
Sirtuin 2 Dysregulates Autophagy in High-Fat-Exposed Immune-Tolerant Macrophages

Abstract
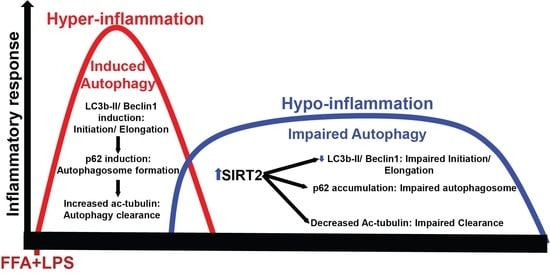
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. RAW 264.7 Cells with Stearic (Free Fatty Acid: FFA) Acid Exposure
2.2. Cytoplasmic Extraction from RAW Cells
2.3. Immunocytochemistry
2.4. Autophagy Flux Assay
2.5. Immunoprecipitation Assay
2.6. Western Blot Analysis
2.7. ELISA
2.8. Statistical Analysis
3. Results
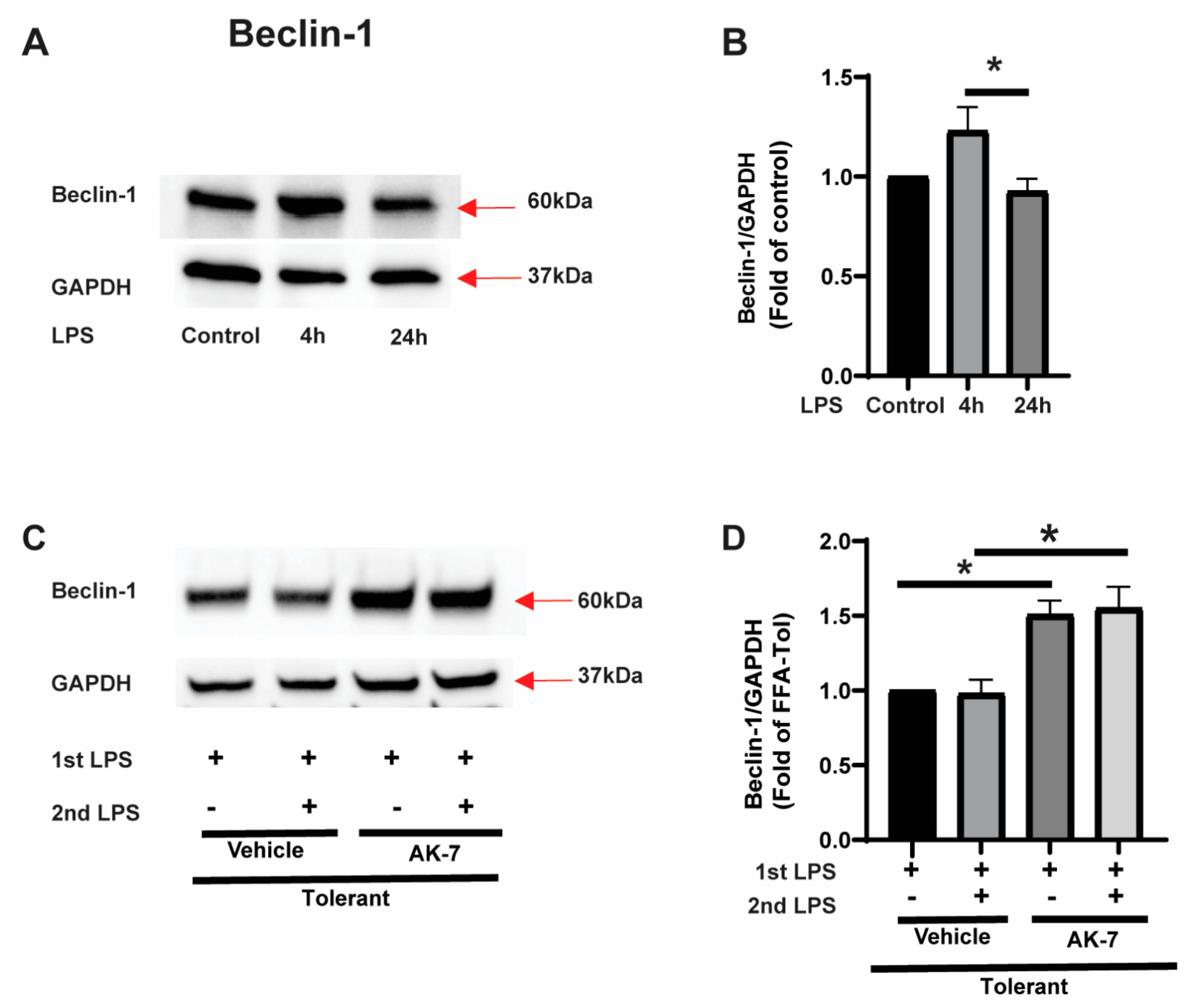
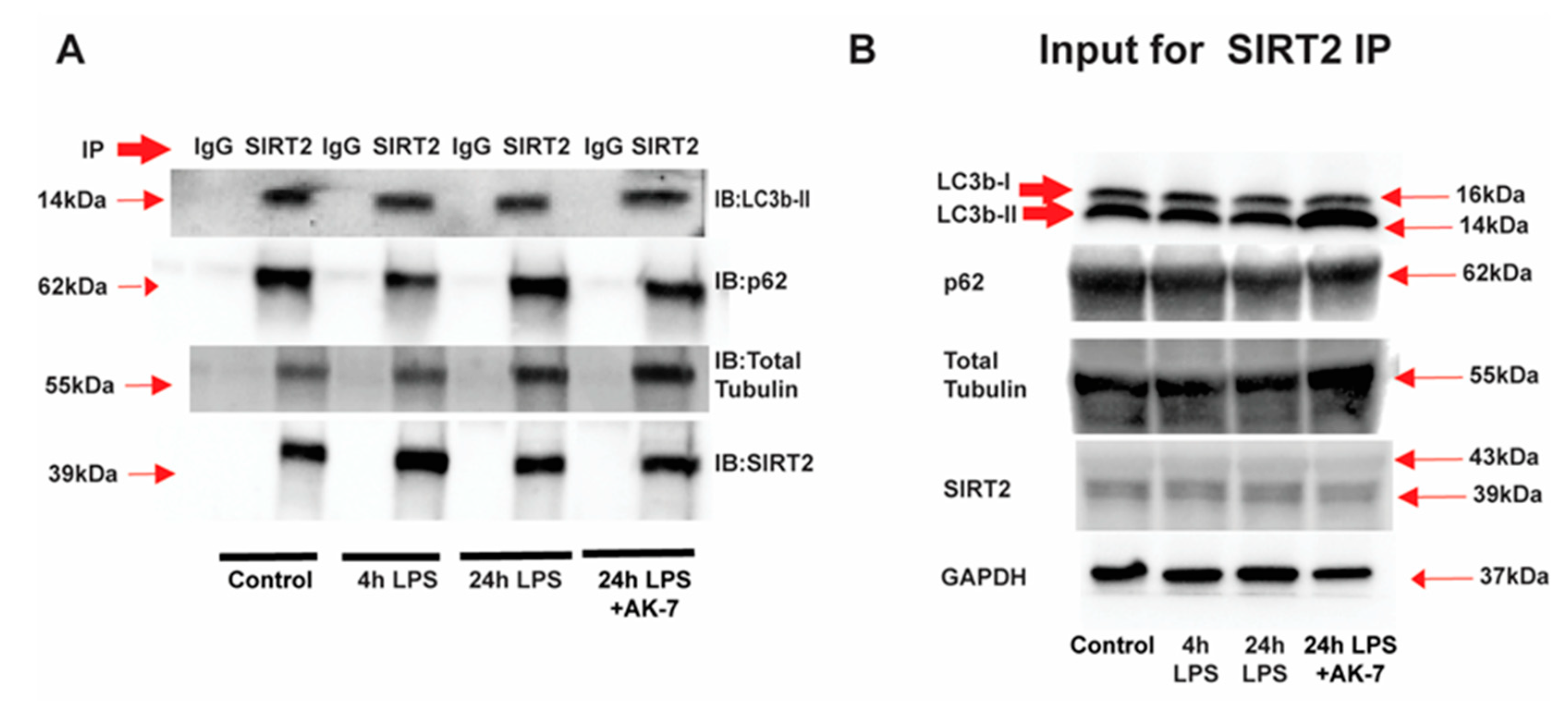
3.1. SIRT2 Expression Is Increased in Tolerant Macrophages
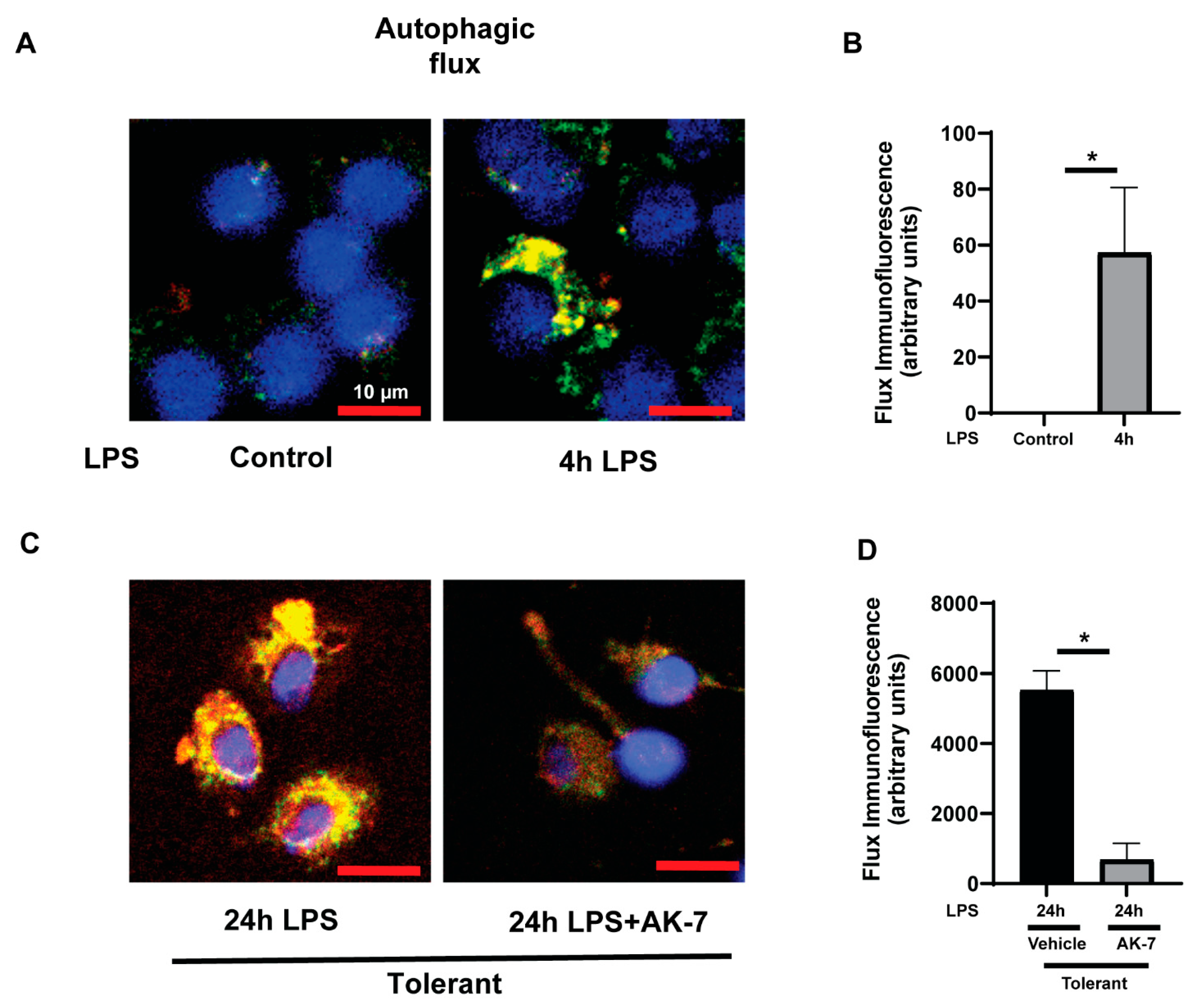
3.2. Free Fatty Acid Exposure Dysregulates Autophagy in Tolerant Macrophages
3.2.1. Effect of SIRT2 Expression on Autophagosome Formation
3.2.2. Autophagy Clearance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SIRT | Sirtuin |
| SIRT2 | Sirtuin 2 |
| FFA | Free fatty acid |
| BSA | Bovine serum albumin |
| LPS | Lipopolysaccharide |
| RAW | RAW 264.7 cells |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| IP | Immunoprecipitation |
| IB | Immunoblot |
| WB | Western blot |
References
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Gaieski, D.F.; Edwards, J.M.; Kallan, M.J.; Carr, B.G. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit. Care Med. 2013, 41, 1167–1174. [Google Scholar] [CrossRef]
- Buchman, T.G.; Simpson, S.Q.; Sciarretta, K.L.; Finne, K.P.; Sowers, N.; Collier, M.; Chavan, S.; Oke, I.; Pennini, M.E.; Santhosh, A.; et al. Sepsis Among Medicare Beneficiaries: 1. The Burdens of Sepsis, 2012–2018. Crit. Care Med. 2020, 48, 276–288. [Google Scholar] [CrossRef]
- Williams, S.C. After Xigris, researchers look to new targets to combat sepsis. Nat. Med. 2012, 18, 1001. [Google Scholar] [CrossRef]
