
Mdm2-Mediated Downmodulation of GRK2 Restricts Centrosome Separation for Proper Chromosome Congression

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture, Transfection and Treatments
2.2. Immunoprecipitation and Western Blot
2.3. In Vitro Ubiquitination Assays
2.4. Polyubiquitinated Protein Pulldown from Synchronized Cells in the G2 Phase
2.5. Immunofluorescence and Confocal Microscopy
2.6. Kinase Activity Assays
2.7. Statistical Analysis
3. Results
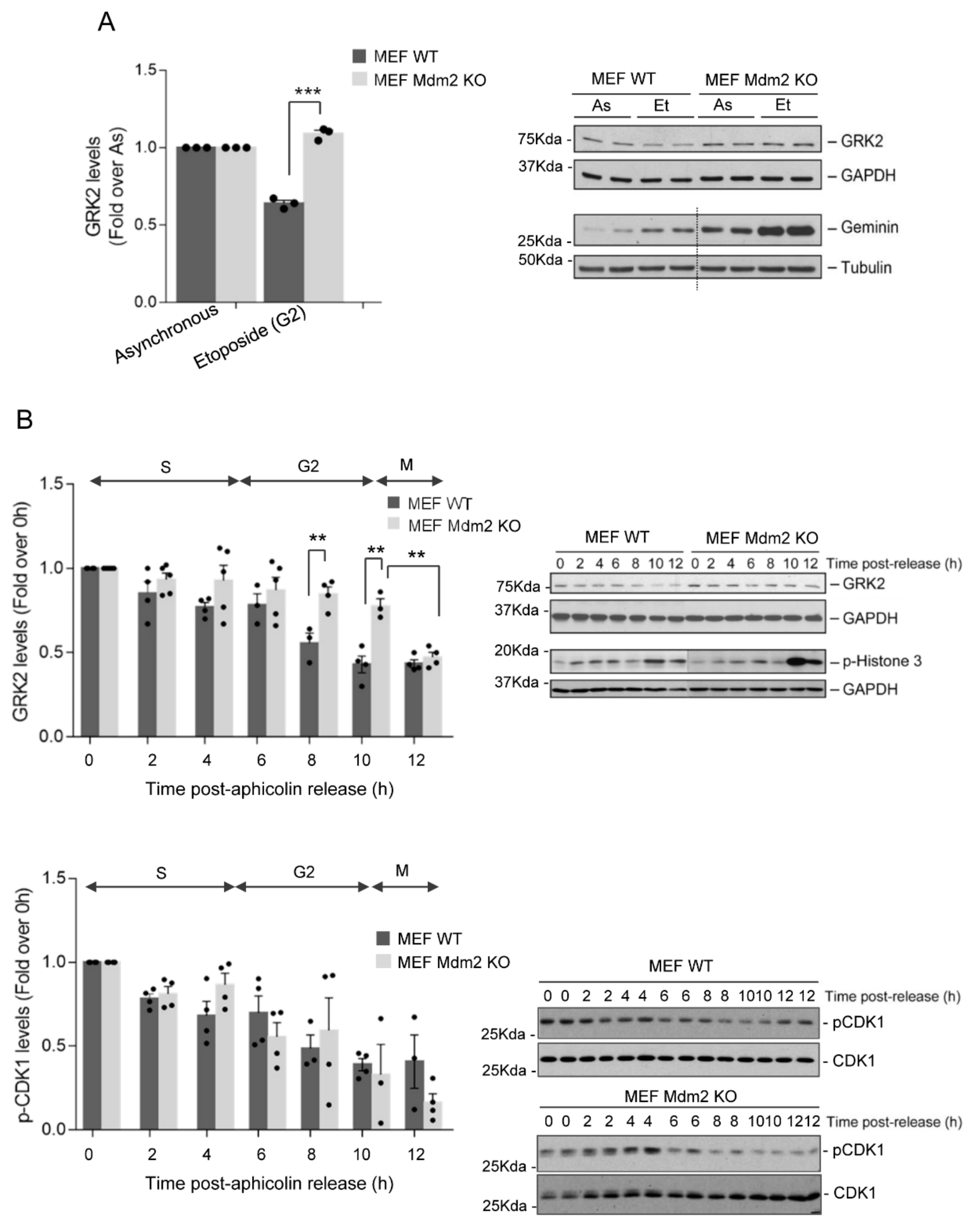
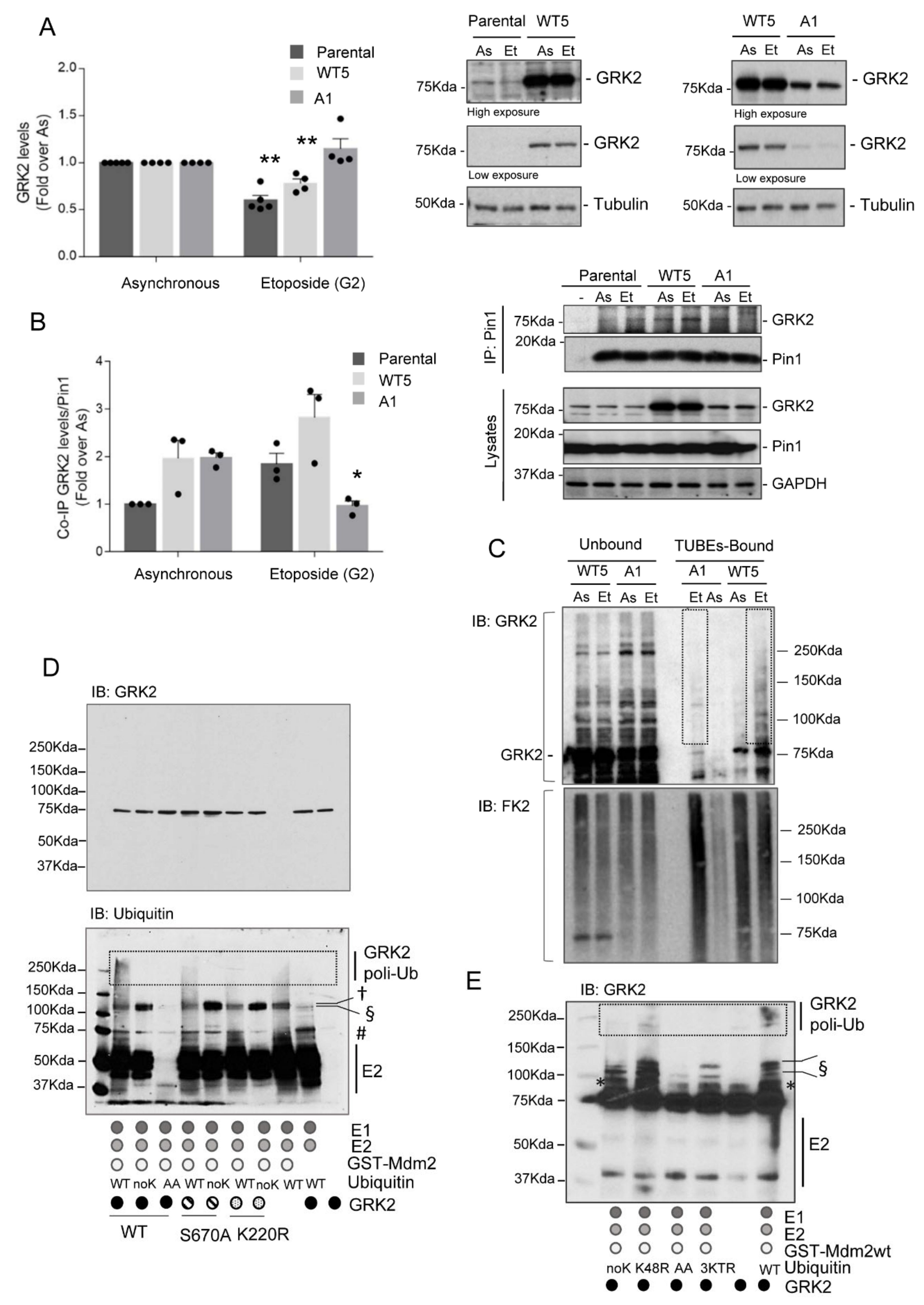
3.1. Mdm2 Mediates GRK2 Down-Modulation in the G2 Phase
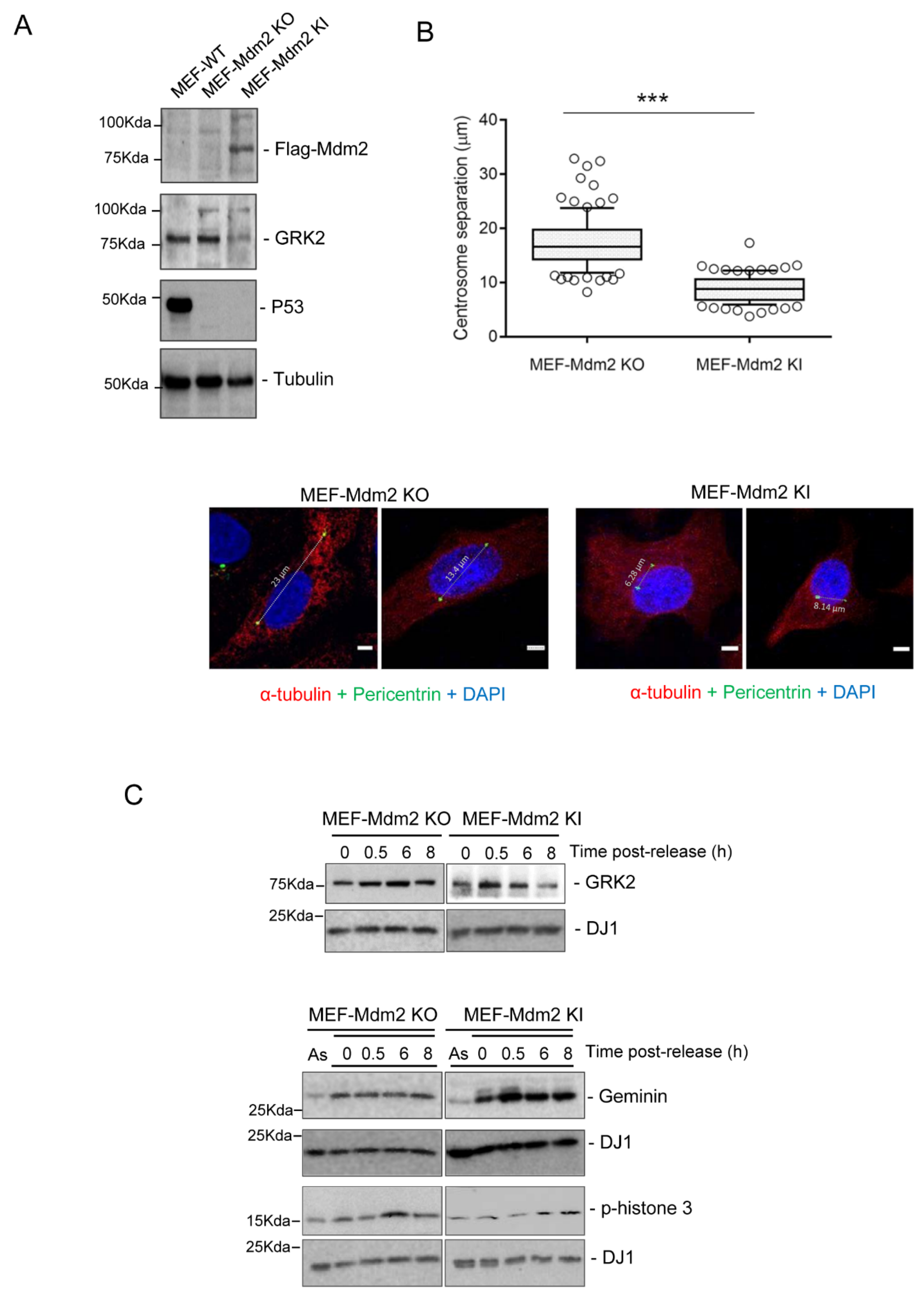
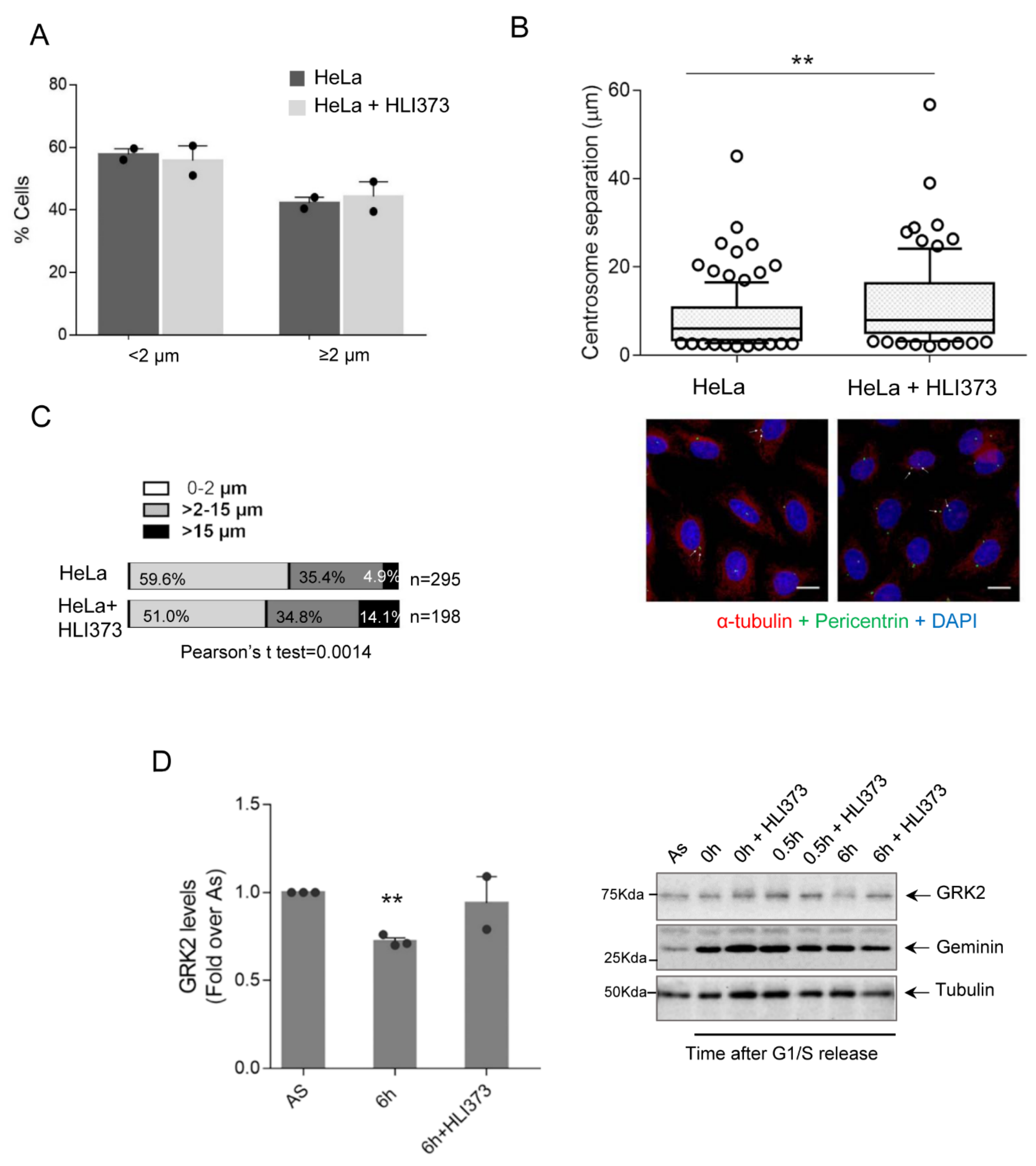
3.2. Mdm2 Regulates Centrosome Separation in the G2 Phase
3.3. GRK2-Associated Defects in Centrosome Separation Correlate with Altered Spindle Lengths and Chromosome Congression
4. Discussion
4.1. The Multifaceted Mdm2 Joins the Control of Centrosome Separation
4.2. GRK2 Fluctuates to Set Centrosome Separation and Spindle Length
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fang, X.; Zhang, P. Aneuploidy and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 595–601. [Google Scholar] [CrossRef]
- Rhys, A.D.; Godinho, S.A. Dividing with Extra centrosomes: A Double edged sword for cancer cells. Adv. Exp. Med. Biol. 2017, 1002, 47–67. [Google Scholar]
- Prosser, S.L.; Pelletier, L. Mitotic spindle assembly in animal cells: A fine balancing act. Nat. Rev. Mol. Cell Biol. 2017, 18, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt-Dias, M.; Glover, D.M. Centrosome biogenesis and function: Centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, I. Centrosomes in mitotic spindle assembly and orientation. Curr. Opin. Struct. Biol. 2020, 66, 193–198. [Google Scholar] [CrossRef]
