
Regulation of TGFβ Signalling by TRPV4 in Chondrocytes

 , , , , , , and
, , , , , , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chondrocyte Isolation and Culture
2.2. Cell Stimulation
2.3. Immunofluorescence
2.4. Calcium Imaging and Quantification
2.5. Generation of SBE Reporter TC28a2 Cell Line
2.6. Luciferase Reporter Assay
2.7. Western Blotting
2.8. SiRNA Transfection
2.9. RNA-Seq
2.10. Pathway Analysis
2.11. Reverse Transcription and qRT-PCR
2.12. Transcription Factor-Binding Motif Analysis
3. Results
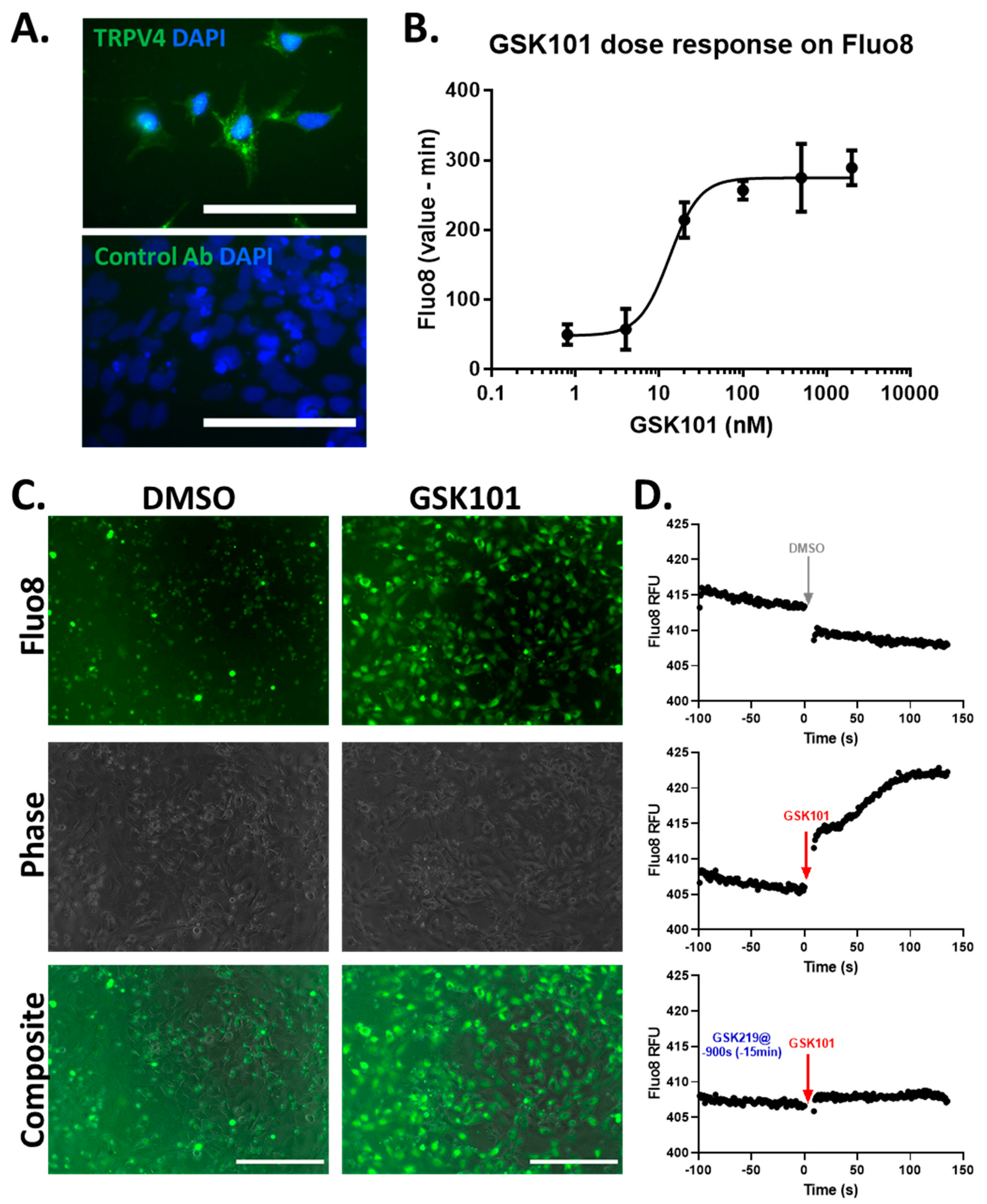
3.1. Expression and Activation of TRPV4 in TC28a2 Chondrocytes
3.2. Validation of SBE Reporter in TC28a2 Chondrocytes
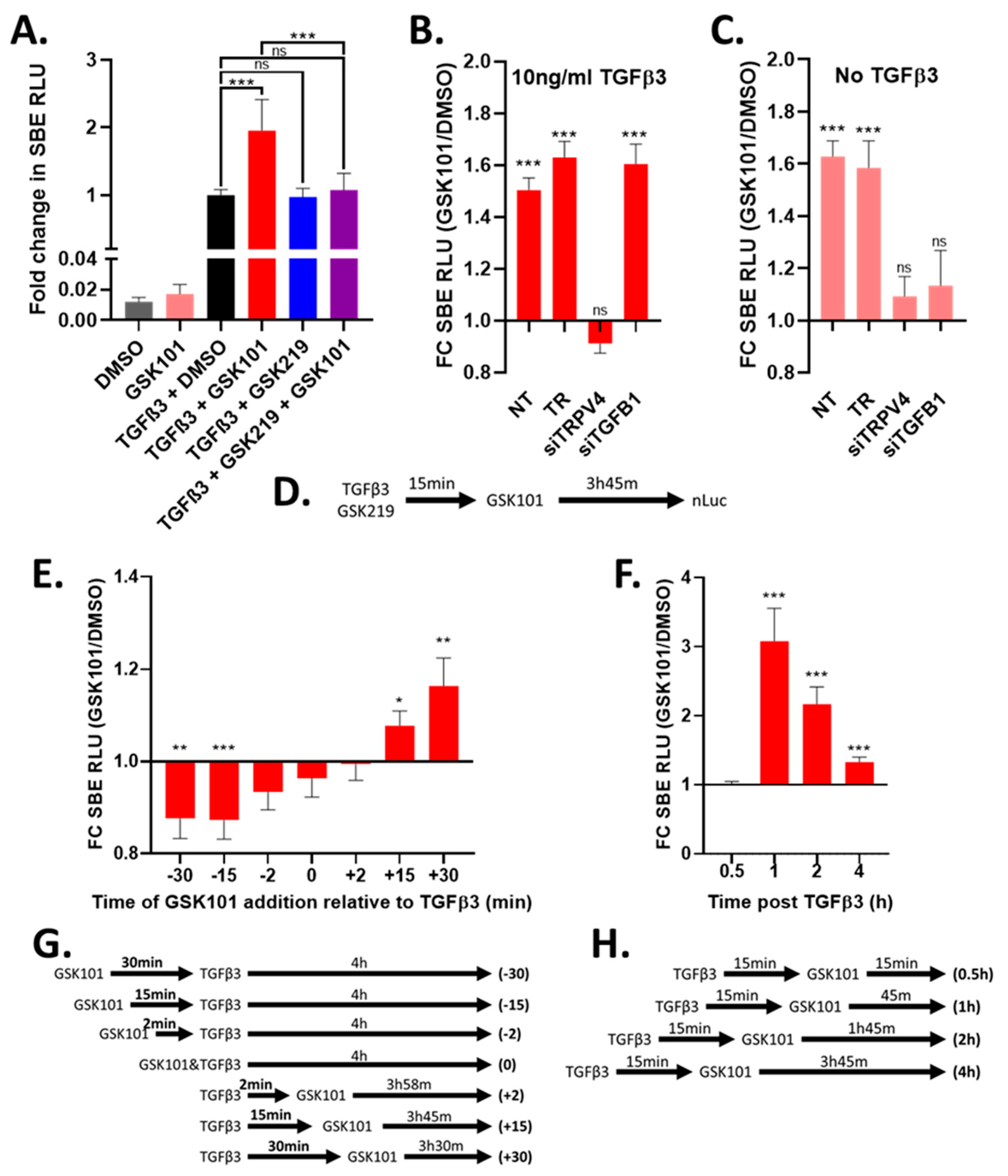
3.3. Activation of TRPV4 Enhances TGFβ Signalling
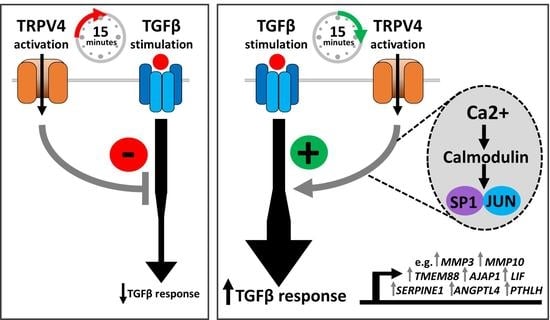
3.4. Timing of TRPV4 Activation and Inhibition Is Important for TGFβ Signalling Activity
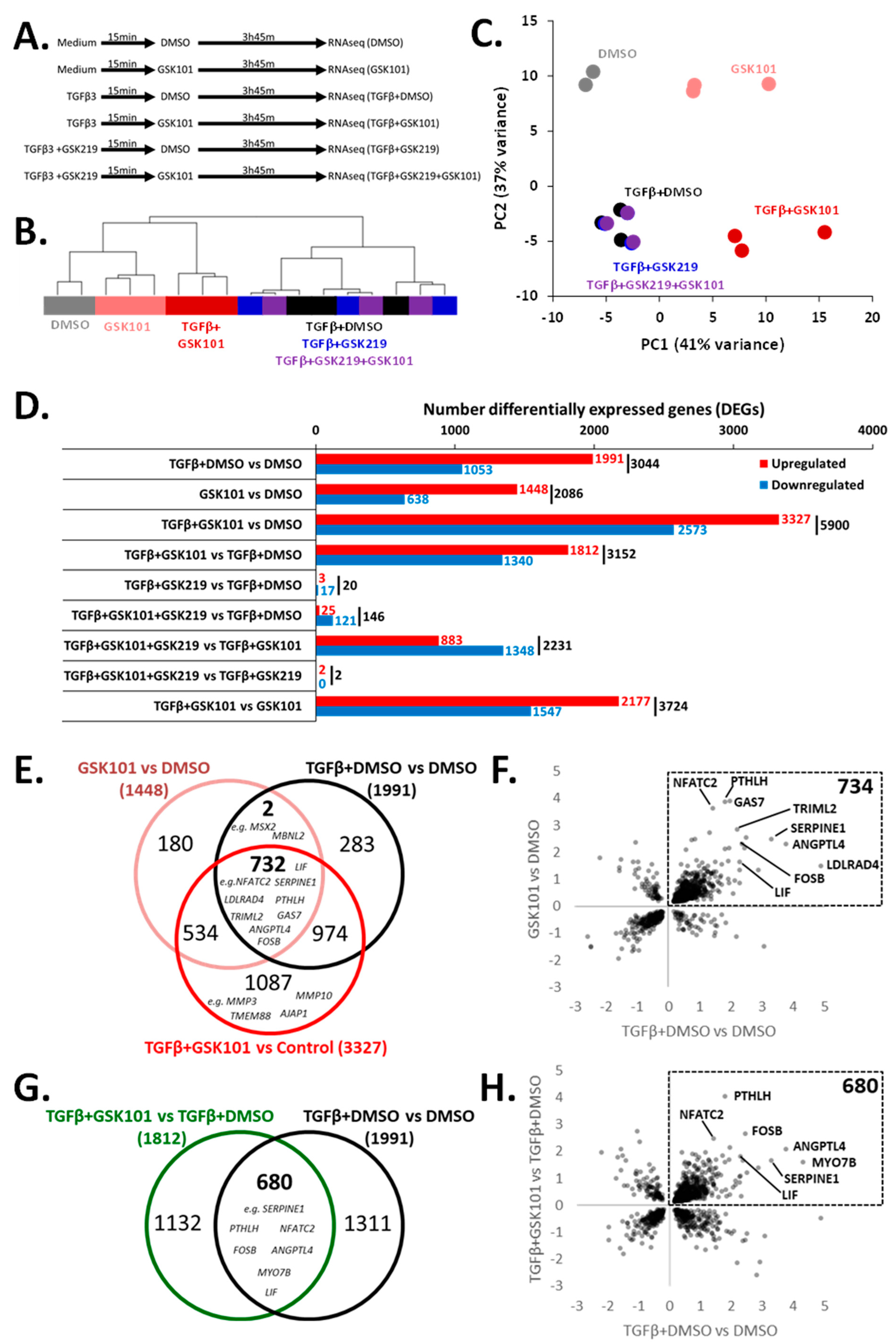
3.5. RNA-Seq Identification of the TGFβ3 Response Genes Enhanced by TRPV4 Activation
3.6. Pathway Analysis
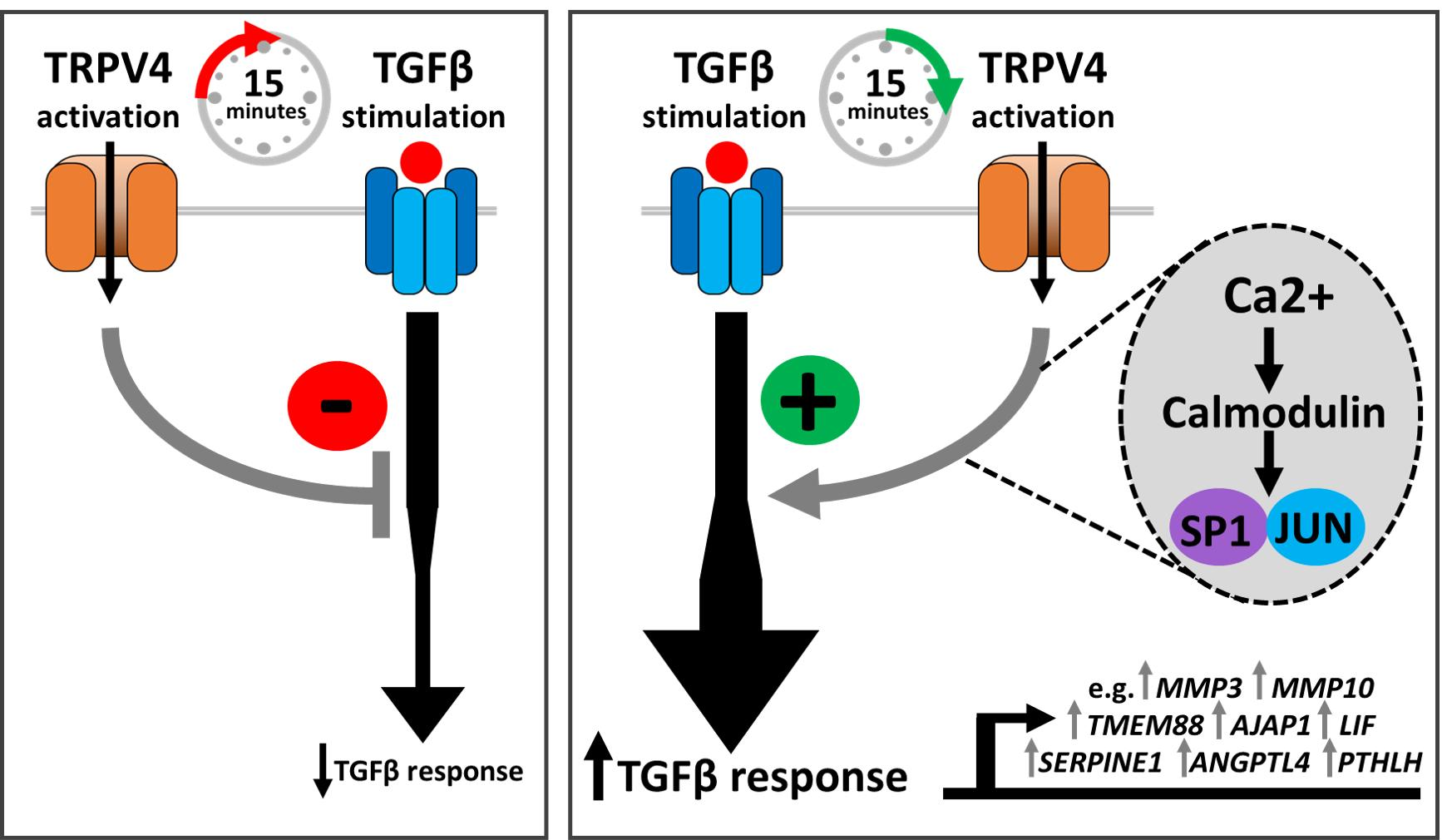
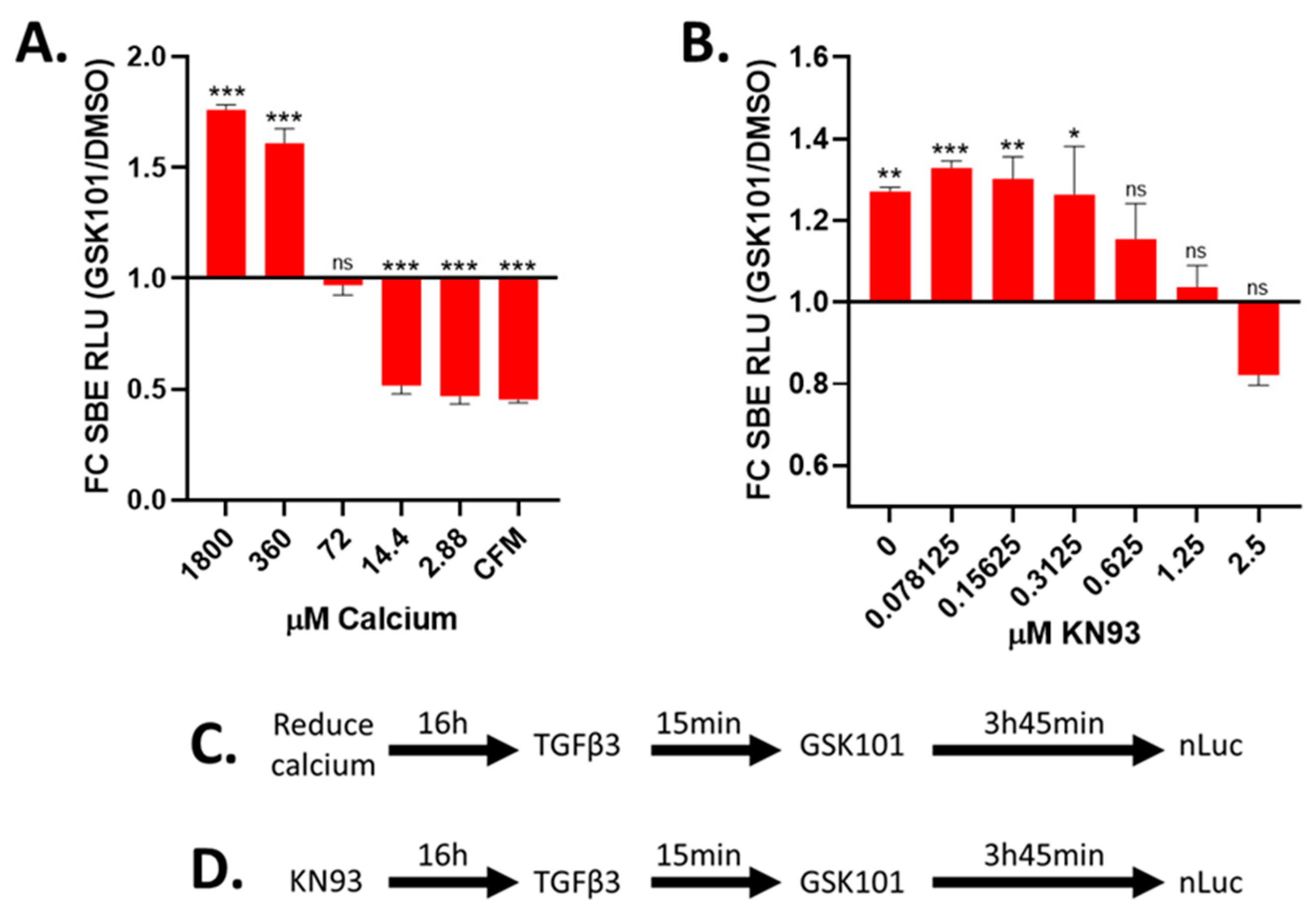
3.7. TRPV4 Activation Enhances TGFβ Signalling through Calcium/Calmodulin
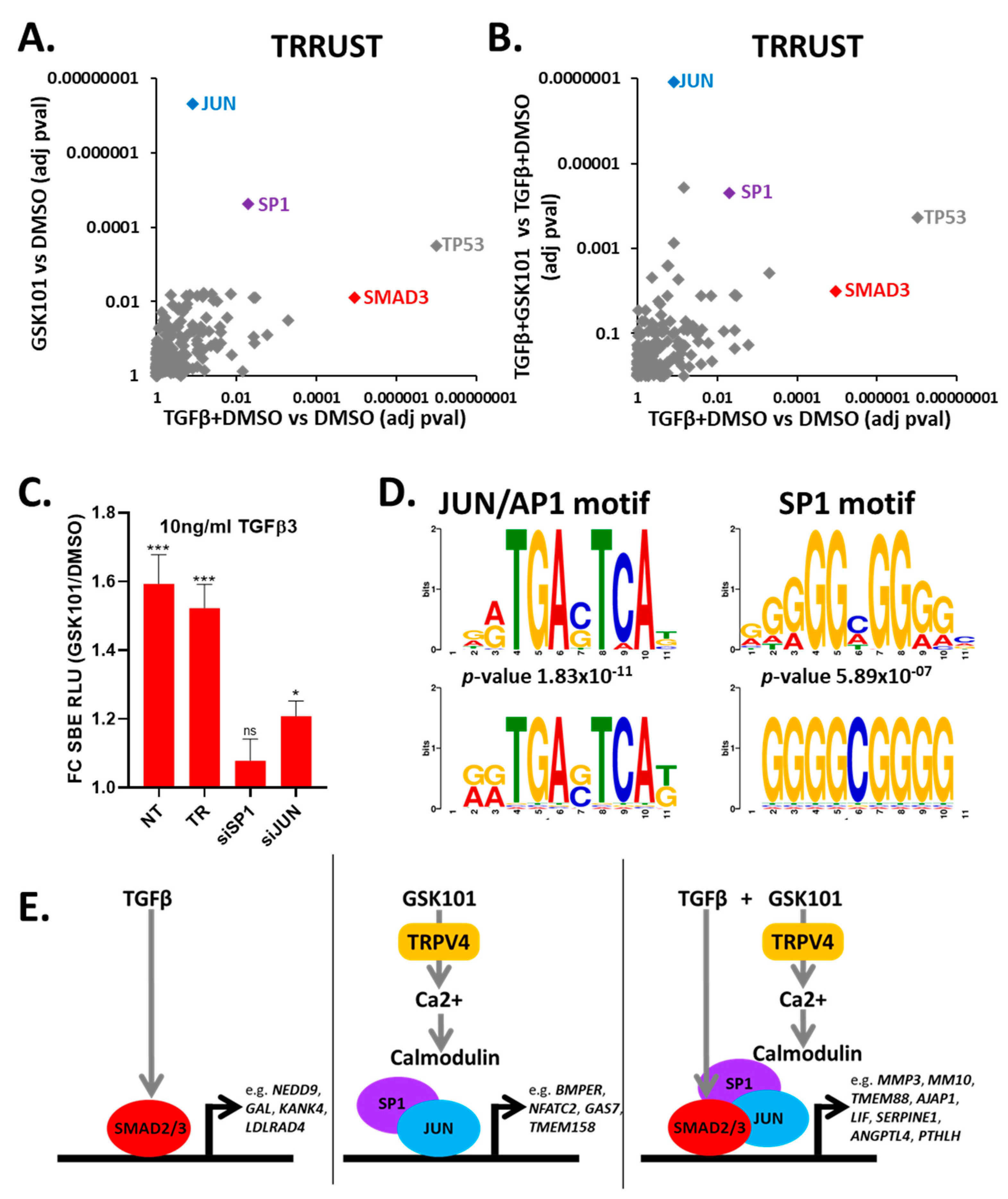
3.8. TRPV4 Activation Enhances TGFβ Signalling through JUN and SP1
4. Discussion
4.1. TRPV4 Activates Canonical TGFβ Signalling
4.2. Mechanism
4.3. TRPV4 Modulation of TGFβ Signalling Is Highly Dependent upon Timing
4.4. Downstream Implications of TRPV4–TGFβ Pathway Interaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liedtke, W.; Choe, Y.; Martí-Renom, M.A.; Bell, A.M.; Denis, C.S.; Sali, A.; Hudspeth, A.J.; Friedman, J.M.; Heller, S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmore-ceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef]