- Vachharajani, V.T.; Fu Liu, T.; Brown, C.M.; Wang, X.; Buechler, N.L.; Wells, J.D.; Yoza, B.K.; McCall, C.E. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J. Leukoc. Biol. 2014, 96, 785–796. [Google Scholar] [CrossRef]
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D., 2nd; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef]
- Otto, G.P.; Sossdorf, M.; Claus, R.A.; Rodel, J.; Menge, K.; Reinhart, K.; Bauer, M.; Riedemann, N.C. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit. Care 2011, 15, R183. [Google Scholar] [CrossRef]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernandez-Jimenez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1alpha. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Dara, S.I.; Tamim, H.M.; Rishu, A.H.; Bouchama, A.; Khedr, M.K.; Feinstein, D.; Parrillo, J.E.; Wood, K.E.; Keenan, S.P.; et al. Clinical characteristics, sepsis interventions and outcomes in the obese patients with septic shock: An international multicenter cohort study. Crit. Care 2013, 17, R72. [Google Scholar] [CrossRef]
- Klang, E.; Kassim, G.; Soffer, S.; Freeman, R.; Levin, M.A.; Reich, D.L. Severe Obesity as an Independent Risk Factor for COVID-19 Mortality in Hospitalized Patients Younger than 50. Obesity 2020, 28, 1595–1599. [Google Scholar] [CrossRef]
- Wang, X.; Buechler, N.L.; Martin, A.; Wells, J.; Yoza, B.; McCall, C.E.; Vachharajani, V. Sirtuin-2 Regulates Sepsis Inflammation in ob/ob Mice. PLoS ONE 2016, 11, e0160431. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Buechler, N.L.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Adiponectin treatment attenuates inflammatory response during early sepsis in obese mice. J. Inflamm. Res. 2016, 9, 167–174. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Nowell, M.; Lahni, P.; O’Connor, M.P.; Hake, P.W.; Zingarelli, B. Short-term high fat feeding increases organ injury and mortality after polymicrobial sepsis. Obesity 2012, 20, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins—Emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef]
- Chien, W.S.; Chen, Y.H.; Chiang, P.C.; Hsiao, H.W.; Chuang, S.M.; Lue, S.I.; Hsu, C. Suppression of autophagy in rat liver at late stage of polymicrobial sepsis. Shock 2011, 35, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Pu, Q.; Gan, C.; Li, R.; Li, Y.; Tan, S.; Li, X.; Wei, Y.; Lan, L.; Deng, X.; Liang, H.; et al. Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis in Pseudomonas Sepsis. J. Immunol. 2017, 198, 3205–3213. [Google Scholar] [CrossRef]
- Ju, L.; Han, J.; Zhang, X.; Deng, Y.; Yan, H.; Wang, C.; Li, X.; Chen, S.; Alimujiang, M.; Li, X.; et al. Obesity-associated inflammation triggers an autophagy-lysosomal response in adipocytes and causes degradation of perilipin 1. Cell Death Dis. 2019, 10, 121. [Google Scholar] [CrossRef]
- Pabon, M.A.; Ma, K.C.; Choi, A.M. Autophagy and Obesity-Related Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 636–646. [Google Scholar] [CrossRef]
- Ouimet, M. Autophagy in obesity and atherosclerosis: Interrelationships between cholesterol homeostasis, lipoprotein metabolism and autophagy in macrophages and other systems. Biochim. Biophys. Acta 2013, 1831, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Williams, J.A.; Ding, W.X. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp. Biol. Med. 2013, 238, 525–538. [Google Scholar] [CrossRef]