- Nigg, E.A.; Stearns, T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 2011, 13, 1154–1160. [Google Scholar] [CrossRef]
- Wang, G.; Jiang, Q.; Zhang, C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J. Cell Sci. 2014, 127, 4111–4122. [Google Scholar] [CrossRef]
- Mardin, B.R.; Schiebel, E.J. Breaking the ties that bind: New advances in centrosome biology. Cell Biol. 2012, 197, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Agircan, F.G.; Schiebel, E.; Mardin, B.R. Separate to operate: Control of centrosome positioning and separation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130461. [Google Scholar] [CrossRef] [PubMed]
- Remo, A.; Li, X.; Schiebel, E.; Pancione, M. The Centrosome Linker and Its Role in Cancer and Genetic Disorders. Trends Mol. Med. 2020, 26, 380–393. [Google Scholar] [CrossRef]
- Faragher, A.J.; Fry, A.M. Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol. Biol. Cell 2003, 14, 2876–2889. [Google Scholar] [CrossRef]
- Van Ree, J.H.; Nam, H.J.; van Deursen, J.M. Mitotic kinase cascades orchestrating timely disjunction and movement of centrosomes maintain chromosomal stability and prevent cancer. Chromosome Res. 2016, 24, 67–76. [Google Scholar] [CrossRef][Green Version]
- Nam, H.J.; Naylor, R.; van Deursen, J.M. Centrosome dynamics as a source of chromosomal instability. Trends Cell Biol. 2015, 23, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Silkworth, W.T.; Nardi, I.K.; Paul, R.; Mogilner, A.; Cimini, D. Timing of centrosome separation is important for accurate chromosome segregation. Mol. Biol. Cell 2012, 23, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.M.; Phillips, B.T. The benefits of local depletion: The centrosome as a scaffold for ubiquitin-proteasome-mediated degradation. Cell Cycle 2016, 15, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Galardy, P.J. Ubiquitin, the centrosome, and chromosome segregation. Chromosome Res. 2016, 24, 77–91. [Google Scholar] [CrossRef]
