
Cell Surface GRP94 as a Novel Emerging Therapeutic Target for Monoclonal Antibody Cancer Therapy



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Structure and Physiological Roles of GRP94 in Cells
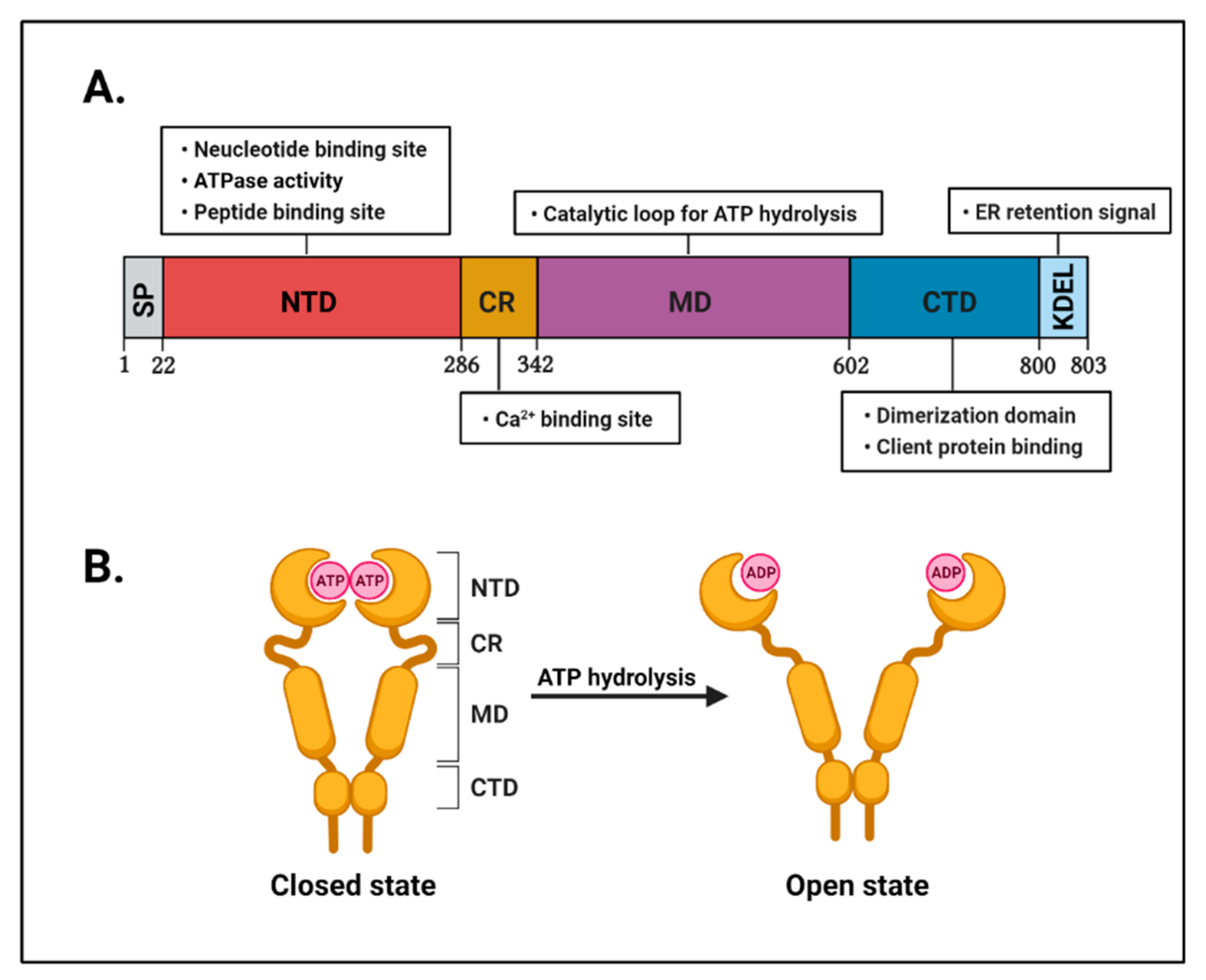
2.1. The Structure of GRP94
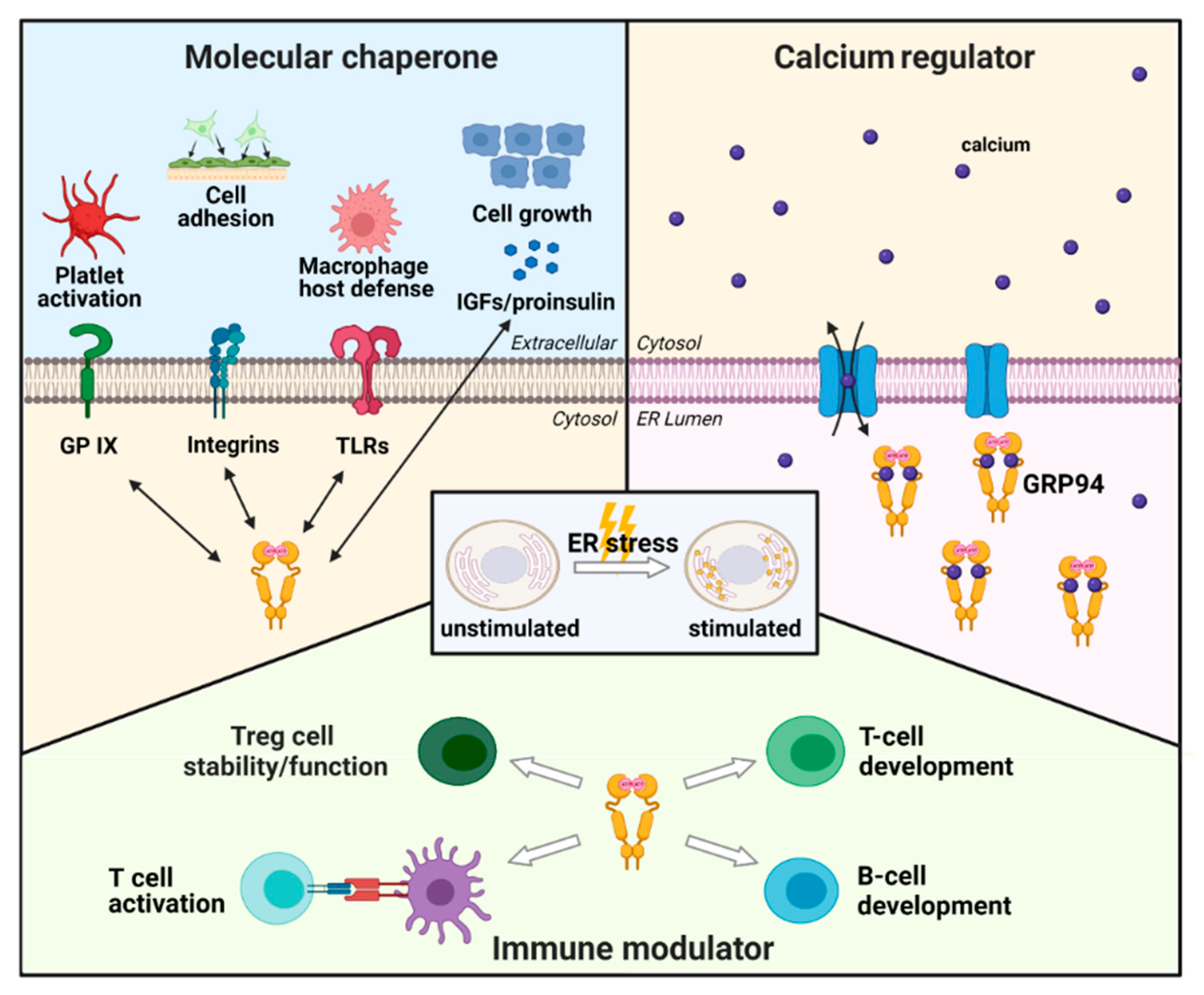
2.2. Physiological Role of GRP94 in Cells
2.2.1. GRP94 as a Molecular Chaperone
2.2.2. GRP94 as a Calcium Regulator
2.2.3. GRP94 as an Immune Modulator
3. Role and Relevance of GRP94 in Cancer
3.1. Clinical Relevance of GRP94 in Cancer
3.2. Role of GRP94 in Cancer Progression and Metastasis
3.3. Role of GRP94 in Tumor Resistance
4. The Development of GRP94-Specific Inhibitors
4.1. GRP94 Small Molecule Inhibitors for Cancer Therapy
4.1.1. Benzoquinone Ansamycin Class
4.1.2. Resorcinol Class
4.1.3. Purine Class
4.2. GRP94 Monoclonal Antibodies for Cancer Therapy
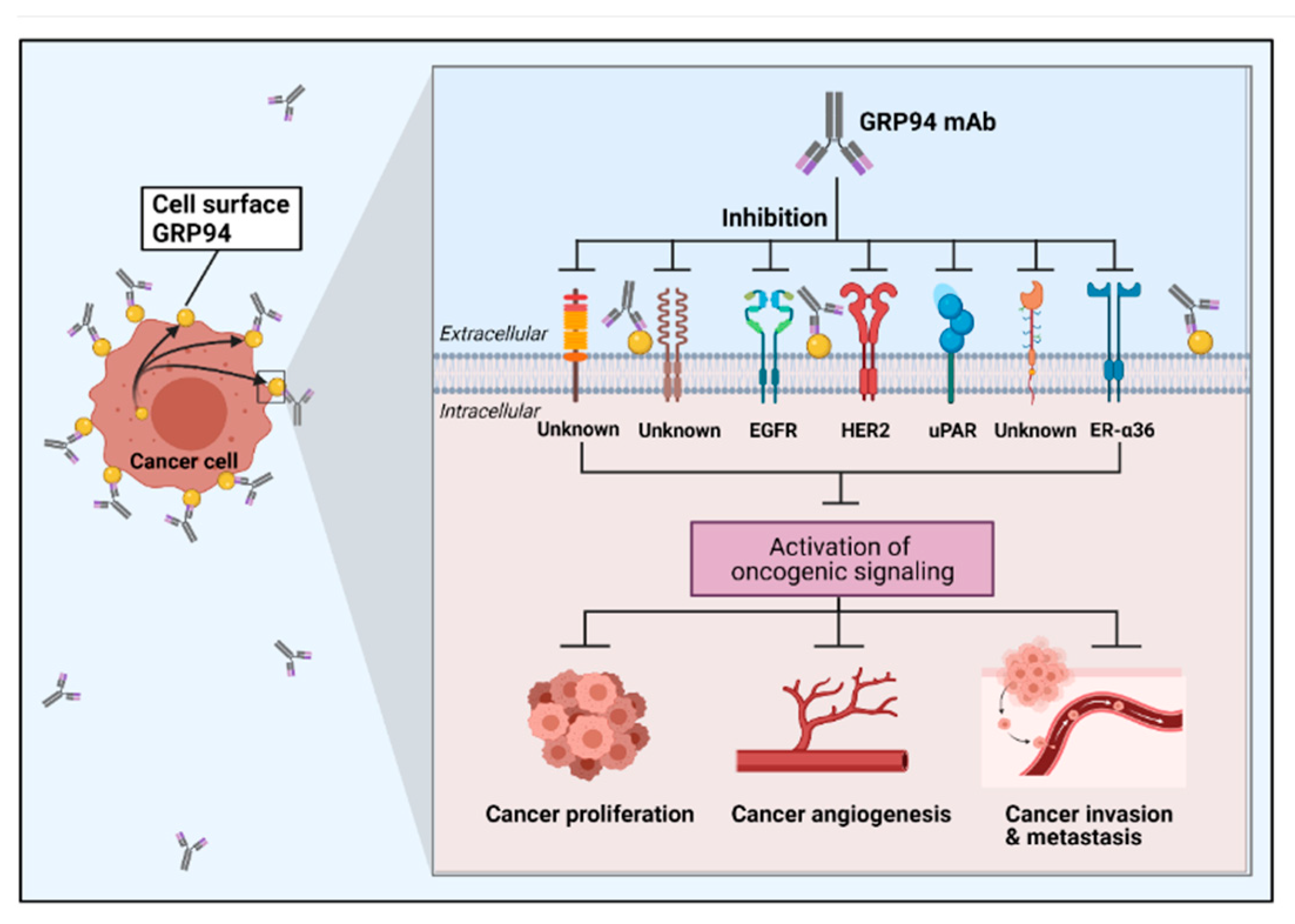
4.2.1. Cell Surface GRP94 in Cancers
4.2.2. Mouse Monoclonal Antibody
4.2.3. Human Monoclonal Antibody
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 17-AAG | 17-Allylamino-17-demethoxygeldanamycin |
| 17-DMAG | 17-Desmethoxy-17-N,N-dimethylaminoethyl amino geldanamycin |
| AKT | Protein kinase B |
| APC | Antigen-presenting cell |
| ATP | Adenosine triphosphate |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1 |
| BSDL | Bile-salt-dependent lipase |
| CHOP | C/EBP homologous protein |
| CR | Charged linker region |
| CRC | Colorectal cancer |
| CTD | C-terminal domain |
| EGFR | Epidermal growth factor receptor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ER-α36 | Estrogen receptor-α36 |
| FAK | Focal adhesion kinase |
| GARP | Glycoprotein A repetitions predominant |
| GDA | Geldanamycin |
| GRP94 | Glucose-regulated protein 94 |
| GP | Glycoprotein |
| HCC | Hepatocellular carcinoma |
| HER2 | Human epidermal growth factor receptor 2 |
| HSP90 | Heat shock protein 90 |
| HSP90B1 | Heat shock protein 90 kDa beta member 1 |
| IGFs | Insulin-like growth factors |
| IgG | Immunoglobulin G |
| LRP6 | Lipoprotein receptor-related protein 6 |
