
Nicotinamide Treatment Facilitates Mitochondrial Fission through Drp1 Activation Mediated by SIRT1-Induced Changes in Cellular Levels of cAMP and Ca2+



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemicals
2.2. Western Blot Analysis
2.3. Flow Cytometry for Determination of Cellular Mitochondrial Content
2.4. Confocal Microscopy and Immunofluorescence
2.5. Transfection with siRNA
2.6. Measurement of Cellular cAMP Level
2.7. Determination of Cytosolic Ca2+ Level
2.8. Statistical Analysis
3. Results
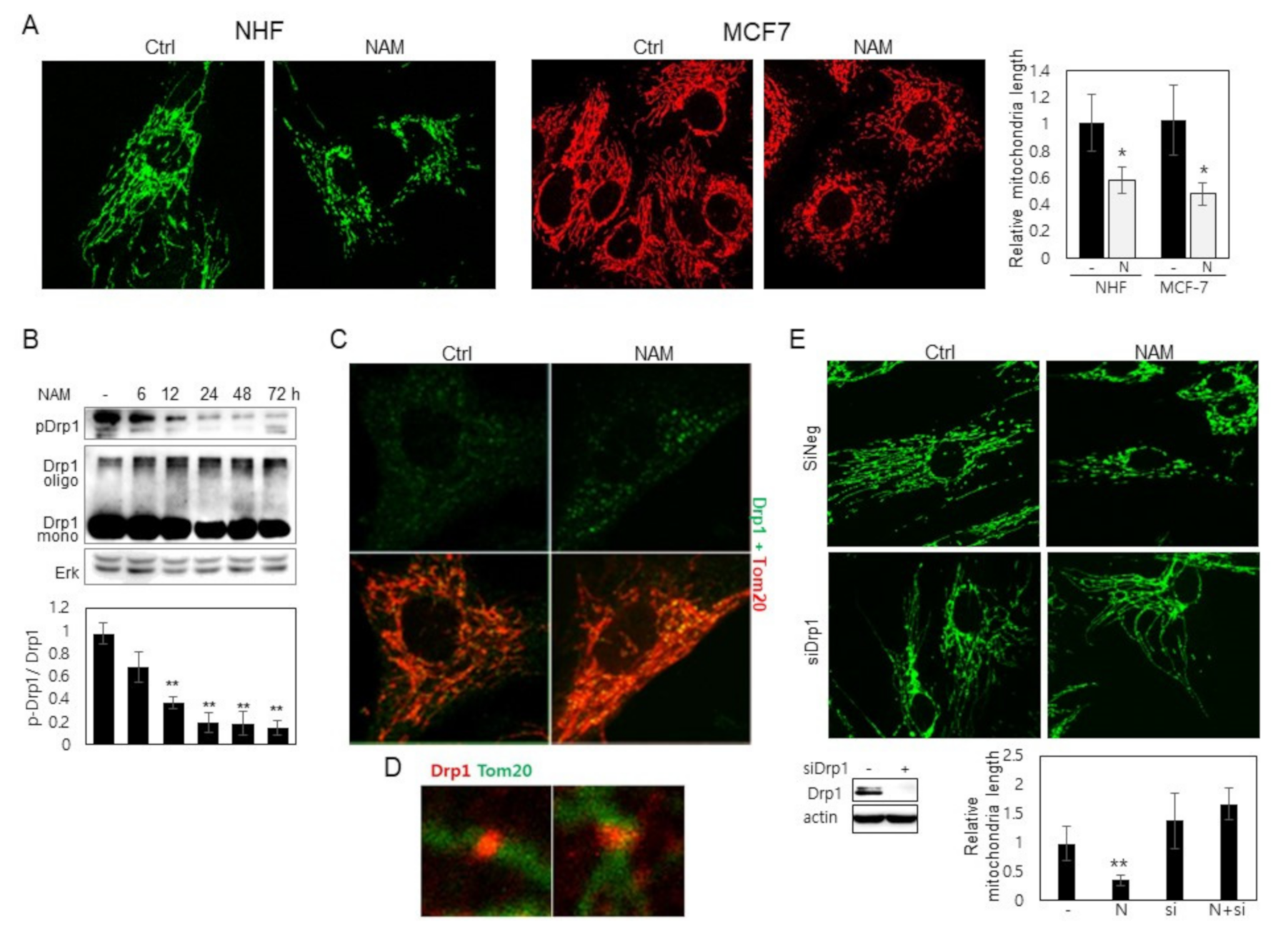
3.1. Drp1 Is Activated and Induces Mitochondrial Fragmentation upon NAM Treatment
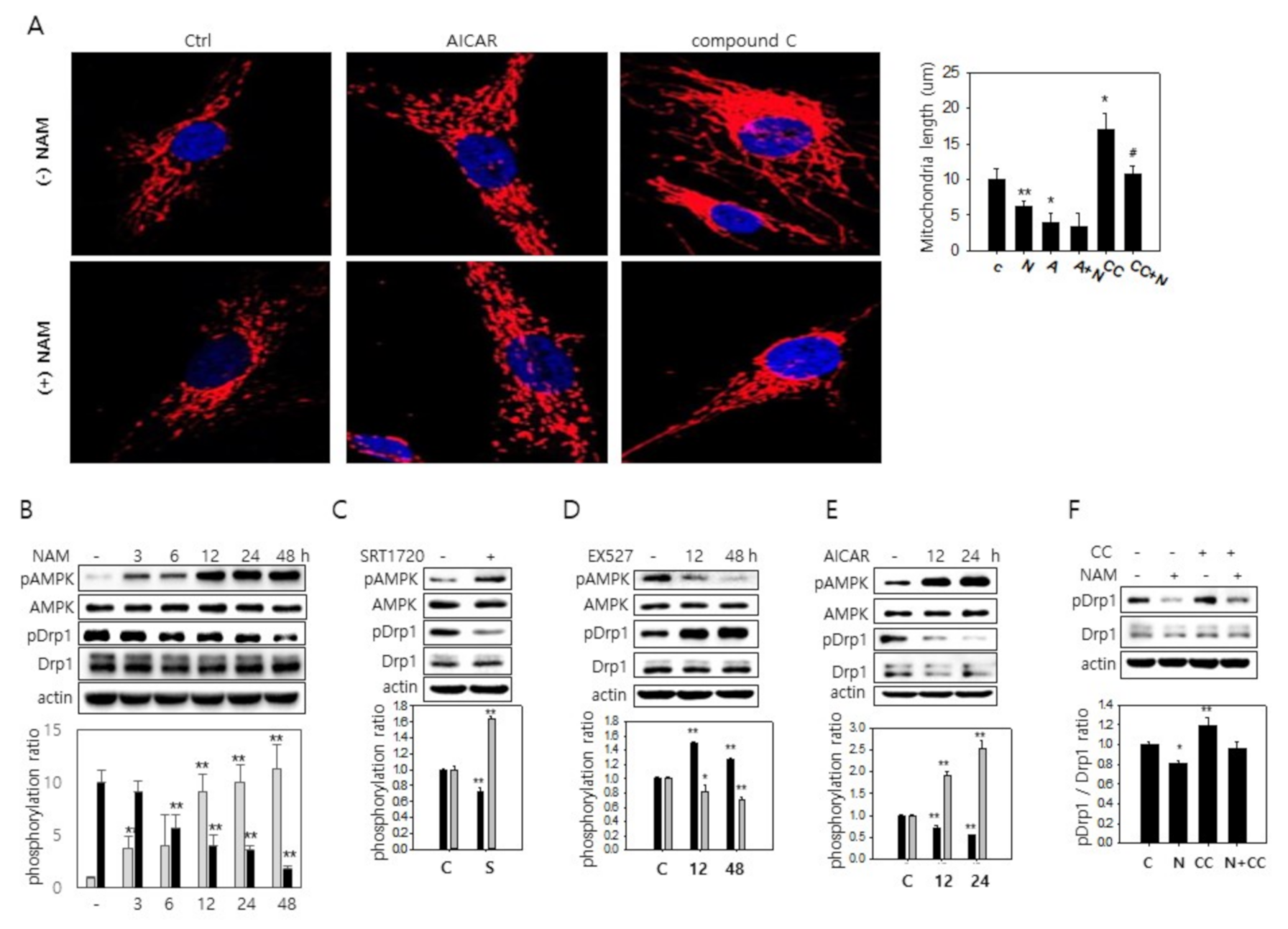
3.2. NAM-Induced Mitochondrial Fragmentation Is Mediated by SIRT1 Activation