- Hames, R.S.; Wattam, S.L.; Yamano, H.; Bacchieri, R.; Fry, A.M. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 2001, 20, 7117–7127. [Google Scholar] [CrossRef]
- Eguren, M.; Álvarez-Fernández, M.; García, F.; López-Contreras, A.J.; Fujimitsu, K.; Yaguchi, H.; Luque-García, J.L.; Fernández-Capetillo, O.; Muñoz, J.; Yamano, H.; et al. A synthetic lethal interaction between APC/C and topoisomerase poisons uncovered by proteomic screens. Cell Rep. 2014, 6, 670–683. [Google Scholar] [CrossRef]
- Meraldi, P.; Nigg, E.A. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J. Cell Sci. 2001, 114, 3749–3757. [Google Scholar]
- Mardin, B.R.; Agircan, F.G.; Lange, C.; Schiebel, E. Plk1 controls the Nek2A-PP1γ Antagonism in centrosome disjunction. Curr. Biol. 2011, 21, 1145–1151. [Google Scholar] [CrossRef]
- Mardin, B.R.; Lange, C.; Baxter, J.E.; Hardy, T.; Scholz, S.R.; Fry, A.M.; Schiebel, E. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat. Cell Biol. 2010, 12, 1166–1176. [Google Scholar] [CrossRef]
- Mardin, B.R.; Isokane, M.; Cosenza, M.R.; Krämer, A.; Ellenberg, J.; Fry, A.M.; Schiebel, E. EGF-induced centrosome separation promotes mitotic progression and cell survival. Dev. Cell. 2013, 25, 229–240. [Google Scholar] [CrossRef] [PubMed]
- So, C.H.; Michal, A.; Komolov, K.E.; Luo, J.; Benovic, J.L. G protein-coupled receptor kinase 2 (GRK2) is localized to centrosomes and mediates epidermal growth factor-promoted centrosomal separation. Mol. Biol. Cell. 2013, 24, 2795–2806. [Google Scholar] [CrossRef]
- Watari, K.; Nakaya, M.; Kurose, H. Multiple functions of G protein-coupled receptor kinases. J. Mol. Signal 2014, 9, 1–9. [Google Scholar] [CrossRef]
