
The Inherited and Familial Component of Early-Onset Colorectal Cancer

 , , ,
, , ,  , and
, and 
Abstract
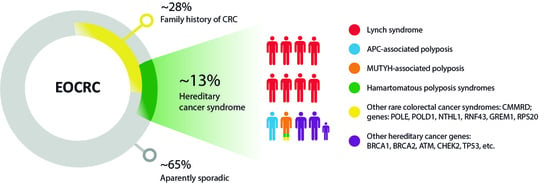
1. Introduction
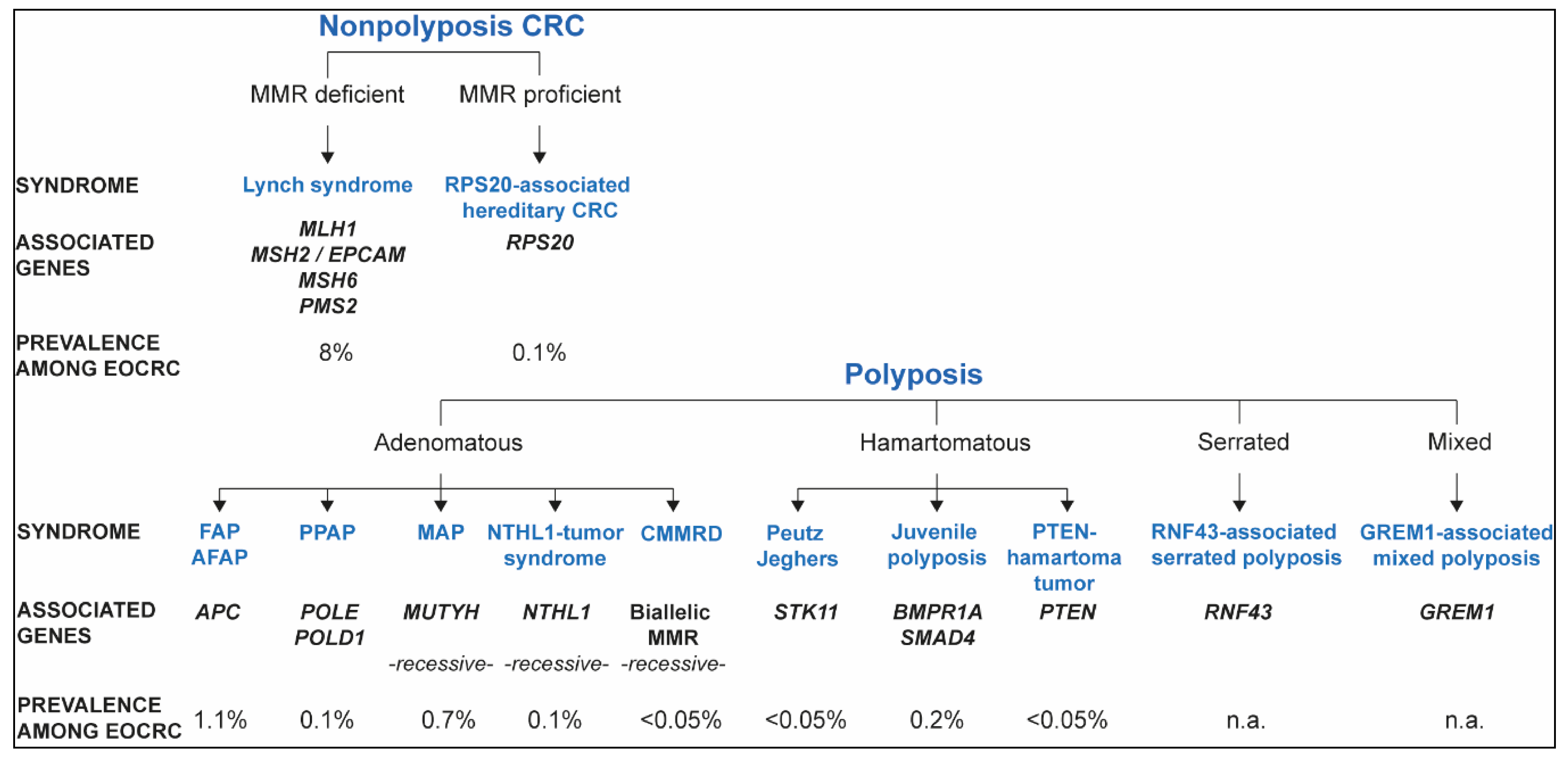
2. Inherited Syndromes That Predispose to EOCRC
2.1. Lynch Syndrome and Constitutional Mismatch Repair Deficiency
2.1.1. Lynch Syndrome
2.1.2. Constitutional Mismatch Repair Deficiency (CMMRD)
2.2. Nonpolyposis Mismatch Repair Proficient EOCRC: RPS20 and Other Candidate Genes
2.3. APC-Associated Polyposis
2.4. Polymerase Proofreading-Associated Polyposis (PPAP)
2.5. MUTYH-Associated Polyposis (MAP)
2.6. NTHL1 Tumor Syndrome
2.7. Hamartomatous Polyposis
2.7.1. Peutz–Jeghers Syndrome (PJS)
2.7.2. Juvenile Polyposis
2.7.3. PTEN Hamartoma Tumor Syndrome
2.8. RNF43-Associated Serrated Polyposis
2.9. GREM1-Associated Mixed Polyposis
2.10. Clinical Consequences of having a Hereditary CRC Syndrome
3. EOCRC in Non-CRC Cancer Syndromes
4. Familial Aggregation of CRC in EOCRC
5. Low Risk Alleles and EOCRC
6. Genetic Counseling and Genetic Testing Recommendations for EOCRC
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stoffel, E.M.; Murphy, C.C. Epidemiology and Mechanisms of the Increasing Incidence of Colon and Rectal Cancers in Young Adults. Gastroenterology 2020, 158, 341–353. [Google Scholar] [CrossRef]
- Siegel, R.L.; Torre, L.A.; Soerjomataram, I.; Hayes, R.B.; Bray, F.; Weber, T.K.; Jemal, A. Global patterns and trends in colorectal cancer incidence in young adults. Gut 2019, 68, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Boland, C.R. Colorectal Cancer in Persons Under Age 50: Seeking Causes and Solutions. Gastrointest Endosc. Clin. N. Am. 2020, 30, 441–455. [Google Scholar] [CrossRef]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- Stoffel, E.M.; Koeppe, E.; Everett, J.; Ulintz, P.; Kiel, M.; Osborne, J.; Williams, L.; Hanson, K.; Gruber, S.B.; Rozek, L.S. Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology 2018, 154, 897–905.e1. [Google Scholar] [CrossRef]
- Chang, D.T.; Pai, R.K.; A Rybicki, L.; A Dimaio, M.; Limaye, M.; Jayachandran, P.; Koong, A.C.; A Kunz, P.; A Fisher, G.; Ford, J.M.; et al. Clinicopathologic and molecular features of sporadic early-onset colorectal adenocarcinoma: An adenocarcinoma with frequent signet ring cell differentiation, rectal and sigmoid involvement, and adverse morphologic features. Mod. Pathol. 2012, 25, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Mork, M.E.; You, Y.N.; Ying, J.; Bannon, S.A.; Lynch, P.M.; Rodriguez-Bigas, M.A.; Vilar, E. High Prevalence of Hereditary Cancer Syndromes in Adolescents and Young Adults With Colorectal Cancer. J. Clin. Oncol. 2015, 33, 3544–3549. [Google Scholar] [CrossRef] [PubMed]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef]
- DeRycke, M.S.; Gunawardena, S.; Balcom, J.R.; Pickart, A.M.; Waltman, L.A.; French, A.J.; McDonnell, S.; Riska, S.M.; Fogarty, Z.C.; Larson, M.C.; et al. Targeted sequencing of 36 known or putative colorectal cancer susceptibility genes. Mol. Genet. Genom. Med. 2017, 5, 553–569. [Google Scholar] [CrossRef]
- LaDuca, H.; Polley, E.C.; Yussuf, A.; Hoang, L.; Bs, S.G.; Hart, S.N.; Yadav, S.; Hu, C.; Na Ms, J.; Goldgar, D.E.; et al. A clinical guide to hereditary cancer panel testing: Evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet. Med. 2020, 22, 407–415. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, F.; Wang, Y.; Hu, J.; Ding, P.; Lin, J.; Pan, Z.; Chen, G.; Shao, J.; Xu, R.; et al. Germline mutational profile of Chinese patients under 70 years old with colorectal cancer. Cancer Commun. 2020, 40, 620–632. [Google Scholar] [CrossRef]
- Archambault, A.N.; Su, Y.R.; Jeon, J.; Thomas, M.; Lin, Y.; Conti, D.V.; Win, A.K.; Sakoda, L.C.; Lansdorp-Vogelaar, I.; Peterse, E.F.P.; et al. Cumulative Burden of Colorectal Cancer-Associated Genetic Variants Is More Strongly Associated with Early-Onset vs Late-Onset Cancer. Gastroenterology 2020, 158, 1274–1286.e12. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; Ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef]