3.3. SIRT1-Mediated AMPK Activation Drives Drp1 Hypo-Phosphorylation
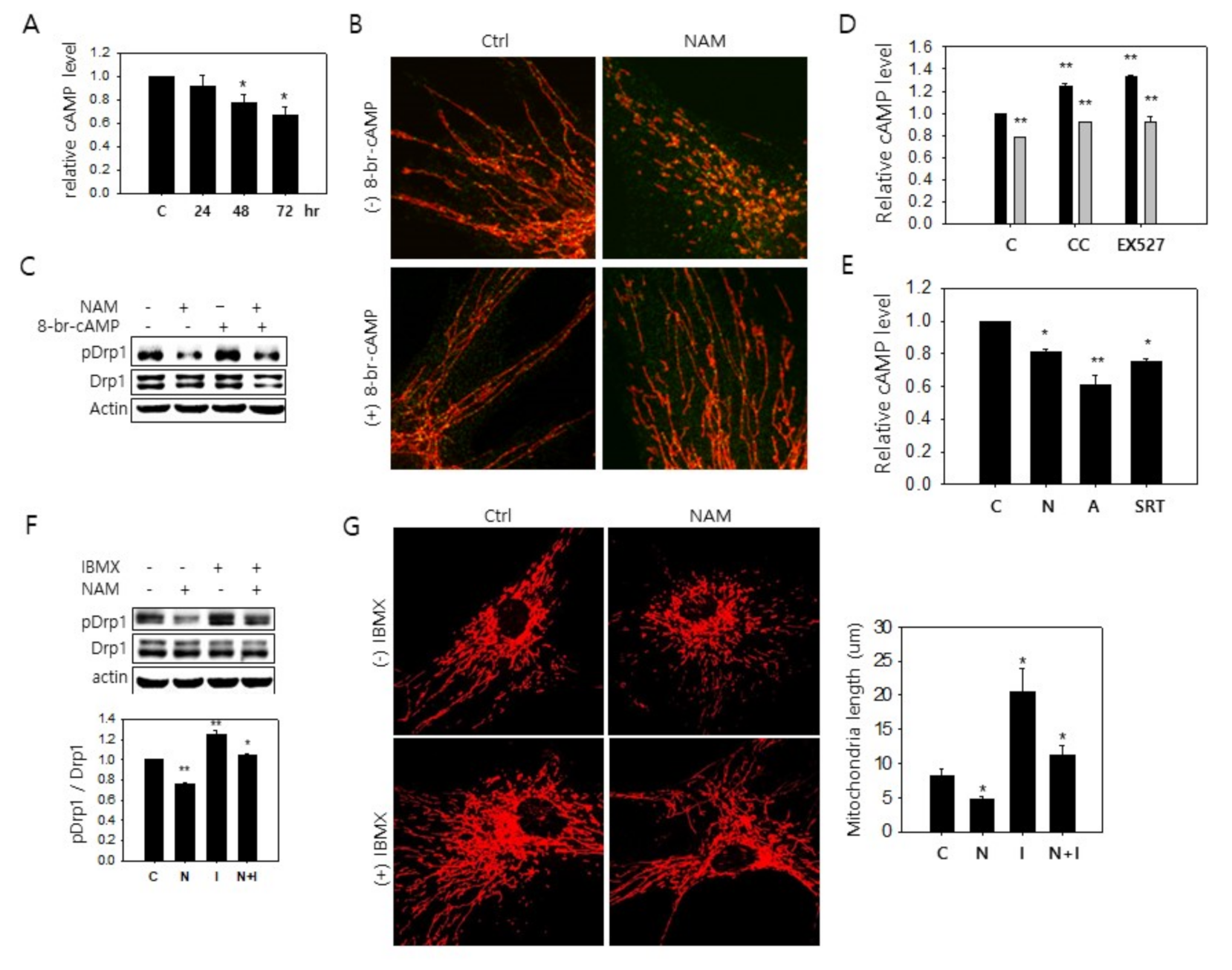
3.4. Possible Downregulation of PKA Activity and Drp1 Hypo-Phosphorylation through a Decrease in cAMP Level in NAM-Treated Cells
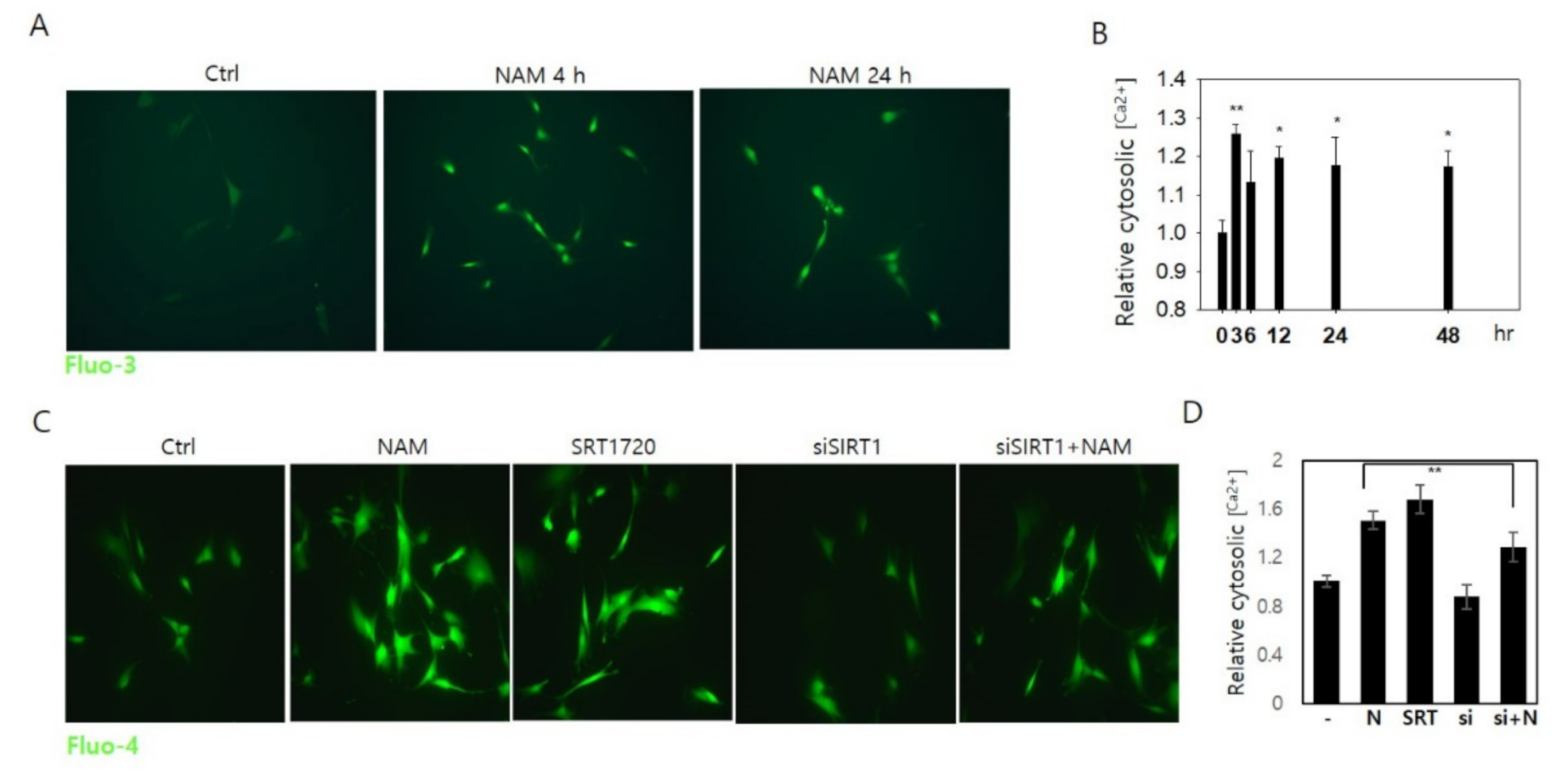
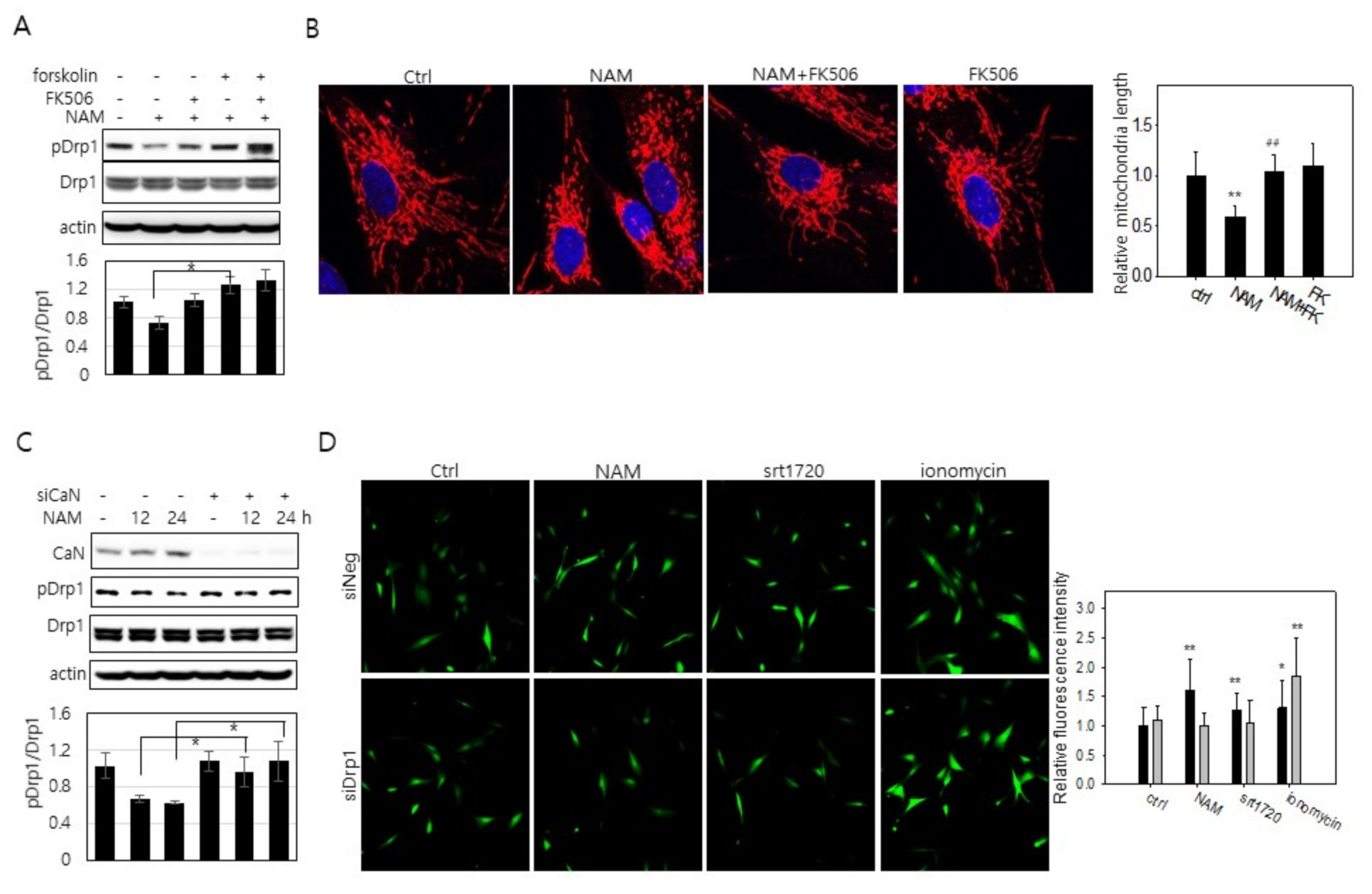
3.5. Increase in Cytosolic Ca2+ and Calcineurin Activation May Also Be Involved in Drp1 Hypo-Phsophorylation in NAM-Treated Cells
3.6. Drp1 Activation Is Necessary for the Increase in Cytosolic Ca2+
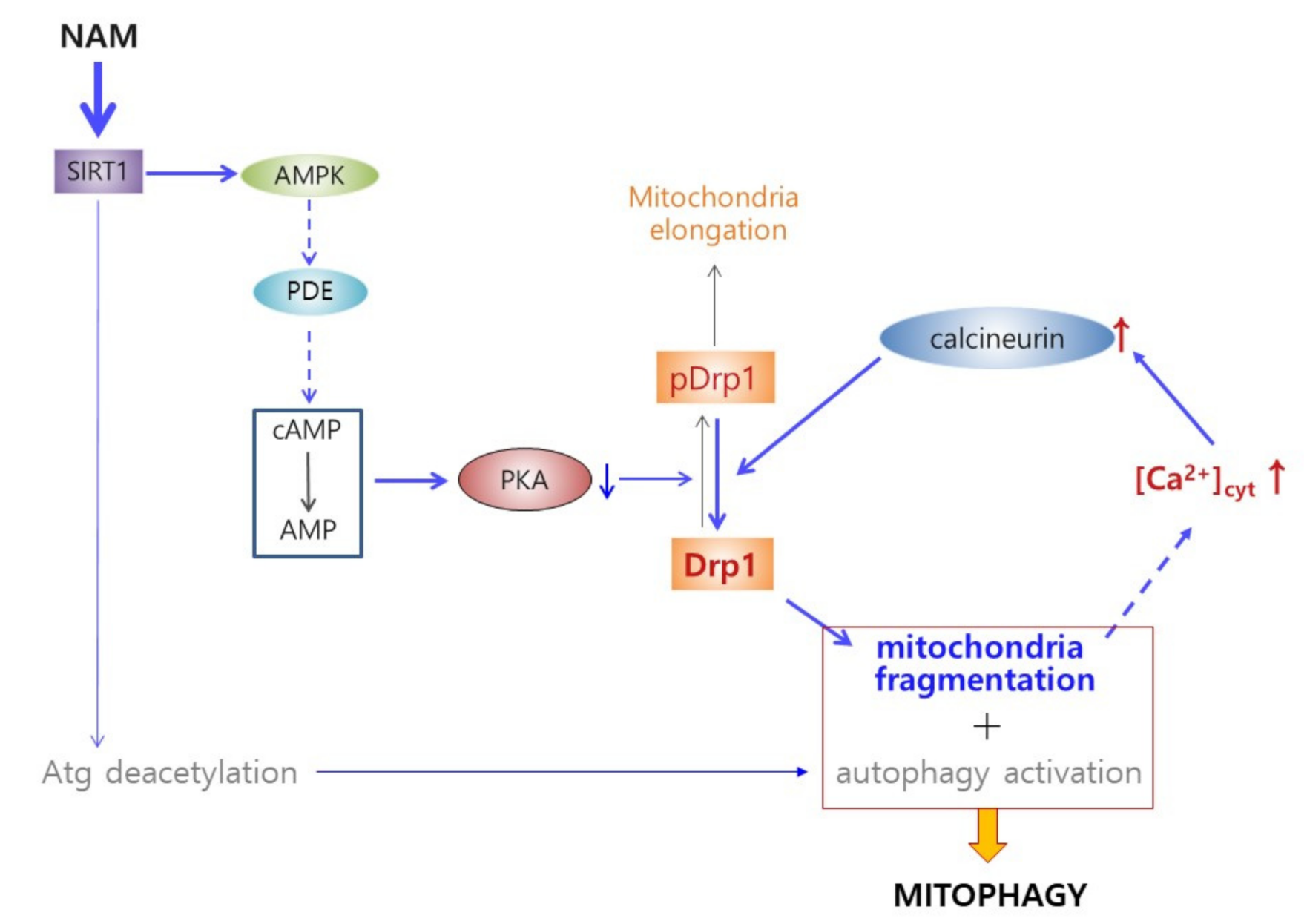
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guarente, L. Mitochondrial—A nexus for aging, calorie restriction, and sirtuins? Cell 2008, 132, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondriall segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. The mitochondriall-lysosomal axis theory of aging: Accumulation of damaged mitochondrial as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef]
- Kang, H.T.; Hwang, E.S. Nicotinamide enhances mitochondrial quality through autophagy activation in human cells. Aging Cell 2009, 8, 426–438. [Google Scholar] [CrossRef]
- Lee, C.S.; Han, J.H.; Jang, Y.Y.; Song, J.H.; Han, E.S. Differential effect of catecholamines and mpp(+) on membrane permeability in brain mitochondrial and cell viability in pc12 cells. Neurochem. Int. 2002, 40, 361–369. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins mfn1 and mfn2 coordinately regulate mitochondriall fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. Hfis1, a novel component of the mammalian mitochondriall fission machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondrial. J. Cell Biol. 1998, 143, 351–358. [Google Scholar] [CrossRef]
- Jendrach, M.; Pohl, S.; Voth, M.; Kowald, A.; Hammerstein, P.; Bereiter-Hahn, J. Morpho-dynamic changes of mitochondrial during ageing of human endothelial cells. Mech. Ageing Dev. 2005, 126, 813–821. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant mitochondrial in dfo-induced cellular senescence: Involvement of enhanced fusion process through modulation of fis1. J. Cell. Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Carreira, R.S. Autophagy in health and disease. 5. Mitophagy as a way of life. Am. J. Physiol. Cell Physiol. 2010, 299, C203–C210. [Google Scholar] [CrossRef]