| KRAS | Kirsten rat sarcoma 2 viral oncogene homolog |
| mAbs | Monoclonal antibodies |
| MD | Middle domain |
| NECA | 5′-N-ethylcarboxamidoadenosine |
| NTD | N-terminal domain |
| PD-1 | Programmed cell death protein-1 |
| PD-L1 | Programmed death-ligand 1 |
| PI3KCA | Phosphoinositide-3-kinase catalytic subunit alpha |
| PTEN | Phosphatase and tensin homolog |
| RDA | Radamide |
| RDC | Radicicol |
| scFv | Single-chain variable fragment |
| shRNA | Short hairpin RNA |
| TLRs | Toll-like receptors |
| TRA1 | Tumor rejection antigen 1 |
| uPAR | Urokinase-type plasminogen activator receptor |
| USFDA | The United States Food and Drug Administration |
| VEGF-A | Vascular endothelial growth factor-A |
References
- Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Henneberg, M. Cancer incidence increasing globally: The role of relaxed natural selection. Evol. Appl. 2017, 11, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Spring, B.; King, A.C.; Pagoto, S.L.; Van Horn, L.; Fisher, J.D. Fostering multiple healthy lifestyle behaviors for primary prevention of cancer. Am. Psychol. 2015, 70, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A. High throughput screening in drug discovery. Clin. Transl. Oncol. 2006, 8, 482–490. [Google Scholar] [CrossRef]
- Albanese, S.K.; Chodera, J.D.; Volkamer, A.; Keng, S.; Abel, R.; Wang, L. Is Structure-Based Drug Design Ready for Selectivity Optimization? J. Chem. Inf. Modeling 2020, 60, 6211–6227. [Google Scholar] [CrossRef]
- Mazanetz, M.P.; Goode, C.H.F.; Chudyk, E.I. Ligand- and Structure-Based Drug Design and Optimization using KNIME. Curr. Med. Chem. 2020, 27, 6458–6479. [Google Scholar] [CrossRef]
- Downey, G.P.; Waddell, T.K.; Fukushima, T.; Sue, A.Q.A. Current techniques in cell and molecular biology. J. Crit. Care 1995, 10, 136–149. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimers Dement. 2017, 3, 651–657. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Fact Sheet: FDA at a Glance. Available online: https://www.fda.gov/about-fda/fda-basics/fact-sheet-fda-glance (accessed on 28 November 2020).