- Vasen, H.F.; Boland, C.R. Progress in genetic testing, classification, and identification of Lynch syndrome. JAMA 2005, 293, 2028–2030. [Google Scholar] [CrossRef]
- Moreira, L.; Balaguer, F.; Lindor, N.; De La Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch Syndrome Among Patients With Colorectal Cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef]
- Lynch, H.T.; Smyrk, T.C.; Watson, P.; Lanspa, S.J.; Lynch, J.F.; Lynch, P.M.; Cavalieri, R.; Boland, C. Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: An updated review. Gastroenterology 1993, 104, 1535–1549. [Google Scholar] [CrossRef]
- Mendelsohn, R.B.; Herzog, K.; Shia, J.; Rahaman, N.; Stadler, Z.K.; Shike, M. Molecular Screening for Lynch Syndrome in Young Patients With Colorectal Adenomas. Clin. Colorectal. Cancer 2017, 16, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Lynch, J.F.; Lynch, P.M.; Attard, T. Hereditary colorectal cancer syndromes: Molecular genetics, genetic counseling, diagnosis and management. Fam. Cancer 2008, 7, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on Genetic Evaluation and Management of Lynch Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, Y.M.; Wagner, A.; Morreau, H.; Menko, F.; Stormorken, A.; Quehenberger, F.; Sandkuijl, L.; Møller, P.; Genuardi, M.; van Houwelingen, H.; et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: Impact on counseling and surveillance. Gastroenterology 2004, 127, 17–25. [Google Scholar] [CrossRef]
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.D.; Lindblom, A.; Lagerstedt, K.; Thibodeau, S.N.; Lindor, N.M.; et al. The Clinical Phenotype of Lynch Syndrome Due to Germ-Line PMS2 Mutations. Gastroenterology 2008, 135, 419–428.e1. [Google Scholar] [CrossRef]
- Jenkins, M.A.; Baglietto, L.; Dowty, J.G.; Van Vliet, C.M.; Smith, L.; Mead, L.J.; Macrae, F.A.; John, D.J.B.S.; Jass, J.R.; Giles, G.G. Cancer Risks For Mismatch Repair Gene Mutation Carriers: A Population-Based Early Onset Case-Family Study. Clin. Gastroenterol. Hepatol. 2006, 4, 489–498. [Google Scholar] [CrossRef]
- Pearlman, R.; Haraldsdottir, S.; de la Chapelle, A.; Jonasson, J.G.; Liyanarachchi, S.; Frankel, W.L.; Rafnar, T.; Stefansson, K.; Pritchard, C.C.; Hampel, H. Clinical characteristics of patients with colorectal cancer with double somatic mismatch repair mutations compared with Lynch syndrome. J. Med. Genet. 2019, 56, 462–470. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef]
- Seguí, N.; Navarro, M.; Pineda, M.; Köger, N.; Bellido, F.; González, S.; Campos, O.; Iglesias, S.; Valdés-Mas, R.; López-Doriga, A.; et al. Exome sequencing identifies MUTYH mutations in a family with colorectal cancer and an atypical phenotype. Gut 2015, 64, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.; Heidenreich, B.; Keller, G.; Hampel, H.; Laner, A.; De La Chapelle, A.; Holinski-Feder, E. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur. J. Hum. Genet. 2014, 22, 1334–1337. [Google Scholar] [CrossRef] [PubMed]
- A Elsayed, F.; Kets, C.M.; Ruano, D.; Akker, B.V.D.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2015, 23, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Limburg, P.J.; Harmsen, W.S.; Chen, H.H.; Gallinger, S.; Haile, R.W.; Baron, J.A.; Casey, G.; Woods, M.O.; Thibodeau, S.N.; Lindor, N.M. Prevalence of Alterations in DNA Mismatch Repair Genes in Patients With Young-Onset Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2011, 9, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Giráldez, M.D.; Balaguer, F.; Bujanda, L.; Cuatrecasas, M.; Muñoz, J.; Alonso-Espinaco, V.; Larzabal, M.; Petit, A.; Gonzalo, V.; Ocaña, T.; et al. MSH6 and MUTYH deficiency is a frequent event in early-onset colorectal cancer. Clin. Cancer Res. 2010, 16, 5402–5413. [Google Scholar] [CrossRef]
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J. Med. Genet. 2014, 51, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Suerink, M.; Wimmer, K.; Brugieres, L.; Colas, C.; Gallon, R.; Ripperger, T.; Benusiglio, E.M.; Bleiker, A.; Ghorbanoghli, Z.; Goldberg, Y.; et al. Report of the fifth meeting of the European Consortium ‘Care for CMMRD’ (C4CMMRD), Leiden, The Netherlands, July 6th 2019. Fam. Cancer 2020, 20, 1–7. [Google Scholar] [CrossRef]