- Penela, P.; Ribas, C.; Sánchez-Madrid, F.; Mayor, F., Jr. G protein-coupled receptor kinase 2 (GRK2) as a multifunctional signaling hub. Cell Mol. Life Sci. 2019, 76, 4423–4446. [Google Scholar] [CrossRef]
- Nogués, L.; Palacios-García, J.; Reglero, C.; Rivas, V.; Neves, M.; Ribas, C.; Penela, P.; Mayor, F., Jr. G protein-coupled receptor kinases (GRKs) in tumorigenesis and cancer progression: GPCR regulators and signaling hubs. Semin. Cancer Biol. 2018, 48, 78–90. [Google Scholar] [CrossRef]
- Murga, C.; Arcones, A.C.; Cruces-Sande, M.; Briones, A.M.; Salaices, M.; Mayor, F., Jr. G Protein-coupled receptor kinase 2 (GRK2) as a potential therapeutic target in cardiovascular and metabolic diseases. Front. Pharmacol. 2019, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Murga, C.; Ribas, C.; Lafarga, V.; Mayor, F., Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br. J. Pharmacol. 2010, 160, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Illario, M.; Trimarco, B.; Iaccarino, G. Trafficking GRK2: Cellular and metabolic consequences of GRK2 subcellular localization. Transl. Med. UniSa 2014, 10, 3–7. [Google Scholar] [PubMed]
- Penela, P. Ubiquitination and protein turnover of G-protein-coupled receptor kinases in GPCR signaling and cellular regulation. Prog. Mol. Biol. Transl. Sci. 2016, 141, 85–140. [Google Scholar] [PubMed]
- Penela, P.; Rivas, V.; Salcedo, A.; Mayor, F., Jr. G protein-coupled receptor kinase 2 (GRK2) modulation and cell cycle progression. Proc. Natl. Acad. Sci. USA 2010, 107, 1118–1123. [Google Scholar] [CrossRef]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, A.; Mayor, F.; Penela, P. Mdm2 is involved in the ubiquitination and degradation of G-protein-coupled receptor kinase 2. EMBO J. 2006, 25, 4752–4762. [Google Scholar] [CrossRef]