- Strotmann, R.; Harteneck, C.; Nunnenmacher, K.; Schultz, G.; Plant, T.D. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2000, 2, 695–702. [Google Scholar] [CrossRef]
- O’Conor, C.J.; Leddy, H.A.; Benefield, H.C.; Liedtke, W.B.; Guilak, F. TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc. Natl. Acad. Sci. USA 2014, 111, 1316–1321. [Google Scholar] [CrossRef]
- Güler, A.D.; Lee, H.; Iida, T.; Shimizu, I.; Tominaga, M.; Caterina, M. Heat-Evoked Activation of the Ion Channel, TRPV4. J. Neurosci. 2002, 22, 6408–6414. [Google Scholar] [CrossRef] [PubMed]
- Phan, M.N.; Leddy, H.A.; Votta, B.J.; Kumar, S.; Levy, D.S.; Lipshutz, D.B.; Lee, S.H.; Liedtke, W.; Guilak, F. Functional characterization of TRPV4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis Rheum. 2009, 60, 3028–3037. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, S.; Wakabayashi, M.; Ohno, T.; Amano, K.; Ooishi, R.; Sugahara, T.; Shiojiri, S.; Tashiro, K.; Suzuki, Y.; Nishimura, R.; et al. Functional Gene Screening System Identified TRPV4 as a Regulator of Chondrogenic Differentiation. J. Biol. Chem. 2007, 282, 32158–32167. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Votta, B.J.; Kumar, S.; Liedtke, W.; Guilak, F. Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: Age- and sex-dependent progression of osteoarthritis in Trpv4-deficient mice. Arthritis Rheum. 2010, 62, 2973–2983. [Google Scholar] [CrossRef]
- Cameron, T.L.; Belluoccio, D.; Farlie, P.G.; Brachvogel, B.; Bateman, J.F. Global comparative transcriptome analysis of cartilage formation in vivo. BMC Dev. Biol. 2009, 9, 17–20. [Google Scholar] [CrossRef]