- Terradas, M.; Capellá, G.; Valle, L. Dominantly Inherited Hereditary Nonpolyposis Colorectal Cancer Not Caused by MMR Genes. J. Clin. Med. 2020, 9, 1954. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, T.T.; O’Donohue, M.F.; Wu, Y.; Lohi, H.; Scherer, S.W.; Paterson, A.D.; Ellonen, P.; Abdel-Rahman, W.M.; Valo, S.; Mecklin, J.P.; et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 2014, 147, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Broderick, P.; Dobbins, S.E.; Chubb, D.; Kinnersley, B.; Dunlop, M.G.; Tomlinson, I.; Houlston, R.S. Validation of Recently Proposed Colorectal Cancer Susceptibility Gene Variants in an Analysis of Families and Patients—a Systematic Review. Gastroenterology 2017, 152, 75–77.e4. [Google Scholar] [CrossRef]
- Belhadj, S.; Terradas, M.; Munoz-Torres, P.M.; Aiza, G.; Navarro, M.; Capellá, G.; Valle, L. Candidate genes for hereditary colorectal cancer: Mutational screening and systematic review. Hum. Mutat. 2020, 41, 1963–1976. [Google Scholar] [CrossRef]
- Thompson, B.A.; Snow, A.K.; Koptiuch, C.; Kohlmann, W.K.; Mooney, R.; Johnson, S.; Huff, C.D.; Yu, Y.; Teerlink, C.C.; Feng, B.; et al. A novel ribosomal protein S20 variant in a family with unexplained colorectal cancer and polyposis. Clin. Genet. 2020, 97, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Brosens, L.A.A.; Offerhaus, G.J.A.; Giardiello, F.M.; de Leng, W.W.J.; Montgomery, E.A. Pathology and genetics of hereditary colorectal cancer. Pathology 2018, 50, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, P.; Davaro, E.P.; Doan, J.V.; Ising, M.E.; Evans, N.R.; Phillips, N.J.; Lai, V.; Guzman, M.A. Familial Adenomatous Polyposis Syndrome: An Update and Review of Extraintestinal Manifestations. Arch. Pathol. Lab. Med. 2019, 143, 1382–1398. [Google Scholar] [CrossRef]
- Morin, P.J. Colorectal cancer: The APC-lncRNA link. J. Clin. Invest 2019, 129, 503–505. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Z.; Huang, X.; Ye, J. [Clinical and molecular characteristics of a child with familial adenomatous polyposis]. Zhonghua Er Ke Za Zhi 2016, 54, 205–208. [Google Scholar] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Guarino Almeida, E.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef]
- Palles, C.; Latchford, A.; Valle, L. Adenomatous polyposis syndromes: Polymerase proofreading-associated polyposis. In Hereditary Colorectal Cancer Genetic Basis and Clinical Implications; Valle, L., Gruber, S.B., Capellá, G., Eds.; Pringer International Publishing AG: Cham, Switzerland, 2018. [Google Scholar]
- Wimmer, K.; Beilken, A.; Nustede, R.; Ripperger, T.; Lamottke, B.; Ure, B.; Steinmann, D.; Reineke-Plaass, T.; Lehmann, U.; Zschocke, J.; et al. A novel germline POLE mutation causes an early onset cancer prone syndrome mimicking constitutional mismatch repair deficiency. Fam. Cancer 2017, 16, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, H.; Scollon, S.; Reuther, J.; Voicu, H.; Rednam, S.P.; Lin, F.Y.; Fisher, K.E.; Chintagumpala, M.; Adesina, A.M.; Parsons, D.W.; et al. Germline POLE mutation in a child with hypermutated medulloblastoma and features of constitutional mismatch repair deficiency. Cold Spring Harb. Mol. Case Stud. 2019, 5, a004499. [Google Scholar] [CrossRef] [PubMed]
- van Gool, I.C.; Bosse, T.; Church, D.N. proofreading mutation, immune response and prognosis in endometrial cancer. Oncoimmunology 2016, 5, e1072675. [Google Scholar] [CrossRef]
- Mur, P.; Ms, S.G.-M.; Del Valle, J.; Ms, L.M.-P.; Vidal, A.; Pineda, M.; Cinnirella, G.; Ms, E.M.-R.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Achatz, M.I.; Porter, C.C.; Brugières, L.; Druker, H.; Frebourg, T.; Foulkes, W.D.; Kratz, C.P.; Kuiper, R.P.; Hansford, J.R.; Hernandez, H.S.; et al. Cancer Screening Recommendations and Clinical Management of Inherited Gastrointestinal Cancer Syndromes in Childhood. Clin. Cancer Res. 2017, 23, e107–e114. [Google Scholar] [CrossRef]
- Nielsen, M.; Joerink-van de Beld, M.C.; Jones, N.; Vogt, S.; Tops, C.M.; Vasen, H.F.; Sampson, J.R.; Aretz, S.; Hes, F.J. Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis. Gastroenterology 2009, 136, 471–476. [Google Scholar] [CrossRef]