- Chen, H.H.W.; Kuo, M.T. Improving radiotherapy in cancer treatment: Promises and challenges. Oncotarget 2017, 8, 62742–62758. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Carelle, N.; Piotto, E.; Bellanger, A.; Germanaud, J.; Thuillier, A.; Khayat, D. Changing patient perceptions of the side effects of cancer chemotherapy. Cancer 2002, 95, 155–163. [Google Scholar] [CrossRef]
- Coates, A.; Abraham, S.; Kaye, S.B.; Sowerbutts, T.; Frewin, C.; Fox, R.M.; Tattersall, M.H. On the receiving end--patient perception of the side-effects of cancer chemotherapy. Eur. J. Cancer Clin. Oncol. 1983, 19, 203–208. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Mohammed, R.; Milne, A.; Kayani, K.; Ojha, U. How the discovery of rituximab impacted the treatment of B-cell non-Hodgkin’s lymphomas. J. Blood Med. 2019, 10, 71–84. [Google Scholar] [CrossRef]
- Ranieri, G.; Patruno, R.; Ruggieri, E.; Montemurro, S.; Valerio, P.; Ribatti, D. Vascular endothelial growth factor (VEGF) as a target of bevacizumab in cancer: From the biology to the clinic. Curr. Med. Chem. 2006, 13, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J. The EGFR as a target for anticancer therapy—Focus on cetuximab. Eur. J. Cancer 2001, 37 (Suppl. 4), S16–S22. [Google Scholar] [CrossRef]
- Harbeck, N.; Beckmann, M.W.; Rody, A.; Schneeweiss, A.; Müller, V.; Fehm, T.; Marschner, N.; Gluz, O.; Schrader, I.; Heinrich, G.; et al. HER2 Dimerization Inhibitor Pertuzumab—Mode of Action and Clinical Data in Breast Cancer. Breast Care 2013, 8, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Lemery, S.J.; Zhang, J.; Rothmann, M.D.; Yang, J.; Earp, J.; Zhao, H.; McDougal, A.; Pilaro, A.; Chiang, R.; Gootenberg, J.E.; et al. U.S. Food and Drug Administration approval: Ofatumumab for the treatment of patients with chronic lymphocytic leukemia refractory to fludarabine and alemtuzumab. Clin. Cancer Res. 2010, 16, 4331–4338. [Google Scholar] [CrossRef]
- Morschhauser, F.; Marlton, P.; Vitolo, U.; Lindén, O.; Seymour, J.F.; Crump, M.; Coiffier, B.; Foà, R.; Wassner, E.; Burger, H.U.; et al. Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann. Oncol. 2010, 21, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.I.; Molina, A.; Witzig, T.; Emmanouilides, C.; Raubtischek, A.; Darif, M.; Schilder, R.J.; Wiseman, G.; White, C.A. Durable responses after ibritumomab tiuxetan radioimmunotherapy for CD20+ B-cell lymphoma: Long-term follow-up of a phase 1/2 study. Blood 2004, 103, 4429–4431. [Google Scholar] [CrossRef]
- Wiseman, G.A.; Gordon, L.I.; Multani, P.S.; Witzig, T.E.; Spies, S.; Bartlett, N.L.; Schilder, R.J.; Murray, J.L.; Saleh, M.; Allen, R.S.; et al. Ibritumomab tiuxetan radioimmunotherapy for patients with relapsed or refractory non-Hodgkin lymphoma and mild thrombocytopenia: A phase II multicenter trial. Blood 2002, 99, 4336–4342. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Damico, L.; Shams, N.; Lowman, H.; Kim, R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006, 26, 859–870. [Google Scholar] [CrossRef]
- Yannuzzi, N.A.; Freund, K.B. Brolucizumab: Evidence to date in the treatment of neovascular age-related macular degeneration. Clin. Ophthalmol. 2019, 13, 1323–1329. [Google Scholar] [CrossRef]
- Messersmith, W.A.; Hidalgo, M. Panitumumab, a monoclonal anti epidermal growth factor receptor antibody in colorectal cancer: Another one or the one? Clin. Cancer Res. 2007, 13, 4664–4666. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.; Haidar, J.N.; Eastman, S.W.; Vieth, M.; Topper, M.; Iacolina, M.D.; Walker, J.M.; Forest, A.; Shen, Y.; Novosiadly, R.D.; et al. Molecular Basis for Necitumumab Inhibition of EGFR Variants Associated with Acquired Cetuximab Resistance. Mol. Cancer Ther. 2018, 17, 521. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef]
- Shah, N.J.; Kelly, W.J.; Liu, S.V.; Choquette, K.; Spira, A. Product review on the Anti-PD-L1 antibody atezolizumab. Hum. Vaccin Immunother. 2018, 14, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.M.; Gulley, J.L. Product review: Avelumab, an anti-PD-L1 antibody. Hum. Vaccin Immunother. 2019, 15, 891–908. [Google Scholar] [CrossRef]
- Faiena, I.; Cummings, A.L.; Crosetti, A.M.; Pantuck, A.J.; Chamie, K.; Drakaki, A. Durvalumab: An investigational anti-PD-L1 monoclonal antibody for the treatment of urothelial carcinoma. Drug Des. Dev. Ther. 2018, 12, 209–215. [Google Scholar] [CrossRef]
- Grosso, J.; Horak, C.E.; Inzunza, D.; Cardona, D.M.; Simon, J.S.; Gupta, A.K.; Sankar, V.; Park, J.-S.; Kollia, G.; Taube, J.M.; et al. Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients (pts) with advanced solid tumors treated with nivolumab (anti-PD-1; BMS-936558; ONO-4538). J. Clin. Oncol. 2013, 31, 3016. [Google Scholar] [CrossRef]
- Hui, R.; Garon, E.B.; Goldman, J.W.; Leighl, N.B.; Hellmann, M.D.; Patnaik, A.; Gandhi, L.; Eder, J.P.; Ahn, M.J.; Horn, L.; et al. Pembrolizumab as first-line therapy for patients with PD-L1-positive advanced non-small cell lung cancer: A phase 1 trial. Ann. Oncol. 2017, 28, 874–881. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 2012, 1823, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.G.; Old, L.J.; Srivastava, P.K. Human homologue of murine tumor rejection antigen gp96: 5’-regulatory and coding regions and relationship to stress-induced proteins. Proc. Natl. Acad. Sci. USA 1990, 87, 5658–5662. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Srivastava, P.K. Tumor rejection antigen gp96/grp94 is an ATPase: Implications for protein folding and antigen presentation. EMBO J. 1993, 12, 3143–3151. [Google Scholar] [CrossRef]
- Chen, B.; Piel, W.H.; Gui, L.; Bruford, E.; Monteiro, A. The HSP90 family of genes in the human genome: Insights into their divergence and evolution. Genomics 2005, 86, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Bell, J.; Ting, J. Biochemical characterization of the 94- and 78-kilodalton glucose-regulated proteins in hamster fibroblasts. J. Biol. Chem. 1984, 259, 4616–4621. [Google Scholar] [CrossRef]