- Walter, B.A.; Purmessur, D.; Moon, A.; Occhiogrosso, J.; Laudier, D.M.; Hecht, A.C.; Iatridis, J.C. Reduced tissue osmolarity increases TRPV4 expression and pro-inflammatory cytokines in intervertebral disc cells. Eur. Cells Mater. 2016, 32, 123–136. [Google Scholar] [CrossRef]
- Leddy, H.A.; McNulty, A.L.; Lee, S.H.; Rothfusz, N.E.; Gloss, B.; Kirby, M.L.; Hutson, M.R.; Cohn, D.H.; Guilak, F.; Liedtke, W. Follistatin in chondrocytes: The link between TRPV4 channelopathies and skeletal malformations. FASEB J. 2014, 28, 2525–2537. [Google Scholar] [CrossRef]
- Lamande, S.R.; Yuan, Y.; Gresshoff, I.L.; Rowley, L.; Belluoccio, D.; Kaluarachchi, K.; Little, C.B.; Botzenhart, E.; Zerres, K.; Amor, D.J.; et al. Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat. Genet. 2011, 43, 1142–1146. [Google Scholar] [CrossRef]
- Soul, J.; Dunn, S.L.; Anand, S.; Serracino-Inglott, F.; Schwartz, J.-M.; Boot-Handford, R.P.; Hardingham, T.E. Stratification of knee osteoarthritis: Two major patient subgroups identified by genome-wide expression analysis of articular cartilage. Ann. Rheum. Dis. 2017, 77, 423. [Google Scholar] [CrossRef]
- Masuyama, R.; Vriens, J.; Voets, T.; Karashima, Y.; Owsianik, G.; Vennekens, R.; Lieben, L.; Torrekens, S.; Moermans, K.; Bosch, A.V.; et al. TRPV4-Mediated Calcium Influx Regulates Terminal Differentiation of Osteoclasts. Cell Metab. 2008, 8, 257–265. [Google Scholar] [CrossRef]
- Allen, J.L.; Cooke, M.E.; Alliston, T. ECM stiffness primes the TGFbeta pathway to promote chondrocyte differentiation. Mol. Biol. Cell 2012, 23, 3731–3742. [Google Scholar] [CrossRef]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.; Miyazono, K.; et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef]