- Castillejo, A.; Vargas, G.; Castillejo, M.I.; Navarro, M.; Barberá, V.M.; González, S.; Hernández-Illán, E.; Brunet, J.; Cajal, T.R.Y.; Balmaña, J.; et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur. J. Cancer 2014, 50, 2241–2250. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Shinde, J.; Alexandrov, L.B.; Assie, G.; Andre, T.; Helias-Rodzewicz, Z.; Ducoudray, R.; Le Corre, D.; Zucman-Rossi, J.; Emile, J.F.; et al. Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J. Pathol. 2017, 242, 10–15. [Google Scholar] [CrossRef]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef]
- Win, A.K.; Dowty, J.G.; Cleary, S.P.; Kim, H.; Buchanan, D.D.; Young, J.P.; Clendenning, M.; Rosty, C.; MacInnis, R.J.; Giles, G.G.; et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 2014, 146, e1–e5. [Google Scholar] [CrossRef] [PubMed]
- Theodoratou, E.; Campbell, H.; Tenesa, A.; Houlston, R.; Webb, E.; Lubbe, S.; Broderick, P.; Gallinger, S.; Croitoru, E.M.; Jenkins, M.A.; et al. A large-scale meta-analysis to refine colorectal cancer risk estimates associated with MUTYH variants. Br. J. Cancer 2010, 103, 1875–1884. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (version 1.2020). Genetic/Familial High-Risk Assessment: Colorectal [Internet]. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf (accessed on 21 January 2021).
- A Weren, R.D.; Ligtenberg, M.J.L.; Kets, C.M.; De Voer, R.M.; Verwiel, E.T.P.; Spruijt, L.; Zelst-Stams, W.A.G.V.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, R.P.; Nielsen, M.; De Voer, R.M.; Hoogerbrugge, N. NTHL1 Tumor Syndrome; GeneReviews ® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Rivera, B.; Castellsagué, E.; Bah, I.; van Kempen, L.C.; Foulkes, W.D. Biallelic NTHL1 Mutations in a Woman with Multiple Primary Tumors. N. Engl. J. Med. 2015, 373, 1985–1986. [Google Scholar] [CrossRef]
- Belhadj, S.; Mur, P.; Navarro, M.; González, S.; Moreno, V.; Capellá, G.; Valle, L. Delineating the Phenotypic Spectrum of the NTHL1-Associated Polyposis. Clin. Gastroenterol. Hepatol. 2017, 15, 461–462. [Google Scholar] [CrossRef]
- Fostira, F.; Kontopodis, E.; Apostolou, P.; Fragkaki, M.; Androulakis, N.; Yannoukakos, D.; Konstantopoulou, I.; Saloustros, E. Extending the clinical phenotype associated with biallelic NTHL1 germline mutations. Clin. Genet. 2018, 94, 588–589. [Google Scholar] [CrossRef] [PubMed]
- Belhadj, S.; Quintana, I.; Mur, P.; Munoz-Torres, P.M.; Alonso, M.H.; Navarro, M.; Terradas, M.; Piñol, V.; Brunet, J.; Moreno, V.; et al. NTHL1 biallelic mutations seldom cause colorectal cancer, serrated polyposis or a multi-tumor phenotype, in absence of colorectal adenomas. Sci. Rep. 2019, 9, 9020. [Google Scholar] [CrossRef]
- Altaraihi, M.; Gerdes, A.M.; Wadt, K. A new family with a homozygous nonsense variant in NTHL1 further delineated the clinical phenotype of NTHL1-associated polyposis. Hum. Genome Var 2019, 6, 46. [Google Scholar] [CrossRef]
- Groves, A.; Gleeson, M.; Spigelman, A.D. NTHL1-associate polyposis: First Australian case report. Fam. Cancer 2019, 18, 179–182. [Google Scholar] [CrossRef]
- Grolleman, J.E.; de Voer, R.M.; Elsayed, F.A.; Nielsen, M.; Weren, R.D.A.; Palles, C.; Ligtenberg, M.J.L.; Vos, J.R.; Ten Broeke, S.W.; de Miranda, N.F.C.C.; et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-tumor Phenotype. Cancer Cell 2019, 35, 256–266. [Google Scholar] [CrossRef]
- Weren, R.D.; Ligtenberg, M.J.; Geurts van Kessel, A.; De Voer, R.M.; Hoogerbrugge, N.; Kuiper, R.P. NTHL1 and MUTYH polyposis syndromes: Two sides of the same coin? J. Pathol. 2018, 244, 135–142. [Google Scholar] [CrossRef]
- Terradas, M.; Munoz-Torres, P.M.; Belhadj, S.; Aiza, G.; Navarro, M.; Brunet, J.; Capellá, G.; Valle, L. Contribution to colonic polyposis of recently proposed predisposing genes and assessment of the prevalence of NTHL1 - and MSH3 -associated polyposes. Hum. Mutat. 2019, 40, 1910–1923. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef]