- Van, P.N.; Peter, F.; Söling, H.D. Four intracisternal calcium-binding glycoproteins from rat liver microsomes with high affinity for calcium. No indication for calsequestrin-like proteins in inositol 1,4,5-trisphosphate-sensitive calcium sequestering rat liver vesicles. J. Biol. Chem. 1989, 264, 17494–17501. [Google Scholar] [CrossRef]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Dollins, D.E.; Warren, J.J.; Immormino, R.M.; Gewirth, D.T. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell 2007, 28, 41–56. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Böttcher, U.M.; Southworth, D.R.; Agard, D.A. Grp94, the endoplasmic reticulum Hsp90, has a similar solution conformation to cytosolic Hsp90 in the absence of nucleotide. Protein Sci. 2009, 18, 1815–1827. [Google Scholar] [CrossRef]
- Frey, S.; Leskovar, A.; Reinstein, J.; Buchner, J. The ATPase cycle of the endoplasmic chaperone Grp94. J. Biol. Chem. 2007, 282, 35612–35620. [Google Scholar] [CrossRef]
- Schulte, T.W.; Akinaga, S.; Soga, S.; Sullivan, W.; Stensgard, B.; Toft, D.; Neckers, L.M. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998, 3, 100–108. [Google Scholar] [CrossRef]
- Schulte, T.W.; Akinaga, S.; Murakata, T.; Agatsuma, T.; Sugimoto, S.; Nakano, H.; Lee, Y.S.; Simen, B.B.; Argon, Y.; Felts, S.; et al. Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol. Endocrinol. 1999, 13, 1435–1448. [Google Scholar] [CrossRef]
- Vogen, S.; Gidalevitz, T.; Biswas, C.; Simen, B.B.; Stein, E.; Gulmen, F.; Argon, Y. Radicicol-sensitive peptide binding to the N-terminal portion of GRP94. J. Biol. Chem. 2002, 277, 40742–40750. [Google Scholar] [CrossRef]
- Biswas, C.; Ostrovsky, O.; Makarewich, C.A.; Wanderling, S.; Gidalevitz, T.; Argon, Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem. J. 2007, 405, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Yamada, S.; Ono, T.; Mizuno, A.; Nemoto, T.K. A hydrophobic segment within the C-terminal domain is essential for both client-binding and dimer formation of the HSP90-family molecular chaperone. Eur. J. Biochem. 2003, 270, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- Joslin, G.; Hafeez, W.; Perlmutter, D.H. Expression of stress proteins in human mononuclear phagocytes. J. Immunol. 1991, 147, 1614. [Google Scholar]
- Yang, Y.; Li, Z. Roles of heat shock protein gp96 in the ER quality control: Redundant or unique function? Mol. Cells 2005, 20, 173–182. [Google Scholar] [PubMed]
- Lee, A.S. Mammalian stress response: Induction of the glucose-regulated protein family. Curr. Opin. Cell Biol. 1992, 4, 267–273. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed]
- Melnick, J.; Dul, J.L.; Argon, Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature 1994, 370, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Randow, F.; Seed, B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat. Cell Biol. 2001, 3, 891–896. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, B.; Dai, J.; Srivastava, P.K.; Zammit, D.J.; Lefrançois, L.; Li, Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 2007, 26, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Staron, M.; Wu, S.; Hong, F.; Stojanovic, A.; Du, X.; Bona, R.; Liu, B.; Li, Z. Heat-shock protein gp96/grp94 is an essential chaperone for the platelet glycoprotein Ib-IX-V complex. Blood 2011, 117, 7136–7144. [Google Scholar] [CrossRef]
- Ostrovsky, O.; Ahmed, N.T.; Argon, Y. The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol. Biol. Cell 2009, 20, 1855–1864. [Google Scholar] [CrossRef]
- Barton, E.R.; Park, S.; James, J.K.; Makarewich, C.A.; Philippou, A.; Eletto, D.; Lei, H.; Brisson, B.; Ostrovsky, O.; Li, Z.; et al. Deletion of muscle GRP94 impairs both muscle and body growth by inhibiting local IGF production. FASEB J. 2012, 26, 3691–3702. [Google Scholar] [CrossRef]
- Ghiasi, S.M.; Dahlby, T.; Hede Andersen, C.; Haataja, L.; Petersen, S.; Omar-Hmeadi, M.; Yang, M.; Pihl, C.; Bresson, S.E.; Khilji, M.S.; et al. Endoplasmic Reticulum Chaperone Glucose-Regulated Protein 94 Is Essential for Proinsulin Handling. Diabetes 2019, 68, 747–760. [Google Scholar] [CrossRef]
- Wu, S.; Hong, F.; Gewirth, D.; Guo, B.; Liu, B.; Li, Z. The molecular chaperone gp96/GRP94 interacts with Toll-like receptors and integrins via its C-terminal hydrophobic domain. J. Biol. Chem. 2012, 287, 6735–6742. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Tseng, C.C.; Pfaffenbach, K.; Kanel, G.; Luo, B.; Stiles, B.L.; Lee, A.S. Liver-specific knockout of GRP94 in mice disrupts cell adhesion, activates liver progenitor cells, and accelerates liver tumorigenesis. Hepatology 2014, 59, 947–957. [Google Scholar] [CrossRef]
- Muresan, Z.; Arvan, P. Thyroglobulin Transport along the Secretory Pathway: Investigation of the role of molecular chaperone, grp94, in protein export from the endoplasmic reticulum. J. Biol. Chem. 1997, 272, 26095–26102. [Google Scholar] [CrossRef]
- Bruneau, N.; Lombardo, D.; Bendayan, M. Participation of GRP94-related protein in secretion of pancreatic bile salt-dependent lipase and in its internalization by the intestinal epithelium. J. Cell Sci. 1998, 111 Pt 17, 2665–2679. [Google Scholar]
- Zhang, Y.; Wu, B.X.; Metelli, A.; Thaxton, J.E.; Hong, F.; Rachidi, S.; Ansa-Addo, E.; Sun, S.; Vasu, C.; Yang, Y.; et al. GP96 is a GARP chaperone and controls regulatory T cell functions. J. Clin. Investig. 2015, 125, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Staron, M.; Hong, F.; Wu, B.X.; Sun, S.; Morales, C.; Crosson, C.E.; Tomlinson, S.; Kim, I.; Wu, D.; et al. Essential roles of grp94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 6877–6882. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca(2+) Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Izquierdo, J.H.; Bonilla-Abadía, F.; Cañas, C.A.; Tobón, G.J. Calcium, channels, intracellular signaling and autoimmunity. Reumatol. Clin. 2014, 10, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Drummond, I.A.; Lee, A.S.; Resendez, E., Jr.; Steinhardt, R.A. Depletion of intracellular calcium stores by calcium ionophore A23187 induces the genes for glucose-regulated proteins in hamster fibroblasts. J. Biol. Chem. 1987, 262, 12801–12805. [Google Scholar] [CrossRef]
