Biol. 1998, 18, 5082–5090. [Google Scholar] [CrossRef]
- Iwahara, T.; Akagi, T.; Shishido, T.; Hanafusa, H. CrkII induces serum response factor activation and cellular transformation through its function in Rho activation. Oncogene 2003, 22, 5946–5957. [Google Scholar] [CrossRef][Green Version]
- Senechal, K.; Halpern, J.; Sawyers, C.L. The CRKL Adaptor Protein Transforms Fibroblasts and Functions in Transformation by the BCR-ABL Oncogene. J. Biol. Chem. 1996, 271, 23255–23261. [Google Scholar] [CrossRef]
- Cheung, H.W.; Du, J.; Boehm, J.S.; He, F.; Weir, B.A.; Wang, X.; Butaney, M.; Sequist, L.V.; Luo, B.; Engelman, J.A.; et al. Amplification of CRKL Induces Transformation and Epidermal Growth Factor Receptor Inhibitor Resistance in Human Non–Small Cell Lung Cancers. Cancer Discov. 2011, 1, 608–625. [Google Scholar] [CrossRef]
- Zheng, J.; Machida, K.; Antoku, S.; Ng, K.Y.; Claffey, K.P.; Mayer, B.J. Proteins that bind the Src homology 3 domain of CrkI have distinct roles in Crk transformation. Oncogene 2010, 29, 6378–6389. [Google Scholar] [CrossRef]
- Ng, K.Y.; Yin, T.; Machida, K.; Wu, Y.I.; Mayer, B.J. Phosphorylation of Dok1 by Abl family kinases inhibits CrkI transforming activity. Oncogene 2015, 34, 2650–2659. [Google Scholar] [CrossRef][Green Version]
- Koptyra, M.; Park, T.-J.; Curran, T. Crk and CrkL are required for cell transformation by v-fos and v-ras. Mol. Carcinog. 2015, 55, 97–104. [Google Scholar] [CrossRef]
- Li, L.; Guris, D.L.; Okura, M.; Imamoto, A. Translocation of CrkL to Focal Adhesions Mediates Integrin-Induced Migration Downstream of Src Family Kinases. Mol. Cell. Biol. 2003, 23, 2883–2892. [Google Scholar] [CrossRef]
- Toffoli, S.; Delaive, E.; Dieu, M.; Feron, O.; Raes, M.; Michiels, C. NDRG1 and CRK-I/II are regulators of endothelial cell migration under intermittent hypoxia. Angiogenesis 2009, 12, 339–354. [Google Scholar] [CrossRef]
- Klemke, R.L.; Leng, J.; Molander, R.; Brooks, P.C.; Vuori, K.; Cheresh, D.A. CAS/Crk Coupling Serves as a “Molecular Switch” for Induction of Cell Migration. J. Cell Biol. 1998, 140, 961–972. [Google Scholar] [CrossRef]
- Cho, S.Y.; Klemke, R.L. Extracellular-Regulated Kinase Activation and Cas/Crk Coupling Regulate Cell Migration and Suppress Apoptosis during Invasion of the Extracellular Matrix. J. Cell Biol. 2000, 149, 223–236. [Google Scholar] [CrossRef]
- Uemura, N.; Griffin, J.D. The Adapter Protein Crkl Links Cbl to C3G after Integrin Ligation and Enhances Cell Migration. J. Biol. Chem. 1999, 274, 37525–37532. [Google Scholar] [CrossRef]
- Mudduluru, G.; Large, N.; Park, T. Impedance-based Real-time Measurement of Cancer Cell Migration and Invasion. J. Vis. Exp. 2020, 158, e60997. [Google Scholar] [CrossRef] [PubMed]
- Noren, N.K.; Foos, G.; Hauser, C.A.; Pasquale, E.B. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl–Crk pathway. Nat. Cell Biol. 2006, 8, 815–825. [Google Scholar] [CrossRef]
- Cipres, A.; Abassi, Y.A.; Vuori, K. Abl functions as a negative regulator of Met-induced cell motility via phosphorylation of the adapter protein CrkII. Cell. Signal. 2007, 19, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.; Knudsen, B.; Hanafusa, H. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J. 1994, 13, 2341–2351. [Google Scholar] [CrossRef]
- Rosen, M.K.; Yamazaki, T.; Gish, G.D.; Kay, C.M.; Pawson, T.; Kay, L.E. Direct demonstration of an intramolecular SH2—Phosphotyrosine interaction in the Crk protein. Nat. Cell Biol. 1995, 374, 477–479. [Google Scholar] [CrossRef]