- Hearle, N.; Schumacher, V.; Menko, F.H.; Olschwang, S.; Boardman, L.A.; Gille, J.J.; Keller, J.J.; Westerman, A.M.; Scott, R.J.; Lim, W.; et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin. Cancer Res. 2006, 12, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- Latchford, A.; Cohen, S.; Auth, M.; Scaillon, M.; Viala, J.; Daniels, R.; Talbotec, C.; Attard, T.; Durno, C.; Hyer, W.; et al. Management of Peutz-Jeghers Syndrome in Children and Adolescents: A Position Paper From the ESPGHAN Polyposis Working Group. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 442–452. [Google Scholar] [CrossRef]
- Volikos, E.; Robinson, J.; Aittomäki, K.; Mecklin, J.P.; Järvinen, H.; Westerman, A.M.; de Rooji, F.W.; Vogel, T.; Moeslein, G.; Launonen, V.; et al. LKB1 exonic and whole gene deletions are a common cause of Peutz-Jeghers syndrome. J. Med. Genet. 2006, 43, e18. [Google Scholar] [CrossRef] [PubMed]
- Gammon, A.; Jasperson, K.; Kohlmann, W.; Burt, R.W. Hamartomatous polyposis syndromes. Best Pr. Res. Clin. Gastroenterol. 2009, 23, 219–231. [Google Scholar] [CrossRef]
- Kidambi, T.D.; Kohli, D.R.; Samadder, N.J.; Singh, A. Hereditary Polyposis Syndromes. Curr. Treat Options Gastroenterol. 2019, 17, 650–665. [Google Scholar] [CrossRef]
- Larsen Haidle, J.; Howe, J.R. Juvenile Polyposis Syndrome; Genereviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N.; Group, P.G.D.; Genturis, E.R.N. Cancer Surveillance Guideline for individuals with PTEN hamartoma tumour syndrome. Eur. J. Hum. Genet. 2020, 28, 1387–1393. [Google Scholar] [CrossRef]
- Pilarski, R. Cowden syndrome: A critical review of the clinical literature. J. Genet. Couns. 2009, 18, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Carballal, S.; Rodriguez-Alcalde, D.; Moreira, L.; Hernandez, L.; Rodriguez, L.; Rodriguez-Moranta, F.; Gonzalo, V.; Bujanda, L.; Bessa, X.; Poves, C.; et al. Colorectal cancer risk factors in patients with serrated polyposis syndrome: A large multicentre study. Gut 2016, 65, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Ijspeert, J.E.G.; Rana, S.A.Q.; Atkinson, N.S.S.; Van Herwaarden, Y.J.; Bastiaansen, B.A.J.; E Van Leerdam, M.; Sanduleanu, S.; Bisseling, T.M.; Spaander, M.C.W.; Clark, S.K.; et al. Clinical risk factors of colorectal cancer in patients with serrated polyposis syndrome: A multicentre cohort analysis. Gut 2017, 66, 278–284. [Google Scholar] [CrossRef]
- Rodríguez-Alcalde, D.; Carballal, S.; Moreira, L.; Hernández, L.; Rodríguez-Alonso, L.; Rodríguez-Moranta, F.; Gonzalo, V.; Bujanda, L.; Bessa, X.; Poves, C.; et al. High incidence of advanced colorectal neoplasia during endoscopic surveillance in serrated polyposis syndrome. Endoscopy 2019, 51, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Bleijenberg, A.G.; Ijspeert, J.E.; Van Herwaarden, Y.J.; Carballal, S.; Pellisé, M.; Jung, G.; Bisseling, T.M.; Nagetaal, I.D.; E Van Leerdam, M.; Van Lelyveld, N.; et al. Personalised surveillance for serrated polyposis syndrome: Results from a prospective 5-year international cohort study. Gut 2019, 69, 112–121. [Google Scholar] [CrossRef] [PubMed]
- IJspeert, J.E.G.; Bevan, R.; Senore, C.; Kaminski, M.F.; Kuipers, E.J.; Mroz, A.; Bessa, X.; Cassoni, P.; Hassan, C.; Repici, A.; et al. Detection rate of serrated polyps and serrated polyposis syndrome in colorectal cancer screening cohorts: A European overview. Gut 2017, 66, 1225–1232. [Google Scholar] [CrossRef]
- Rivero-Sanchez, L.; Lopez-Ceron, M.; Carballal, S.; Moreira, L.; Bessa, X.; Serradesanferm, A.; Pozo, A.; Augé, J.M.; Ocaña, T.; Sánchez, A.; et al. Reassessment colonoscopy to diagnose serrated polyposis syndrome in a colorectal cancer screening population. Endoscopy 2017, 49, 44–53. [Google Scholar] [CrossRef]
- Dekker, E.; Bleijenberg, A.; Balaguer, F.; IJspeert, J.E.; Bleijenberg, A.G.; Pellisé, M.; Carballal, S.; Rivero, L.; Latchford, A. Update on the World Health Organization Criteria for Diagnosis of Serrated Polyposis Syndrome. Gastroenterology 2020, 158, 1520–1523. [Google Scholar] [CrossRef]
- Gala, M.K.; Mizukami, Y.; Le, L.P.; Moriichi, K.; Austin, T.; Yamamoto, M.; Lauwers, G.Y.; Bardeesy, N.; Chung, D.C. Germline Mutations in Oncogene-Induced Senescence Pathways Are Associated With Multiple Sessile Serrated Adenomas. Gastroenterology 2014, 146, 520–529.e6. [Google Scholar] [CrossRef] [PubMed]