- Macer, D.R.; Koch, G.L. Identification of a set of calcium-binding proteins in reticuloplasm, the luminal content of the endoplasmic reticulum. J. Cell Sci. 1988, 91 Pt 1, 61–70. [Google Scholar]
- Vitadello, M.; Penzo, D.; Petronilli, V.; Michieli, G.; Gomirato, S.; Menabò, R.; Di Lisa, F.; Gorza, L. Overexpression of the stress protein Grp94 reduces cardiomyocyte necrosis due to calcium overload and simulated ischemia. FASEB J. 2003, 17, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.; Katayama, T.; Aleshin, A.N.; Manabe, T.; Tohyama, M. GRP94 reduces cell death in SH-SY5Y cells perturbated calcium homeostasis. Apoptosis 2004, 9, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Staron, M.; Yang, Y.; Liu, B.; Li, J.; Shen, Y.; Zúñiga-Pflücker, J.C.; Aguila, H.L.; Goldschneider, I.; Li, Z. gp96, an endoplasmic reticulum master chaperone for integrins and Toll-like receptors, selectively regulates early T and B lymphopoiesis. Blood 2010, 115, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Dai, J.; Stoilova, D.; Li, Z. Cell surface targeting of heat shock protein gp96 induces dendritic cell maturation and antitumor immunity. J. Immunol. 2001, 167, 6731–6735. [Google Scholar] [CrossRef] [PubMed]
- Suto, R.; Srivastava, P.K. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 1995, 269, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- Reed, R.C.; Nicchitta, C.V. Chaperone-mediated cross-priming: A hitchhiker’s guide to vesicle transport (review). Int. J. Mol. Med. 2000, 6, 259–264. [Google Scholar] [CrossRef]
- Tramentozzi, E.; Zamarchi, R.; Pagetta, A.; Brunati, A.M.; Rossi, E.; Tibaldi, E.; Finotti, P. Effects of glucose-regulated protein94 (Grp94) on Ig secretion from human blood mononuclear cells. Cell Stress Chaperones 2011, 16, 329–338. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, B.; Alessandra, M.; Hong, F.; Ansa-Addo, E.; Sun, S.; Liu, B.; Li, Z. Molecular chaperone gp96/grp94 is critical for immunosuppressive functions of regulatory T cells (IRM15P.458). J. Immunol. 2015, 194 (Suppl. 1), 199.6. [Google Scholar]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Chipurupalli, S.; Kannan, E.; Tergaonkar, V.; D’Andrea, R.; Robinson, N. Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer. Int. J. Mol. Sci. 2019, 20, 749. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Rachidi, S.; Sun, S.; Wu, B.X.; Jones, E.; Drake, R.R.; Ogretmen, B.; Cowart, L.A.; Clarke, C.J.; Hannun, Y.A.; Chiosis, G.; et al. Endoplasmic reticulum heat shock protein gp96 maintains liver homeostasis and promotes hepatocellular carcinogenesis. J. Hepatol. 2015, 62, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, R.; Zuo, S.; Luo, R.; Fang, W.; Xie, Y. GRP94 overexpression as an indicator of unfavorable outcomes in breast cancer patients. Int. J. Clin. Exp. Pathol. 2018, 11, 3061–3067. [Google Scholar] [PubMed]
- Chen, X.; Ding, Y.; Liu, C.G.; Mikhail, S.; Yang, C.S. Overexpression of glucose-regulated protein 94 (Grp94) in esophageal adenocarcinomas of a rat surgical model and humans. Carcinogenesis 2002, 23, 123–130. [Google Scholar] [CrossRef]
- Hu, T.; Xie, N.; Qin, C.; Wang, J.; You, Y. Glucose-regulated protein 94 is a novel glioma biomarker and promotes the aggressiveness of glioma via Wnt/β-catenin signaling pathway. Tumor Biol. 2015, 36, 9357–9364. [Google Scholar] [CrossRef]
- Dejeans, N.; Glorieux, C.; Guenin, S.; Beck, R.; Sid, B.; Rousseau, R.; Bisig, B.; Delvenne, P.; Buc Calderon, P.; Verrax, J. Overexpression of GRP94 in breast cancer cells resistant to oxidative stress promotes high levels of cancer cell proliferation and migration: Implications for tumor recurrence. Free Radic. Biol. Med. 2012, 52, 993–1002. [Google Scholar] [CrossRef]
- Duan, X.F.; Xin, Y.W. Overexpression of molecule GRP94 favors tumor progression in lung adenocarcinoma by interaction with regulatory T cells. Thorac. Cancer 2020, 11, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, H.W.; Lee, E.H.; Park, M.I.; Lee, J.S.; Kim, M.S.; Kim, K.; Roh, M.S.; Pak, M.G.; Oh, J.E.; et al. Differential expression of HSP90 isoforms and their correlations with clinicopathologic factors in patients with colorectal cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 978–986. [Google Scholar] [PubMed]
- Nomura, H.; Uzawa, K.; Yamano, Y.; Fushimi, K.; Ishigami, T.; Kato, Y.; Saito, K.; Nakashima, D.; Higo, M.; Kouzu, Y.; et al. Network-based analysis of calcium-binding protein genes identifies Grp94 as a target in human oral carcinogenesis. Br. J. Cancer 2007, 97, 792–801. [Google Scholar] [CrossRef]
- Fu, Z.; Zhen, H.; Zou, F.; Wang, X.; Chen, Y.; Liu, L. Involvement of the Akt signaling pathway in ER-α36/GRP94-mediated signaling in gastric cancer. Oncol. Lett. 2014, 8, 2077–2080. [Google Scholar] [CrossRef]
- Zheng, H.C.; Takahashi, H.; Li, X.H.; Hara, T.; Masuda, S.; Guan, Y.F.; Takano, Y. Overexpression of GRP78 and GRP94 are markers for aggressive behavior and poor prognosis in gastric carcinomas. Hum. Pathol. 2008, 39, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Brzozowa-Zasada, M.; Kurek, J.; Piecuch, A.; Wyrobiec, G. The clinical and prognostic evaluation of GRP94 immunoexpression in Caucasian patients with colorectal adenocarcinoma. Prz. Gastroenterol. 2019, 14, 140–147. [Google Scholar] [CrossRef]