- Yan, J.; Fang, Y.; Ding, L.; Zhu, J.; Lu, Q.; Huang, C.; Yang, X.; Ye, Q. Regulation of large-scale chromatin unfolding by Smad4. Biochem. Biophys. Res. Commun. 2004, 315, 330–335. [Google Scholar] [CrossRef]
- Pouponnot, C.; Jayaraman, L.; Massagué, J. Physical and Functional Interaction of SMADs and p300/CBP. J. Biol. Chem. 1998, 273, 22865–22868. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, X.H.; Derynck, R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced tran-scription. Nature 1998, 394, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Attisano, L. The TGFbeta Superfamily Signaling Pathway. Wiley Interdiscip. Rev. Dev. Boil. 2013, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Rigueur, D.; Lyons, K.M. TGFbeta signaling in cartilage development and maintenance. Birth Defects Res. C Embryo Today 2014, 102, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-F.; Darowish, M.; Zuscik, M.J.; Chen, D.; Schwarz, E.M.; Rosier, R.N.; Drissi, H.; O’Keefe, R.J. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J. Bone Miner. Res. 2005, 21, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.; Yang, T.; Chen, Y.; Munivez, E.; Bertin, T.; Zabel, B.; Lee, B. Interaction of TGFbeta and BMP signaling pathways during chondrogenesis. PLoS ONE 2011, 6, e16421. [Google Scholar] [CrossRef]
- Davidson, E.N.B.; Remst, D.F.G.; Vitters, E.L.; Van Beuningen, H.M.; Blom, A.B.; Goumans, M.-J.; Berg, W.B.V.D.; Van Der Kraan, P.M. Increase in ALK1/ALK5 Ratio as a Cause for Elevated MMP-13 Expression in Osteoarthritis in Humans and Mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Birkhead, J.R.; Suen, L.F.; Yamin, R.; Mizuno, S.; Glowacki, J.; Arbiser, J.L.; Apperley, J.F. Interleukin-1 beta-modulated gene expression in immortalized human chondrocytes. J. Clin. Investig. 