- Taupin, D.; Lam, W.; Rangiah, D.; McCallum, L.; Whittle, B.; Zhang, Y.; Andrews, D.; Field, M.; Goodnow, C.C.; Cook, M.C. A deleterious RNF43 germline mutation in a severely affected serrated polyposis kindred. Hum. Genome Var. 2015, 2, 15013. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.; Lai, J.C.; Ho, S.L.; Leung, W.K.; Law, W.L.; Lee, J.F.; Chan, A.K.W.; Tsui, W.Y.; Chan, A.S.Y.; Lee, B.C.H.; et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2016, 66, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, D.D.; Clendenning, M.; Zhuoer, L.; Stewart, J.R.; Joseland, S.; Woodall, S.; Arnold, J.; Semotiuk, K.; Aronson, M.; Holter, S.; et al. Lack of evidence for germline RNF43 mutations in patients with serrated polyposis syndrome from a large multinational study. Gut 2017, 66, 1170–1172. [Google Scholar] [CrossRef] [PubMed]
- Quintana, I.; Mejías-Luque, R.; Terradas, M.; Navarro, M.; Piñol, V.; Mur, P.; Belhadj, S.; Grau, E.; Darder, E.; Solanes, A.; et al. Evidence suggests that germline RNF43 mutations are a rare cause of serrated polyposis. Gut 2018, 67, 2230–2232. [Google Scholar] [CrossRef]
- Yu, J.; Mohamed Yuso, P.A.B.; Woutersen, D.T.J.; Goh, P.; Harmston, N.; Smits, R.; Harmston, N.; Smits, R.; Epstein, D.M.; Virshup, D.M.; et al. The functional landscape of patient-derived RNF43 mutations predicts sensitivity to Wnt inhibiton. Cancer Res. 2020, 80, 5619–5632. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, E.; Woodford-Richens, K.; Lockett, M.; Rowan, A.; Sawyer, E.; Heinimann, K.; Rozen, P.; Murday, V.; Whitelaw, S.; Ginsberg, A.; et al. An Ancestral Ashkenazi Haplotype at the HMPS/CRAC1 Locus on 15q13–q14 Is Associated with Hereditary Mixed Polyposis Syndrome. Am. J. Hum. Genet. 2003, 72, 1261–1267. [Google Scholar] [CrossRef][Green Version]
- Whitelaw, S.C.; A Murday, V.; Tomlinson, I.P.; Thomas, H.J.; Cottrell, S.; Ginsberg, A.; Bukofzer, S.; Hodgson, S.V.; Skudowitz, R.B.; Jass, J.R.; et al. Clinical and molecular features of the hereditary mixed polyposis syndrome. Gastroenterology 1997, 112, 327–334. [Google Scholar] [CrossRef]
- Thomas, H.J.; Whitelaw, S.C.; Cottrell, S.E.; Murday, V.A.; Tomlinson, I.P.; Markie, D.; Jones, T.; Bishop, D.T.; Hodgson, S.V.; Sheer, D.; et al. Genetic mapping of hereditary mixed polyposis syndrome to chromosome 6q. Am. J. Hum. Genet. 1996, 58, 770–776. [Google Scholar]
- Jaeger, E.; Leedham, S.J.; Lewis, A.; Segditsas, S.; Becker, M.; Cuadrado, P.R.; Davis, H.; Kaur, K.; Heinimann, K.; Howarth, K.; et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat. Genet. 2012, 44, 699–703. [Google Scholar] [CrossRef]
- Lieberman, S.; Walsh, T.; Schechter, M.; Adar, T.; Goldin, E.; Beeri, R.; Baris, H.; Avi, L.B.; Half, E.; Lerer, I.; et al. Features of Patients With Hereditary Mixed Polyposis Syndrome Caused by Duplication of GREM1 and Implications for Screening and Surveillance. Gastroenterology 2017, 152, 1876–1880.e1. [Google Scholar] [CrossRef]
- Rohlin, A.; Eiengard, F.; Lundstam, U.; Zagoras, T.; Nilsson, S.; Edsjo, A.; Pedersen, J.; Svensson, J.; Skullman, S.; Karlsson, B.G.; et al. GREM1 and POLE variants in hereditary colorectal cancer syndromes. Genes Chromosomes Cancer 2016, 55, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, R.; Verwiel, E.T.; Kamping, E.J.; Hoenselaar, E.; Gorgens, H.; Schackert, H.K.; van Krieken, J.H.J.M.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; van Kessel, A.G.; et al. Identification of candidate predisposing copy number variants in familial and early-onset colorectal cancer patients. Int. J. Cancer 2011, 129, 1635–1642. [Google Scholar] [CrossRef]
- Monahan, K.J.; Bradshaw, N.; Dolwani, S.; Desouza, B.; Dunlop, M.G.; E East, J.; Ilyas, M.; Kaur, A.; Lalloo, F.; Latchford, A.; et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut 2019, 69, 411–444. [Google Scholar] [CrossRef]
- Idos, G.; Valle, L. Lynch Syndrome; GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Seppälä, T.T.; Latchford, A.; Negoi, I.; Soares, A.S.; Jimenez-Rodriguez, R.; Sánchez-Guillén, L.; Evans, D.G.; Ryan, N.; Crosbie, E.J.; Dominguez-Valentin, M.; et al. European guidelines from the EHTG and ESCP for Lynch syndrome: An updated third edition of the Mallorca guidelines based on gene and gender. Br. J. Surg. 2020. [Google Scholar] [CrossRef]