- Huang, C.Y.; Lee, C.H.; Tu, C.C.; Wu, C.H.; Huang, M.T.; Wei, P.L.; Chang, Y.J. Glucose-regulated protein 94 mediates progression and metastasis of esophageal squamous cell carcinoma via mitochondrial function and the NF-kB/COX-2/VEGF axis. Oncotarget 2018, 9, 9425–9441. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Zhang, X.; Zhang, L.; Wang, S.; Wu, D.; Yang, W. Decreased functional expression of Grp78 and Grp94 inhibits proliferation and attenuates apoptosis in a human gastric cancer cell line in vitro. Oncol. Lett. 2015, 9, 1181–1186. [Google Scholar] [CrossRef]
- Jeoung, M.H.; Kim, T.K.; Kim, J.W.; Cho, Y.B.; Na, H.J.; Yoo, B.C.; Shim, H.; Song, D.K.; Heo, K.; Lee, S. Antibody-Based Targeting of Cell Surface GRP94 Specifically Inhibits Cetuximab-Resistant Colorectal Cancer Growth. Biomolecules 2019, 9, 681. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Batzorig, U.; Cheng, W.L.; Huang, M.T.; Chen, W.; Wei, P.L.; Chang, Y.J. Glucose-regulated protein 94 mediates cancer progression via AKT and eNOS in hepatocellular carcinoma. Tumour Biol. 2016, 37, 4295–4304. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, B.; Sun, S.; Li, Z. Essential roles of heat shock protein gp96 (Hsp90b1, grp94) in melanoma growth and melanosome development (46.1). J. Immunol. 2012, 188 (Suppl. 1), 46.1. [Google Scholar]
- Buc Calderon, P.; Sennesael, A.L.; Glorieux, C. Glucose-regulated protein of 94 kDa contributes to the development of an aggressive phenotype in breast cancer cells. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 105, 115–120. [Google Scholar] [CrossRef]
- Wei, P.L.; Huang, C.Y.; Tai, C.J.; Batzorig, U.; Cheng, W.L.; Hunag, M.T.; Chang, Y.J. Glucose-regulated protein 94 mediates metastasis by CCT8 and the JNK pathway in hepatocellular carcinoma. Tumor Biol. 2016, 37, 8219–8227. [Google Scholar] [CrossRef]
- Hong, F.; Liu, B.; Chiosis, G.; Gewirth, D.T.; Li, Z. α7 helix region of αI domain is crucial for integrin binding to endoplasmic reticulum chaperone gp96: A potential therapeutic target for cancer metastasis. J. Biol. Chem. 2013, 288, 18243–18248. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Qiu, F.R.; Liu, G.Z.; Chen, R.F. Correlation between clinicopathology and expression of heat shock protein 70 and glucose-regulated protein 94 in human colonic adenocarcinoma. World J. Gastroenterol. 2005, 11, 1056–1059. [Google Scholar] [CrossRef]
- Sanz-Pamplona, R.; Aragüés, R.; Driouch, K.; Martín, B.; Oliva, B.; Gil, M.; Boluda, S.; Fernández, P.L.; Martínez, A.; Moreno, V.; et al. Expression of endoplasmic reticulum stress proteins is a candidate marker of brain metastasis in both ErbB-2+ and ErbB-2- primary breast tumors. Am. J. Pathol. 2011, 179, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.; Brown, J. The Evolution and Ecology of Resistance in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2018, 8, a033415. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Chang, J.T.; Wang, H.M.; Chan, S.H.; Chiu, C.C.; Lin, C.Y.; Fan, K.H.; Liao, C.T.; Chen, I.H.; Liu, T.Z.; et al. Proteomics of the radioresistant phenotype in head-and-neck cancer: Gp96 as a novel prediction marker and sensitizing target for radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 246–256. [Google Scholar] [CrossRef]
- Kubota, H.; Suzuki, T.; Lu, J.; Takahashi, S.; Sugita, K.; Sekiya, S.; Suzuki, N. Increased expression of GRP94 protein is associated with decreased sensitivity to X-rays in cervical cancer cell lines. Int. J. Radiat. Biol. 2005, 81, 701–709. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Ferrone, C.R.; Schwab, J.H.; Ferrone, S. Intracellular antigens as targets for antibody based immunotherapy of malignant diseases. Mol. Oncol. 2015, 9, 1982–1993. [Google Scholar] [CrossRef]
- McLaughlin, M.; Vandenbroeck, K. The endoplasmic reticulum protein folding factory and its chaperones: New targets for drug discovery? Br. J. Pharm. 2011, 162, 328–345. [Google Scholar] [CrossRef]
- Huck, J.D.; Que, N.L.; Hong, F.; Li, Z.; Gewirth, D.T. Structural and Functional Analysis of GRP94 in the Closed State Reveals an Essential Role for the Pre-N Domain and a Potential Client-Binding Site. Cell Rep. 2017, 20, 2800–2809. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Rüdiger, S.G.D. Hsp90 interaction with clients. Trends Biochem. Sci. 2015, 40, 117–125. [Google Scholar] [CrossRef]
- Khandelwal, A.; Crowley, V.M.; Blagg, B.S.J. Resorcinol-Based Grp94-Selective Inhibitors. ACS Med. Chem. Lett. 2017, 8, 1013–1018. [Google Scholar] [CrossRef]
- Wang, L.; Xu, X.; Jiang, Z.; You, Q. Modulation of protein fate decision by small molecules: Targeting molecular chaperone machinery. Acta Pharm. Sin. B 2020, 10, 1904–1925. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Miyata, Y. Hsp90 inhibitor geldanamycin and its derivatives as novel cancer chemotherapeutic agents. Curr. Pharm. Des. 2005, 11, 1131–1138. [Google Scholar] [CrossRef]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Ishii, H.; Hyodo, F.; Samuni, U.; Krishna, M.C.; Goldstein, S.; Mitchell, J.B. Reactive oxygen species mediate hepatotoxicity induced by the Hsp90 inhibitor geldanamycin and its analogs. Free Radic. Biol. Med. 2010, 48, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Price, J.T.; Quinn, J.M.W.; Sims, N.A.; Vieusseux, J.; Waldeck, K.; Docherty, S.E.; Myers, D.; Nakamura, A.; Waltham, M.C.; Gillespie, M.T.; et al. The Heat Shock Protein 90 Inhibitor, 17-Allylamino-17-demethoxygeldanamycin, Enhances Osteoclast Formation and Potentiates Bone Metastasis of a Human Breast Cancer Cell Line. Cancer Res. 2005, 65, 4929. [Google Scholar] [CrossRef] [PubMed]
- Talaei, S.; Mellatyar, H.; Asadi, A.; Akbarzadeh, A.; Sheervalilou, R.; Zarghami, N. Spotlight on 17-AAG as an Hsp90 inhibitor for molecular targeted cancer treatment. Chem. Biol. Drug Des. 2019, 93, 760–786. [Google Scholar] [CrossRef]
- Schulte, T.W.; Neckers, L.M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 1998, 42, 273–279. [Google Scholar] [CrossRef]