- Nogués, L.; Salcedo, A.; Mayor, F.; Penela, P. Multiple scaffolding functions of β-arrestins in the degradation of G protein-coupled receptor kinase. J. Biol. Chem. 2011, 286, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Jean-Charles, P.Y.; Yu, S.M.; Abraham, D.; Kommaddi, R.P.; Mao, L.; Strachan, R.T.; Zhang, Z.S.; Bowles, D.E.; Brian, L.; Stiber, J.A.; et al. Mdm2 regulates cardiac contractility by inhibiting GRK2-mediated desensitization of beta-adrenergic receptor signaling. JCI Insight 2017, 2, e95998. [Google Scholar] [CrossRef]
- Carroll, P.E.; Okuda, M.; Horn, H.F.; Biddinger, P.; Stambrook, P.J.; Gleich, L.L.; Li, Y.Q.; Tarapore, P.; Fukasawa, K. Centrosome hyperamplification in human cancer: Chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene 1999, 18, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Cázares-Ordoñez, V.; Makishima, M.; Yokoyama, A.; Suenaga, Y.; Nagase, H.; Kobayashi, S.; Kamijo, T.; Koshinaga, T.; Wada, S. CEP131 abrogates CHK1 inhibitor-induced replication defects and is associated with unfavorable outcome in neuroblastoma. J. Oncol. 2020, 2020, 2752417. [Google Scholar] [CrossRef]
- Ali, A.; Veeranki, S.N.; Chinchole, A.; Tyagi, S. MLL/WDR5 complex regulates Kif2A localization to ensure chromosome congression and proper spindle assembly during mitosis. Dev. Cell. 2017, 41, 605–622.e7. [Google Scholar] [CrossRef]
- Ruiz-Gomez, A.; Humrich, J.; Murga, C.; Quitterer, U.; Loshe, M.J.; Mayor, F., Jr. Phosphorylation of phosducin and phosducin-like protein by G protein-coupled receptor kinase 2. J. Biol. Chem. 2000, 275, 29724–29730. [Google Scholar] [CrossRef]
- Wohlschlegel, J.A.; Dwyer, B.T.; Dhar, S.K.; Cvetic, C.; Walter, J.C.; Duta, A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 2000, 290, 2309–2312. [Google Scholar] [CrossRef] [PubMed]