- Herzig, D.; Hardimann, K.; Weiser, M.; You, N.; Paquette, I.; Feingold, D.L.; Steele, S.R. The American Society of Colon and Rectal Surgeons Clinical Practice Guidelines for the Management of Inherited Polyposis Syndromes. Dis. Colon Rectum 2017, 60, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2019, 247, 574–588. [Google Scholar] [CrossRef]
- Ulusan, A.M.; Rajendran, P.; Dashwood, W.M.; Yavuz, O.F.; Kapoor, S.; Gustafson, T.A.; Savage, M.I.; Brown, P.H.; Sei, S.; Mohammed, A.; et al. Optimization of Erlotinib Plus Sulindac Dosing Regimens for Intestinal Cancer Prevention in an Apc-Mutant Model of Familial Adenomatous Polyposis (FAP). Cancer Prev. Res. 2021, 14, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.-P.; Möslein, G.; E McRonald, F.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chan, A.T. Aspirin for Lynch syndrome: A legacy of prevention. Lancet 2020, 395, 1817–1818. [Google Scholar] [CrossRef]
- Boardman, L.A.; Vilar, E.; You, Y.N.; Samadder, J. AGA Clinical Practice Update on Young Adult-Onset Colorectal Cancer Diagnosis and Management: Expert Review. Clin. Gastroenterol. Hepatol. 2020, 18, 2415–2424. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. New Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Lau, D.; Kalaitzaki, E.; Church, D.N.; Pandha, H.; Tomlinson, I.; Annels, N.; Gerlinger, M.; Sclafani, F.; Smith, G.; Begum, R.; et al. Rationale and design of the POLEM trial: Avelumab plus fluoropyrimidine-based chemotherapy as adjuvant treatment for stage III mismatch repair deficient or POLE exonuclease domain mutant colon cancer: A phase III randomised study. ESMO Open 2020, 5, e000638. [Google Scholar] [CrossRef] [PubMed]
- Volkov, N.M.; Yanus, G.A.; Ivantsov, A.O.; Moiseenko, F.V.; Matorina, O.G.; Bizin, I.V.; Moiseyenko, V.M.; Imyanitov, E.N. Efficacy of immune checkpoint blockade in MUTYH-associated hereditary colorectal cancer. Investig. New Drugs 2019, 38, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Boland, P.M.; Yurgelun, M.B.; Boland, C.R. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J. Clin. 2018, 68, 217–231. [Google Scholar] [CrossRef]
- Valle, L.; de Voer, R.M.; Goldberg, Y.; Sjursen, W.; Försti, A.; Ruiz-Ponte, C.; Caldés, T.; Garré, P.; Olsen, M.F.; Nordling, M.; et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Aspects Med. 2019, 69, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Terradas, M.; Mur, P.; Belhadj, S.; Woodward, E.R.; Burghel, G.J.; Munoz-Torres, P.M.; Quintana, I.; Navarro, M.; Brunet, J.; Lazaro, C.; et al. TP53, a gene for colorectal cancer predisposition in the absence of Li-Fraumeni-associated phenotypes. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Stanich, P.P.; Pelstring, K.R.; Hampel, H.; Pearlman, R. A High Percentage of Early-Age Onset Colorectal Cancer is Potentially Preventable. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Chen, F.W.; Sundaram, V.; Chew, T.A.; Ladabaum, U. Advanced-Stage Colorectal Cancer in Persons Younger Than 50 Years Not Associated With Longer Duration of Symptoms or Time to Diagnosis. Clin. Gastroenterol. Hepatol. 2017, 15, 728–737.e3. [Google Scholar] [CrossRef]
- Chen, F.W.; Sundaram, V.; Chew, T.A.; Ladabaum, U. Low Prevalence of Criteria for Early Screening in Young-Onset Colorectal Cancer. Am. J. Prev. Med. 2017, 53, 933–934. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.B.; Maggard, M.A.; Livingston, E.H.; Yo, C.K. Colorectal cancer in the young. Am. J. Surg. 2004, 187, 343–348. [Google Scholar] [CrossRef] [PubMed]
- van Leerdam, M.E.; Roos, V.H.; van Hooft, J.E.; Balaguer, F.; Dekker, E.; Kaminski, M.F.; Latchford, A.; Neumann, H.; Ricciardiello, L.; Rupińska, M. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy 2019, 51, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Rex, D.K.; Boland, C.R.; Dominitz, J.A.; Giardiello, F.M.; Johnson, D.A.; Kaltenbach, T.; Levin, T.R.; Lieberman, D.; Robertson, D.J. Colorectal Cancer Screening: Recommendations for Physicians and Patients From the U.S. Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2017, 153, 307–323. [Google Scholar] [CrossRef]