- Schnur, R.C.; Corman, M.L.; Gallaschun, R.J.; Cooper, B.A.; Dee, M.F.; Doty, J.L.; Muzzi, M.L.; DiOrio, C.I.; Barbacci, E.G.; Miller, P.E.; et al. erbB-2 oncogene inhibition by geldanamycin derivatives: Synthesis, mechanism of action, and structure-activity relationships. J. Med. Chem. 1995, 38, 3813–3820. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Gore, M.; Chao, D.; Banerji, U.; Larkin, J.; Sarker, S.; Owen, K.; Asad, Y.; Raynaud, F.; Walton, M.; et al. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Investig. New Drugs 2012, 30, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toft, D.; Reid, J.; Ames, M.; Stensgard, B.; Safgren, S.; Adjei, A.A.; Sloan, J.; Atherton, P.; Vasile, V.; et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.; Sausville, E.A.; Camalier, R.F.; Fiebig, H.H.; Burger, A.M. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: Effects on Hsp90 and client proteins in melanoma models. Cancer Chemother. Pharmacol. 2005, 56, 126–137. [Google Scholar] [CrossRef]
- Pacey, S.; Wilson, R.H.; Walton, M.; Eatock, M.M.; Hardcastle, A.; Zetterlund, A.; Arkenau, H.-T.; Moreno-Farre, J.; Banerji, U.; Roels, B.; et al. A Phase I Study of the Heat Shock Protein 90 Inhibitor Alvespimycin (17-DMAG) Given Intravenously to Patients with Advanced Solid Tumors. Clin. Cancer Res. 2011, 17, 1561. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, P.; Delmotte-Plaque, J. A new antifungal substance of fungal origin. Nature 1953, 171, 344. [Google Scholar] [CrossRef]
- Amolins, M.W.; Blagg, B.S.J. Natural product inhibitors of Hsp90: Potential leads for drug discovery. Mini-Rev. Med. Chem. 2009, 9, 140–152. [Google Scholar] [CrossRef]
- Soga, S.; Shiotsu, Y.; Akinaga, S.; Sharma, S.V. Development of radicicol analogues. Curr. Cancer Drug Targets 2003, 3, 359–369. [Google Scholar] [CrossRef]
- Crowley, V.M.; Khandelwal, A.; Mishra, S.; Stothert, A.R.; Huard, D.J.E.; Zhao, J.; Muth, A.; Duerfeldt, A.S.; Kizziah, J.L.; Lieberman, R.L.; et al. Development of Glucose Regulated Protein 94-Selective Inhibitors Based on the BnIm and Radamide Scaffold. J. Med. Chem. 2016, 59, 3471–3488. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef]
- Sharp, S.Y.; Roe, S.M.; Kazlauskas, E.; Čikotienė, I.; Workman, P.; Matulis, D.; Prodromou, C. Co-Crystalization and In Vitro Biological Characterization of 5-Aryl-4-(5-Substituted-2-4-Dihydroxyphenyl)-1,2,3-Thiadiazole Hsp90 Inhibitors. PLoS ONE 2012, 7, e44642. [Google Scholar] [CrossRef] [PubMed]
- Renouf, D.J.; Hedley, D.; Krzyzanowska, M.K.; Schmuck, M.; Wang, L.; Moore, M.J. A phase II study of the HSP90 inhibitor AUY922 in chemotherapy refractory advanced pan-creatic cancer. Cancer Chemother Pharmacol. 2016, 78, 541–545. [Google Scholar] [CrossRef]
- Bendell, J.C.; Bauer, T.M.; Lamar, R.; Joseph, M.; Penley, W.; Thompson, D.S.; Spigel, D.R.; Owera, R.; Lane, C.M.; Earwood, C.; et al. A Phase 2 Study of the Hsp90 Inhibitor AUY922 as Treatment for Patients with Refractory Gastrointestinal Stromal Tumors. Cancer Investig. 2016, 34, 265–270. [Google Scholar] [CrossRef]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, Y.; Chu, H.; Bing, D.; Wang, S.; Zhou, L.; Chen, J.; Chen, Q.; Pan, C.; Sun, Y.; et al. 17-DMAG induces Hsp70 and protects the auditory hair cells from kanamycin ototoxicity in vitro. Neurosci. Lett. 2015, 588, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, M.J.; Brough, P.A.; Massey, A.; Jensen, M.R.; Schoepfer, J. Targeting Hsp90 for the treatment of cancer. Curr. Opin. Drug Discov. Dev. 2006, 9, 483–495. [Google Scholar]
- Dakappagari, N.; Neely, L.; Tangri, S.; Lundgren, K.; Hipolito, L.; Estrellado, A.; Burrows, F.; Zhang, H. An investigation into the potential use of serum Hsp70 as a novel tumour biomarker for Hsp90 inhibitors. Biomarkers 2010, 15, 31–38. [Google Scholar] [CrossRef]
- Ghoshal, S.; Rao, I.; Earp, J.C.; Jusko, W.J.; Wetzler, M. Down-regulation of heat shock protein 70 improves arsenic trioxide and 17-DMAG effects on constitutive signal transducer and activator of transcription 3 activity. Cancer Chemother. Pharmacol. 2010, 66, 681–689. [Google Scholar] [CrossRef]
- Kühnel, A.; Schilling, D.; Combs, S.E.; Haller, B.; Schwab, M.; Multhoff, G. Radiosensitization of HSF-1 Knockdown Lung Cancer Cells by Low Concentrations of Hsp90 Inhibitor NVP-AUY922. Cells 2019, 8, 1166. [Google Scholar] [CrossRef]
- Speranza, G.; Anderson, L.; Chen, A.P.; Do, K.; Eugeni, M.; Weil, M.; Rubinstein, L.; Majerova, E.; Collins, J.; Horneffer, Y.; et al. First-in-human study of the epichaperome in-hibitor PU-H71: Clinical results and metabolic profile. Investig. New Drugs 2018, 36, 230–239. [Google Scholar]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.; Pillarsetty, N.; Koren, J.; Ger-ecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome net-work that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar]
- Usmani, S.Z.; Bona, R.D.; Chiosis, G.; Li, Z. The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J. Hematol. Oncol. 2010, 3, 40. [Google Scholar] [CrossRef]
- Patel, H.J.; Patel, H.D.; Ochiana, S.O.; Yan, P.; Sun, W.; Patel, M.R.; Shah, S.K.; Tramentozzi, E.; Brooks, J.; Bolaender, A.; et al. Structure–Activity Relationship in a Pu-rine-Scaffold Compound Series with Selectivity for the Endoplasmic Reticulum Hsp90 Pa-ralog Grp94. J. Med. Chem. 2015, 58, 3922–3943. [Google Scholar]
- Huck, J.D.; Que, N.L.S.; Immormino, R.M.; Shrestha, L.; Taldone, T.; Chiosis, G.; Gewirth, D.T. NECA derivatives exploit the paralog-specific properties of the site 3 side pocket of Grp94, the endoplasmic reticulum Hsp90. J. Biol. Chem. 2019, 294, 16010–16019. [Google Scholar]
- Assess the Safety, Tolerability Oral PU-H71 in Subjects Taking Ruxolitinib. Identifier NCT03935555. Available online: https://clinicaltrials.gov/ct2/show/NCT03935555?term=PU-H71&draw=2&rank=1 (accessed on 2 April 2021).