- Takihara, Y.; Ishimaru, N.; Pagano, M.; Takata, T.; Kudo, Y.; Tsunematsu, T. Aurora-A controls pre-replicative complex assembly and DNA replication by stabilizing geminin in mitosis. Nat. Commun. 2013, 4, 1885. [Google Scholar]
- Reglero, C.; Lafarga, V.; Rivas, V.; Albitre, A.; Ramos, P.; Berciano, S.R.; Tapia, O.; Martínez-Chantar, M.L.; Mayor, F., Jr.; Penela, P. GRK2-Dependent HuR Phosphorylation Regulates HIF1α Activation under Hypoxia or Adrenergic Stress. Cancers (Basel) 2020, 12, 1216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Foreman, O.; Wigle, D.A.; Kosari, F.; Vasmatzis, G.; Salisbury, J.L.; van Deursen, J.; Galardy, P.J. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J. Clin. Investig. 2012, 122, 4362–4374. [Google Scholar] [CrossRef]
- Eibes, S.; Gallisà-Suñé, N.; Rosas-Salvans, M.; Martínez-Delgado, P.; Vernos, I.; Roig, J. Nek9 Phosphorylation Defines a New Role for TPX2 in Eg5-dependent centrosome separation before nuclear envelope breakdown. Curr. Biol. 2018, 28, 121–129.e4. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.-J.; van Deursen, J.M. Cyclin B2 and p53 control proper timing of centrosome separation. Nat. Cell Biol. 2014, 16, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Graser, S.; Stierhof, Y.D.; Nigg, E.A. Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J. Cell Sci. 2007, 120, 4321–4331. [Google Scholar] [CrossRef]
- Bertran, M.T.; Sdelci, S.; Regué, L.; Avruch, J.; Caelles, C.; Roig, J. Nek9 is a Plk1-activated kinase that controls early centrosome separation through Nek6/7 and Eg5. EMBO J. 2011, 30, 2634–2647. [Google Scholar] [CrossRef]
- Lafarga, V.; Aymerich, I.; Tapia, O.; Mayor, F., Jr.; Penela, P. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 2012, 31, 856–869. [Google Scholar] [CrossRef]