- Gal, J.; Bang, Y.; Choi, H.J. SIRT2 interferes with autophagy-mediated degradation of protein aggregates in neuronal cells under proteasome inhibition. Neurochem. Int. 2012, 61, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, S.H.; Imbesi, M.; Nathan, W.J.; Zou, X.; Zhu, Y.; Jiang, H.; Parisiadou, L.; Gius, D. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid. Redox Signal. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Buechler, N.L.; Long, D.L.; Furdui, C.M.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Cysteine thiol oxidation on SIRT2 regulates inflammation in obese mice with sepsis. Inflammation 2019, 42, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Poulsen, K.L.; Sanz-Garcia, C.; Huang, E.; McMullen, M.R.; Roychowdhury, S.; Dasarathy, S.; Nagy, L.E. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J. Hepatol. 2020, 73, 616–627. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016, 1, e87748. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.M.; Tomkinson, E.M.; Nobles, J.; Wizeman, J.W.; Amore, A.M.; Quinti, L.; Chopra, V.; Hersch, S.M.; Kazantsev, A.G. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum. Mol. Genet. 2011, 20, 3986–3996. [Google Scholar] [CrossRef]
- Chae, B.S. Comparative study of the endotoxemia and endotoxin tolerance on the production of Th cytokines and macrophage interleukin-6: Differential regulation of indomethacin. Arch. Pharm. Res. 2002, 25, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Dembek, A.; Laggai, S.; Kessler, S.M.; Czepukojc, B.; Simon, Y.; Kiemer, A.K.; Hoppstadter, J. Hepatic interleukin-6 production is maintained during endotoxin tolerance and facilitates lipid accumulation. Immunobiology 2017, 222, 786–796. [Google Scholar] [CrossRef]