1994, 94, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Minogue, B.M.; Richardson, S.M.; Zeef, L.A.; Freemont, A.J.; Hoyland, J.A. Transcriptional profiling of bovine intervertebral disc cells: Implications for identification of normal and degenerate human intervertebral disc cell phenotypes. Arthritis Res. Ther. 2010, 12, R22. [Google Scholar] [CrossRef] [PubMed]
- Zawel, L.; Le Dai, J.; Buckhaults, P.; Zhou, S.; Kinzler, K.W.; Vogelstein, B.; Kern, S.E. Human Smad3 and Smad4 Are Sequence-Specific Transcription Activators. Mol. Cell 1998, 1, 611–617. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, T.; Tew, S.; Murdoch, A. Tissue engineering: Chondrocytes and cartilage. Arthritis Res. 2002, 4, S63–S68. [Google Scholar] [CrossRef]
- Thorneloe, K.S.; Sulpizio, A.C.; Lin, Z.; Figueroa, D.J.; Clouse, A.K.; McCafferty, G.P.; Chendrimada, T.P.; Lashinger, E.S.R.; Gordon, E.; Evans, L.; et al. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1 -piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J. Pharmacol. Exp. Ther. 2008, 326, 432–442. [Google Scholar]
- Thorneloe, K.S.; Cheung, M.; Bao, W.; Alsaid, H.; Lenhard, S.; Jian, M.-Y.; Costell, M.; Maniscalco-Hauk, K.; Krawiec, J.A.; Olzinski, A.; et al. An Orally Active TRPV4 Channel Blocker Prevents and Resolves Pulmonary Edema Induced by Heart Failure. Sci. Transl. Med. 2012, 4, 159ra148. [Google Scholar] [CrossRef] [PubMed]
- Mamuya, F.A.; Duncan, M.K. aV integrins and TGF-beta-induced EMT: A circle of regulation. J. Cell. Mol. Med. 2012, 16, 445–455. [Google Scholar] [CrossRef]
- Sharma, S.; Goswami, R.; Zhang, D.X.; Rahaman, S.O. TRPV4 regulates matrix stiffness and TGFbeta1-induced epithelial-mesenchymal transition. J. Cell. Mol. Med. 2019, 23, 761–774. [Google Scholar] [CrossRef]