- Lafarga, V.; Mayor, F., Jr.; Penela, P. The interplay between G protein-coupled receptor kinase 2 (GRK2) and histone deacetylase 6 (HDAC6) at the crossroads of epithelial cell motility. Cell Adh. Migr. 2012, 6, 495–501. [Google Scholar] [CrossRef][Green Version]
- De Boer, L.; Oakes, V.; Beamish, H.; Giles, N.; Stevens, F.; Somodevilla-Torres, M.; Desouza, C.; Gabrielli, M. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 2008, 27, 4261–4268. [Google Scholar] [CrossRef]
- Hayes, M.J.; Kimata, Y.; Wattam, S.L.; Lindon, C.; Mao, G.; Yamano, H.; Fry, A.M. Early mitotic degradation of Nek2A depends on Cdc20- independent interaction with the APC/C. Nat. Cell Biol. 2006, 8, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Ying, H.; Zheng, H.; Murray, S.A.; Xiao, Z.X.J. The MDM2 RING finger is required for cell cycle-dependent regulation of its protein expression. FEBS Lett. 2003, 544, 218–222. [Google Scholar] [CrossRef]
- Zhang, T.; Prives, C. Cyclin A-CDK Phosphorylation Regulates MDM2 Protein Interactions. J. Biol. Chem. 2001, 276, 29702–29710. [Google Scholar] [CrossRef]
- Kim, H.T.; Kim, K.P.; Lledias, F.; Kisselev, A.F.; Scaglione, K.M.; Skowyra, D.; Gygi, S.P.; Goldberg, A.L. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 2007, 282, 17375–17386. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492. [Google Scholar] [CrossRef]
- Kirkpatrick, D.S.; Hathaway, N.A.; Hanna, J.; Elsasser, S.; Rush, J.; Finley, D.; King, R.W.; Gygi, S.P. Quantitative analysis of In vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat. Cell Biol. 2006, 8, 700–710. [Google Scholar] [CrossRef]
- Manfredi, J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010, 24, 1580–1589. [Google Scholar] [CrossRef]
- Onel, K.; Cordon-Cardo, C. MDM2 and prognosis. Mol. Cancer Res. 2004, 2, 1–8. [Google Scholar]
- Patel, H.; Stavrou, I.; Shrestha, R.L.; Draviam, V.; Frame, M.C.; Brunton, V.G. Kindlin1 regulates microtubule function to ensure normal mitosis. J. Mol. Cell Biol. 2016, 8, 338–348. [Google Scholar] [CrossRef]