- Kobashigawa, Y.; Sakai, M.; Naito, M.; Yokochi, M.; Kumeta, H.; Makino, Y.; Ogura, K.; Tanaka, S.; Inagaki, F. Structural basis for the transforming activity of human cancer-related signaling adaptor protein CRK. Nat. Struct. Mol. Biol. 2007, 14, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Jankowski, W.; Kasikara, C.; Reichman, C.; Saleh, T.; Nguyen, K.-Q.; Li, J.; Hornbeck, P.; Machida, K.; Liu, T.; et al. Iterative tyrosine phosphorylation controls non-canonical domain utilization in Crk. Oncogene 2014, 34, 4260–4269. [Google Scholar] [CrossRef][Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nat. Cell Biol. 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Larsson, H.; Klint, P.; Landgren, E.; Claesson-Welsh, L. Fibroblast Growth Factor Receptor-1-mediated Endothelial Cell Proliferation Is Dependent on the Src Homology (SH) 2/SH3 Domain-containing Adaptor Protein Crk. J. Biol. Chem. 1999, 274, 25726–25734. [Google Scholar] [CrossRef]
- Lundin, L.; Rönnstrand, L.; Cross, M.; Hellberg, C.; Lindahl, U.; Claesson-Welsh, L. Differential tyrosine phosphorylation of fibroblast growth factor (FGF) receptor-1 and receptor proximal signal transduction in response to FGF-2 and heparin. Exp. Cell Res. 2003, 287, 190–198. [Google Scholar] [CrossRef]
- Cao, X.-C.; Zhang, W.-R.; Cao, W.-F.; Liu, B.-W.; Zhang, F.; Zhao, H.-M.; Meng, R.; Zhang, L.; Niu, R.-F.; Hao, X.-S.; et al. Aquaporin3 Is Required for FGF-2-Induced Migration of Human Breast Cancers. PLoS ONE 2013, 8, e56735. [Google Scholar] [CrossRef]
- Abolhassani, A.; Riazi, G.H.; Azizi, E.; Amanpour, S.; Muhammadnejad, S.; Haddadi, M.; Zekri, A.; Shirkoohi, R. FGF10: Type III Epithelial Mesenchymal Transition and Invasion in Breast Cancer Cell Lines. J. Cancer 2014, 5, 537–547. [Google Scholar] [CrossRef]
- Matsuda, Y.; Yoshimura, H.; Suzuki, T.; Uchida, E.; Naito, Z.; Ishiwata, T. Inhibition of fibroblast growth factor receptor 2 attenuates proliferation and invasion of pancreatic cancer. Cancer Sci. 2014, 105, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Huang, S.; Lei, Y.; Zhang, T.; Wang, K.; Liu, B.; Nice, E.C.; Xiang, R.; Xie, K.; Li, J.; et al. FGF8 promotes colorectal cancer growth and metastasis by activating YAP1. Oncotarget 2014, 6, 935–952. [Google Scholar] [CrossRef]
- Knuchel, S.; Anderle, P.; Werfelli, P.; Diamantis, E.; Rüegg, C. Fibroblast surface-associated FGF-2 promotes contact-dependent colorectal cancer cell migration and invasion through FGFR-SRC signaling and integrin αvβ5-mediated adhesion. Oncotarget 2015, 6, 14300–14317. [Google Scholar] [CrossRef]
- Shi, S.; Li, X.; You, B.; Shan, Y.; Cao, X.; You, Y. High Expression of FGFR4 Enhances Tumor Growth and Metastasis in Nasopharyngeal Carcinoma. J. Cancer 2015, 6, 1245–1254. [Google Scholar] [CrossRef]
- Qi, L.; Song, W.; Li, L.; Cao, L.; Yu, Y.; Song, C.; Wang, Y.; Zhang, F.; Li, Y.; Zhang, B.; et al. FGF4 induces epithelial-mesenchymal transition by inducing store-operated calcium entry in lung adenocarcinoma. Oncotarget 2016, 7, 74015–74030. [Google Scholar] [CrossRef]
- Huang, T.; Wang, L.; Liu, D.; Li, P.; Xiong, H.; Zhuang, L.; Sun, L.; Yuan, X.; Qiu, H. FGF7/FGFR2 signal promotes invasion and migration in human gastric cancer through upregulation of thrombospondin-1. Int. J. Oncol. 2017, 50, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Fan, X.; Zhang, Q.; Shi, X.; Xu, G.; Zou, C. Cancer-associated fibroblasts secrete FGF-1 to promote ovarian proliferation, migration, and invasion through the activation of FGF-1/FGFR4 signaling. Tumor Biol. 2017, 39, 1010428317712592. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Xiao, Y.; Liao, X.; Tang, S.; Xie, X.; Liu, R.; Chen, Q. FGF8 induces epithelial-mesenchymal transition and promotes metastasis in oral squamous cell carcinoma. Int. J. Oral Sci. 2021, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Birge, R.B. Emerging Roles for Crk in Human Cancer. Genes Cancer 2010, 1, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.S.; Park, M. Models of Crk Adaptor Proteins in Cancer. Genes Cancer 2012, 3, 341–352. [Google Scholar] [CrossRef]
- Tsuda, M.; Tanaka, S. Roles for Crk in Cancer Metastasis and Invasion. Genes Cancer 2012, 3, 334–340. [Google Scholar] [CrossRef]