- Jie, P.; Hong, Z.; Tian, Y.; Li, Y.; Lin, L.; Zhou, L.; Du, Y.; Chen, L.; Chen, L. Activation of transient receptor potential vanilloid 4 induces apoptosis in hippocampus through downregu-lating PI3K/Akt and upregulating p38 MAPK signaling pathways. Cell Death Dis. 2015, 6, e1775. [Google Scholar] [CrossRef]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef]
- Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Chilian, W.; Thodeti, C.K. TRPV4 deletion protects heart from myocardial infarction-induced adverse remodeling via modula-tion of cardiac fibroblast differentiation. Basic Res. Cardiol. 2020, 115, 14. [Google Scholar] [CrossRef]
- Eslaminejad, M.B.; Karimi, N.; Shahhoseini, M. Chondrogenic differentiation of human bone marrow-derived mes-enchymal stem cells treated by GSK-3 inhibitors. Histochem. Cell Biol. 2013, 140, 623–633. [Google Scholar] [CrossRef]
- Nilius, B.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T. TRPV4 calcium entry channel: A paradigm for gating diversity. Am. J. Physiol. Physiol. 2004, 286, C195–C205. [Google Scholar] [CrossRef] [PubMed]
- Saitta, B.; Elphingstone, J.; Limfat, S.; Shkhyan, R.; Evseenko, D. CaMKII inhibition in human primary and pluripotent stem cell-derived chondrocytes modulates effects of TGFbeta and BMP through SMAD signaling. Osteoarthr. Cartil. 2019, 27, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Wicks, S.J.; Lui, S.; Abdel-Wahab, N.; Mason, R.M.; Chantry, A. Inactivation of smad-transforming growth factor beta signaling by Ca(2+)-calmodulin-dependent protein kinase II. Mol. Cell. Biol. 2000, 20, 8103–8111. [Google Scholar] [CrossRef] [PubMed]