- Gupta, S.; Bharti, B.; Ahnen, D.J.; Buchanan, D.D.; Cheng, I.C.; Cotterchio, M.; Figueiredo, J.C.; Gallinger, S.J.; Haile, R.W.; Jenkins, M.A.; et al. Potential impact of family history–based screening guidelines on the detection of early-onset colorectal cancer. Cancer 2020, 126, 3013–3020. [Google Scholar] [CrossRef]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef]
- Gupta, S.; Provenzale, D.; Llor, X.; Halverson, A.L.; Grady, W.; Chung, D.C.; Haraldsdottir, S.; Markowitz, A.J.; Slavin, T.P.; Hampel, H.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019. J. Natl. Compr. Canc. Netw. 2019, 17, 1032–1041. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study [Population] | EOCRC Age Cutoff | No. Patients Analyzed | Testing Approach | Hereditary Cancer | Gene/Mutation Spectrum in CRC Genes a % P or LP Variant Carriers (n) | Gene/Mutation Spectrum in Other Cancer Genes a,b % P or LP Variant Carriers (n) |
|---|---|---|---|---|---|---|
| Laduca 2020 [11] [Different populations] | <50 | Mean: 2672 (range: 986–4017) | Nine different multigene panels (5–49 genes evaluated) | 362/4017 (9.0%) | 5.3% MMR genes (213/3994) 0.5% APC (20/3884) 0.4% biallelic MUTYH (15/3953) 0.1% SMAD4 (5/3881) 0.1% BMPR1A (3/3881) 0.0% STK11 (1/3954) 0,0% PTEN (1/4014) 0.0% GREM1 (1/2366) | 1.5% BRCA1/2 (21/1387) 1.1% CHEK2 (43/3954) 1.0% ATM (14/,345) 0.2% TP53 (9/4017) 0.1% PALB2 (2/1350) 0.1% BARD1 (1/1339) 0.3% BRIP1 (4/1342) 0.1% RAD51C (1/1342) 0.2% CDKN2A (2/1228) 0.1% SMARCA4 (1/986) 0.1% NBN (2/1339) 0.1% CDH1 (2/3965) 0.1% NF1 (1/1298) |
| Chubb 2016 [9] [UK] | ≤55 | 1006 | Exome sequencing | 158/1006 (15.7%) | 11% MMR genes (111) 1.9% APC (19) 0.9% biallelic MUTYH (9) 0.4% POLE/POLD1 (4) 0.1% biallelic NTHL1 (1) | 0.9% BRCA1/2 (9) 0.3% ATM (3) 0.1% TP53 (1) 0.1% PALB2 (1) |
| Pearlman 2017 [5] [USA] | <50 | 450 | 25-gene panel | 65/450 (14.4%) | 8.2% MMR genes (37) 1.1% APC (5) 0.9% APC p.I1307K (4) 0.9% biallelic MUTYH (4) 0.2% SMAD4 (1) 0.2% APC/PMS2 (1) | 0.45% BRCA1 (2) 0.9% BRCA2 (4) 0.7% ATM (3) 0.2% ATM/CHEK2 (1) 0.45% PALB2 (2) 0.2% CDKN2A (1) |
| DeRycke 2017 [10] [USA, Canada, Australia] | <50 | 333 | 36-gene panel | 88/333 (26.4%) | 13.5% MMR genes (45) 3.3% APC (11) 1.5% biallelic MUTYH (5) 0.9% SMAD4 (3) 0.6% BMPR1A (2) 0.3% PTEN (1) | 1.8% CHEK2 (6) 0.6% TP53 (2) 0.9% CDH1 (3) 2.1% RECQL5 (7) 0.6% FLCN (2) 0.3% biallelic BLM (1) |
| Stoffel 2018 [6] [USA] | <50 | 315 | 124-gene panel 67-gene panel | 76/315 (24.1%) | 17.8% MMR genes (56) 2.5% APC (8) 2.2% biallelic MUTYH (7) 0.6% SMAD4 (2) | 0.3% BRCA1 (1) 0.3% CHEK2 (1) 0.3% TP53 (1) |
| Jiang 2020 [12] [China] | <50 | 261 | 14 genes analyzed by target sequencing | 47/261 (18%) | 15.7% MMR genes (41) 1.5% APC (4) 0.4% biallelic MUTYH (1)c 0.4% STK11 (1) | |
| Mork 2015 [8] [USA] | <35 | 205 | Variety of germline tests | 41/193 (21.2%) | 11.9 % MMR genes (23) 6.7% APC (13) 1% biallelic MUTYH (2) 1% biallelic MSH6/PMS2 (2) | 0.5% TP53 (1) |
| Total | 6587 | 837/6587 (12.7%) | 682/6587 (10.4%) | 155/6587 (2.4%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daca Alvarez, M.; Quintana, I.; Terradas, M.; Mur, P.; Balaguer, F.; Valle, L. The Inherited and Familial Component of Early-Onset Colorectal Cancer. Cells 2021, 10, 710. https://doi.org/10.3390/cells10030710
Daca Alvarez M, Quintana I, Terradas M, Mur P, Balaguer F, Valle L. The Inherited and Familial Component of Early-Onset Colorectal Cancer. Cells. 2021; 10(3):710. https://doi.org/10.3390/cells10030710
Chicago/Turabian StyleDaca Alvarez, Maria, Isabel Quintana, Mariona Terradas, Pilar Mur, Francesc Balaguer, and Laura Valle. 2021. "The Inherited and Familial Component of Early-Onset Colorectal Cancer" Cells 10, no. 3: 710. https://doi.org/10.3390/cells10030710
APA StyleDaca Alvarez, M., Quintana, I., Terradas, M., Mur, P., Balaguer, F., & Valle, L. (2021). The Inherited and Familial Component of Early-Onset Colorectal Cancer. Cells, 10(3), 710. https://doi.org/10.3390/cells10030710