- Guo, C.; Liu, S.; Sun, M.-Z. The role of CT10 regulation of kinase-like in cancer. Futur. Oncol. 2014, 10, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Fajardo, J.E.; Birge, R.B.; Sriram, G. Crk at the Quarter Century Mark: Perspectives in Signaling and Cancer. J. Cell. Biochem. 2014, 115, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Posern, G.; Zheng, J.; Knudsen, B.S.; Kardinal, C.; Müller, K.B.; Voss, J.; Shishido, T.; Cowburn, D.; Cheng, G.; Wang, B.; et al. Development of highly selective SH3 binding peptides for Crk and CRKL which disrupt Crk-complexes with DOCK180, SoS and C3G. Oncogene 1998, 16, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Kardinal, C. Cell-penetrating SH3 domain blocker peptides inhibit proliferation of primary blast cells from CML patients. FASEB J. 2000, 14, 1529–1538. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tumor Cell Function | Crk | CrkL | Crk and CrkL |
|---|---|---|---|
| Cell spreading | Breast [25,26] K | Glioblastoma [27] K | Glioblastoma [27] K |
| In vitro cell proliferation | Ovarian cancer [28,29] K Synovial sarcoma [30] K Hepatocellular [31] O | Lung [32] O Rhabdomyosarcoma [33] K Breast [34] K Gastric [35] K Cervical [36] K, [37] O Endometrial [38] O Hepatocellular [39] K Glioblastoma [27] K | Glioblastoma [27] K Colorectal [40] K |
| Anchorage-independent growth | Ovarian [28] K Glioblastoma [41] K Hepatocellular [31] O | Glioma [42] K Cervical [43] K | Breast [26] K Colorectal [40] K |
| In vivo tumor growth | Ovarian [28] K Glioblastoma [41] K Breast [26,44] K Bladder [45] K | Head and neck [46] K Rhabdomyosarcoma [33] K Hepatocellular [39] K | |
| Metastasis | Breast [26] K Bladder [45] K | Colorectal [47] K | |
| Migration and invasion | Bladder [45] K, [48] O Glioblastoma [27,41] K, [49,50] O Fibrosarcoma [51] O Breast/cervical/lung [25] K Ovarian [28,29] K Synovial sarcoma [52] K Oral squamous [53] K Prostate [54,55] K Breast [26] K, [56] O Lung [57,58] O Gastric [59] K Colorectal [40] K Hepatocellular [31] O Pancreatic [60] K | Head and neck [46] K Glioma [42] K Ovarian [61] K Hepatocellular [62] O Gastric [63] K Cervical [43] K, [37] O Colorectal [40] K Glioblastoma [27] K | Colorectal/pancreatic [40] K Glioblastoma [27] K |
| Epithelial-mesenchymal transition | Bladder [45] K Lung [64] O Breast [44] K Colorectal [40] K | Ovarian [65] K Gastric [63] K Colorectal [40] K | Colorectal/pancreatic [40] K |
| Chemoresistance | Cervical [37] O Endometrial [38] O | Colorectal [40] K | |
| Overexpression in cancer tissues | Lung/gastric/breast [66] Glioblastoma [41,50,56] Lung [67] Breast [25] Oral squamous [53] Bladder [45] Gastric [68,69] Kidney [56] | Lung [32,70] Rhabdomyosarcoma [33] Breast [34] Gastric [35,63] Thyroid [71] Ovarian [65] Cervical [37,72] Endometrial [38] Pancreatic [73] Colorectal [47] | Ovarian [29] Breast [26] |
| Lower survival | Oral squamous [53] Glioblastoma [50] Lung [64] Gastric [68,69] Colorectal [40] | Lung [32] Ovarian [65] Gastric [63] Pancreatic [73] Colorectal [40] | Colorectal [40] |
| Copy Number Alteration | Total Number of Cases | Median Months Overall |
|---|---|---|
| Unaltered group | 21,270 | 118.30 |
| CRK: amplification | 69 | 46.78 |
| CRKL: amplification | 231 | 53.92 |
| CRK: heterozygous deletion | 5072 | 81.93 |
| CRK: homozygous deletion | 108 | 94.00 |
| CRKL: heterozygous deletion | 3542 | 71.17 |
| CRKL: homozygous deletion | 51 | 158.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, T. Crk and CrkL as Therapeutic Targets for Cancer Treatment. Cells 2021, 10, 739. https://doi.org/10.3390/cells10040739
Park T. Crk and CrkL as Therapeutic Targets for Cancer Treatment. Cells. 2021; 10(4):739. https://doi.org/10.3390/cells10040739
Chicago/Turabian StylePark, Taeju. 2021. "Crk and CrkL as Therapeutic Targets for Cancer Treatment" Cells 10, no. 4: 739. https://doi.org/10.3390/cells10040739
APA StylePark, T. (2021). Crk and CrkL as Therapeutic Targets for Cancer Treatment. Cells, 10(4), 739. https://doi.org/10.3390/cells10040739