- Gooch, J.L.; Gorin, Y.; Zhang, B.-X.; Abboud, H.E. Involvement of calcineurin in transforming growth factor-beta-mediated regulation of extracellular matrix accumulation. J. Biol. Chem. 2004, 279, 15561–15570. [Google Scholar] [CrossRef] [PubMed]
- Roach, K.M.; Feghali-Bostwick, C.; Wulff, H.; Amrani, Y.; Bradding, P. Human lung myofibroblast TGFbeta1-dependent Smad2/3 signalling is Ca(2+)-dependent and regulated by KCa3.1 K(+) channels. Fibrogenesis Tissue Repair 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.-W.; Lee, S.-Y.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Backes, T.M.; Rössler, O.G.; Hui, X.; Grötzinger, C.; Lipp, P.; Thiel, G. Stimulation of TRPV1 channels activates the AP-1 transcription factor. Biochem. Pharmacol. 2018, 150, 160–169. [Google Scholar] [CrossRef]
- Peake, M.A.; Cooling, L.M.; Magnay, J.L.; Thomas, P.B.M.; El Haj, A.J. Selected Contribution: Regulatory pathways involved in mechanical induction of c-fos gene expression in bone cells. J. Appl. Physiol. 2000, 89, 2498–2507. [Google Scholar] [CrossRef]
- Datto, M.B.; Yu, Y.; Wang, X.F. Functional analysis of the transforming growth factor beta responsive elements in the WAF1/Cip1/p21 promoter. J. Biol. Chem. 1995, 270, 28623–28628. [Google Scholar] [CrossRef]
- Bachmeier, B.E.; Albini, A.; Vené, R.; Benelli, R.; Noonan, D.; Weigert, C.; Weiler, C.; Lichtinghagen, R.; Jochum, M.; Nerlich, A.G. Cell density-dependent regulation of matrix metalloproteinase and TIMP expression in differently tumorigenic breast cancer cell lines. Exp. Cell Res. 2005, 305, 83–98. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Nie, P.; Lu, J.; Ling, Y.; Guo, J.; Zhang, B.; Hu, J.; Liao, J.; Gu, J.; Dai, B.; et al. Less mechanical loading attenuates osteoarthritis by reducing cartilage degeneration, subchondral bone remodelling, secondary inflammation, and activation of NLRP3 inflammasome. Bone Jt. Res. 2020, 9, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.D.; Thodeti, C.K.; Tytell, J.D.; Mammoto, A.; Overby, D.R.; Ingber, D.E. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr. Biol. (Camb) 2010, 2, 435–442. [Google Scholar] [CrossRef]
- Saitta, B.; Passarini, J.; Sareen, D.; Ornelas, L.; Sahabian, A.; Argade, S.; Krakow, D.; Cohn, D.H.; Svendsen, C.N.; Rimoin, D.L. Patient-derived skeletal dysplasia induced pluripotent stem cells display abnormal chondrogenic marker expression and regulation by BMP2 and TGFbeta1. Stem. Cells Dev. 2014, 23, 1464–1478. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woods, S.; Humphreys, P.A.; Bates, N.; Richardson, S.A.; Kuba, S.Y.; Brooks, I.R.; Cain, S.A.; Kimber, S.J. Regulation of TGFβ Signalling by TRPV4 in Chondrocytes. Cells 2021, 10, 726. https://doi.org/10.3390/cells10040726
Woods S, Humphreys PA, Bates N, Richardson SA, Kuba SY, Brooks IR, Cain SA, Kimber SJ. Regulation of TGFβ Signalling by TRPV4 in Chondrocytes. Cells. 2021; 10(4):726. https://doi.org/10.3390/cells10040726
Chicago/Turabian StyleWoods, Steven, Paul A. Humphreys, Nicola Bates, Sophie Alice Richardson, Shweta Yogesh Kuba, Imogen R. Brooks, Stuart A. Cain, and Susan J. Kimber. 2021. "Regulation of TGFβ Signalling by TRPV4 in Chondrocytes" Cells 10, no. 4: 726. https://doi.org/10.3390/cells10040726
APA StyleWoods, S., Humphreys, P. A., Bates, N., Richardson, S. A., Kuba, S. Y., Brooks, I. R., Cain, S. A., & Kimber, S. J. (2021). Regulation of TGFβ Signalling by TRPV4 in Chondrocytes. Cells, 10(4), 726. https://doi.org/10.3390/cells10040726