- Wang, J.; Koh, H.W.; Zhou, L.; Bae, U.J.; Lee, H.S.; Bang, I.H.; Ka, S.O.; Oh, S.H.; Bae, E.J.; Park, B.H. Sirtuin 2 aggravates postischemic liver injury by deacetylating mitogen-activated protein kinase phosphatase-1. Hepatology 2017, 65, 225–236. [Google Scholar] [CrossRef]
- Ren, C.; Zhang, H.; Wu, T.T.; Yao, Y.M. Autophagy: A Potential Therapeutic Target for Reversing Sepsis-Induced Immunosuppression. Front. Immunol. 2017, 8, 1832. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, B.; Zheng, X.; Chen, L.; Chen, W.; Fang, Z. The protective role of autophagy in sepsis. Microb. Pathog. 2019, 131, 106–111. [Google Scholar] [CrossRef]
- McKnight, N.C.; Zhenyu, Y. Beclin 1, an Essential Component and Master Regulator of PI3K-III in Health and Disease. Curr. Pathobiol. Rep. 2013, 1, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Palma, A.M.; Gomes, R.; Santos, D.; Silva, D.F.; Cardoso, S.M. Acetylation as a major determinant to microtubule-dependent autophagy: Relevance to Alzheimer’s and Parkinson disease pathology. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2008–2023. [Google Scholar] [CrossRef]
- Inoue, T.; Hiratsuka, M.; Osaki, M.; Yamada, H.; Kishimoto, I.; Yamaguchi, S.; Nakano, S.; Katoh, M.; Ito, H.; Oshimura, M. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007, 26, 945–957. [Google Scholar] [CrossRef]
- Jeong, S.G.; Cho, G.W. The tubulin deacetylase sirtuin-2 regulates neuronal differentiation through the ERK/CREB signaling pathway. Biochem. Biophys. Res. Commun. 2017, 482, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Nakayama, Y.; Li, Y.; Matsumori, H.; Takahashi, H.; Kojima, H.; Wanibuchi, H.; Katoh, M.; Oshimura, M. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J. 2014, 281, 2623–2637. [Google Scholar] [CrossRef]