- Goshima, G.; Wollman, R.; Stuurman, N.; Scholey, J.M.; Vale, R.D. Length control of the metaphase spindle. Curr. Biol. 2005, 15, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Lee, J.; Kim, K.; Yoo, J.C.; Rhee, K. Characterization of NIP2/centrobin, a novel substrate of Nek2, and its potential role in microtubule stabilization. J. Cell Sci. 2007, 120, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Steinhäuser, K.; Klöble, P.; Kreis, N.N.; Ritter, A.; Friemel, A.; Roth, S.; Reichel, J.M.; Michaelis, J.; Rieger, M.A.; Louwen, F.; et al. Deficiency of RITA results in multiple mitotic defects by affecting microtubule dynamics. Oncogene 2017, 36, 2146–2159. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reglero, C.; Ortiz del Castillo, B.; Rivas, V.; Mayor, F., Jr.; Penela, P. Mdm2-Mediated Downmodulation of GRK2 Restricts Centrosome Separation for Proper Chromosome Congression. Cells 2021, 10, 729. https://doi.org/10.3390/cells10040729
Reglero C, Ortiz del Castillo B, Rivas V, Mayor F Jr., Penela P. Mdm2-Mediated Downmodulation of GRK2 Restricts Centrosome Separation for Proper Chromosome Congression. Cells. 2021; 10(4):729. https://doi.org/10.3390/cells10040729
Chicago/Turabian StyleReglero, Clara, Belén Ortiz del Castillo, Verónica Rivas, Federico Mayor, Jr., and Petronila Penela. 2021. "Mdm2-Mediated Downmodulation of GRK2 Restricts Centrosome Separation for Proper Chromosome Congression" Cells 10, no. 4: 729. https://doi.org/10.3390/cells10040729
APA StyleReglero, C., Ortiz del Castillo, B., Rivas, V., Mayor, F., Jr., & Penela, P. (2021). Mdm2-Mediated Downmodulation of GRK2 Restricts Centrosome Separation for Proper Chromosome Congression. Cells, 10(4), 729. https://doi.org/10.3390/cells10040729