- PU-H71 With Nab-paclitaxel (Abraxane) in Metastatic Breast Cancer. Identifier NCT03166085. Available online: https://clinicaltrials.gov/ct2/show/NCT03166085?term=PU-H71&draw=2&rank=2 (accessed on 2 April 2021).
- The First-in-human Phase I Trial of PU-H71 in Patients with Advanced Malignan-cies. Identifier NCT01393509. Available online: https://clinicaltrials.gov/ct2/show/NCT01393509?term=PU-H71&draw=2&rank=4 (accessed on 2 April 2021).
- Liu, S.; Street, T.O. 5’-N-ethylcarboxamidoadenosine is not a paralog-specific Hsp90 inhibitor. Protein Sci. 2016, 25, 2209–2215. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Yang, Y.L.; Liao, K.H.; Lai, T.W. Adenosine receptor agonist NECA increases cerebral extravasation of fluorescein and low molecular weight dextran independent of blood-brain barrier modulation. Sci. Rep. 2016, 6, 23882. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Okuno, S.H.; Keohan, M.L.; Maki, R.G.; D’Adamo, D.R.; Akhurst, T.J.; Antonescu, C.R.; Schwartz, G.K. Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann. Oncol. 2013, 24, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.T.; Liu, M.; Zuccola, H.; Neubert, T.; Beaumont, K.; Turnbull, A.; Kallel, A.; Vought, B.; Stamos, D. Correlation between chemotype-dependent binding conformations of HSP90α/β and isoform selectivity-Implications for the structure-based design of HSP90α/β selective inhibitors for treating neurodegenerative diseases. Bioorganic Med. Chem. Lett. 2014, 24, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.; Dersh, D.; Argon, Y. GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol. 2010, 21, 479–485. [Google Scholar] [CrossRef]
- Li, X.; Sun, L.; Hou, J.; Gui, M.; Ying, J.; Zhao, H.; Lv, N.; Meng, S. Cell membrane gp96 facilitates HER2 dimerization and serves as a novel target in breast cancer. Int. J. Cancer 2015, 137, 512–524. [Google Scholar] [CrossRef]
- Hou, J.; Li, X.; Li, C.; Sun, L.; Zhao, Y.; Zhao, J.; Meng, S. Plasma membrane gp96 enhances invasion and metastatic potential of liver cancer via regulation of uPAR. Mol. Oncol. 2015, 9, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Patel, H.J.; Sharma, S.; Corben, A.; Wang, T.; Panchal, P.; Yang, C.; Sun, W.; Araujo, T.L.; Rodina, A.; et al. Molecular Stressors Engender Protein Connectivity Dysfunction through Aberrant N-Glycosylation of a Chaperone. Cell Rep. 2020, 31, 107840. [Google Scholar] [CrossRef]
- Sabbatino, F.; Favoino, E.; Wang, Y.; Wang, X.; Villani, V.; Cai, L.; Yang, L.; Ferrone, S.; Ferrone, C.R. Grp94-specific monoclonal antibody to counteract BRAF inhibitor resistance in BRAF(V600E) melanoma. J. Transl Med. 2015, 13, K12. [Google Scholar] [CrossRef]
- Melendez, K.; Wallen, E.S.; Edwards, B.S.; Mobarak, C.D.; Bear, D.G.; Moseley, P.L. Heat shock protein 70 and glycoprotein 96 are differentially expressed on the surface of malignant and nonmalignant breast cells. Cell Stress Chaperones 2006, 11, 334–342. [Google Scholar] [CrossRef]
- Hou, J.; Deng, M.; Li, X.; Liu, W.; Chu, X.; Wang, J.; Chen, F.; Meng, S. Chaperone gp96 mediates ER-α36 cell membrane expression. Oncotarget 2015, 6, 31857–31867. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, N.K.; Dwiwedi, P.; Charan, J.; Kaur, R.; Sidhu, P.; Chugh, V.K. Monoclonal Antibodies: A Review. Curr. Clin. Pharmacol. 2018, 13, 85–99. [Google Scholar] [CrossRef]
- Yang, H.Y.; Kang, K.J.; Chung, J.E.; Shim, H. Construction of a large synthetic human scFv library with six diversified CDRs and high functional diversity. Mol. Cells 2009, 27, 225–235. [Google Scholar] [CrossRef]
- Santini, D.; Vincenzi, B.; Addeo, R.; Garufi, C.; Masi, G.; Scartozzi, M.; Mancuso, A.; Frezza, A.M.; Venditti, O.; Imperatori, M.; et al. Cetuximab rechallenge in metastatic colorectal cancer patients: How to come away from acquired resistance? Ann. Oncol. 2012, 23, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.A.; Brogden, R.N. Muromonab CD3. A review of its pharmacology and therapeutic potential. Drugs 1989, 37, 871–899. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Tabasinezhad, M.; Talebkhan, Y.; Wenzel, W.; Rahimi, H.; Omidinia, E.; Mahboudi, F. Trends in therapeutic antibody affinity maturation: From in-vitro towards next-generation sequencing approaches. Immunol. Lett. 2019, 212, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.W.; Cho, Y.B.; Lee, S. Cell Surface GRP94 as a Novel Emerging Therapeutic Target for Monoclonal Antibody Cancer Therapy. Cells 2021, 10, 670. https://doi.org/10.3390/cells10030670
Kim JW, Cho YB, Lee S. Cell Surface GRP94 as a Novel Emerging Therapeutic Target for Monoclonal Antibody Cancer Therapy. Cells. 2021; 10(3):670. https://doi.org/10.3390/cells10030670
Chicago/Turabian StyleKim, Ji Woong, Yea Bin Cho, and Sukmook Lee. 2021. "Cell Surface GRP94 as a Novel Emerging Therapeutic Target for Monoclonal Antibody Cancer Therapy" Cells 10, no. 3: 670. https://doi.org/10.3390/cells10030670
APA StyleKim, J. W., Cho, Y. B., & Lee, S. (2021). Cell Surface GRP94 as a Novel Emerging Therapeutic Target for Monoclonal Antibody Cancer Therapy. Cells, 10(3), 670. https://doi.org/10.3390/cells10030670