- You, Z.; Xu, Y.; Wan, W.; Zhou, L.; Li, J.; Zhou, T.; Shi, Y.; Liu, W. TP53INP2 contributes to autophagosome formation by promoting LC3-ATG7 interaction. Autophagy 2019, 15, 1309–1321. [Google Scholar] [CrossRef]
- You, Z.; Jiang, W.X.; Qin, L.Y.; Gong, Z.; Wan, W.; Li, J.; Wang, Y.; Zhang, H.; Peng, C.; Zhou, T.; et al. Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat. Commun. 2019, 10, 5792. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2007, 2, e784. [Google Scholar] [CrossRef] [PubMed]
- Widdrington, J.D.; Gomez-Duran, A.; Pyle, A.; Ruchaud-Sparagano, M.H.; Scott, J.; Baudouin, S.V.; Rostron, A.J.; Lovat, P.E.; Chinnery, P.F.; Simpson, A.J. Exposure of Monocytic Cells to Lipopolysaccharide Induces Coordinated Endotoxin Tolerance, Mitochondrial Biogenesis, Mitophagy, and Antioxidant Defenses. Front. Immunol. 2018, 9, 2217. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roychowdhury, S.; Gandhirajan, A.; Kibler, C.; Wang, X.; Vachharajani, V. Sirtuin 2 Dysregulates Autophagy in High-Fat-Exposed Immune-Tolerant Macrophages. Cells 2021, 10, 731. https://doi.org/10.3390/cells10040731
Roychowdhury S, Gandhirajan A, Kibler C, Wang X, Vachharajani V. Sirtuin 2 Dysregulates Autophagy in High-Fat-Exposed Immune-Tolerant Macrophages. Cells. 2021; 10(4):731. https://doi.org/10.3390/cells10040731
Chicago/Turabian StyleRoychowdhury, Sanjoy, Anugraha Gandhirajan, Christopher Kibler, Xianfeng Wang, and Vidula Vachharajani. 2021. "Sirtuin 2 Dysregulates Autophagy in High-Fat-Exposed Immune-Tolerant Macrophages" Cells 10, no. 4: 731. https://doi.org/10.3390/cells10040731
APA StyleRoychowdhury, S., Gandhirajan, A., Kibler, C., Wang, X., & Vachharajani, V. (2021). Sirtuin 2 Dysregulates Autophagy in High-Fat-Exposed Immune-Tolerant Macrophages. Cells, 10(4), 731. https://doi.org/10.3390/cells10040731