- Biala, A.K.; Dhingra, R.; Kirshenbaum, L.A. Mitochondriall dynamics: Orchestrating the journey to advanced age. J. Mol. Cell. Cardiol. 2015, 83, 37–43. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondriall dynamics and its involvement in disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Lee, H.I.; Hwang, E.S. Nicotinamide extends replicative lifespan of human cells. Aging Cell 2006, 5, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. The vitamin nicotinamide: Translating nutrition into clinical care. Molecules 2009, 14, 3446–3485. [Google Scholar] [CrossRef]
- Ok, J.S.; Song, S.B.; Hwang, E.S. Enhancement of replication and differentiation potential of human bone marrow stem cells by nicotinamide treatment. Int. J. Stem Cells 2018, 11, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high nad+/nadh ratio and sirt1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the nad-dependent deacetylase sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. Sirt1: Regulation of longevity via autophagy. Cell. Signal. 2009, 21, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondriall fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related gtpase drp1 participates in mitochondriall fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Cyclic amp-dependent protein kinase phosphorylation of drp1 regulates its gtpase activity and mitochondriall morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of drp1 by cyclic amp-dependent protein kinase and calcineurin regulates mitochondriall fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of drp1 to mitochondrial. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Huang, Y.; Li, L. Drp1-dependent mitochondriall fission plays critical roles in physiological and pathological progresses in mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondriall fission through modification of the dynamin-related protein drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific sirt1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Nahimana, A.; Attinger, A.; Aubry, D.; Greaney, P.; Ireson, C.; Thougaard, A.V.; Tjornelund, J.; Dawson, K.M.; Dupuis, M.; Duchosal, M.A. The nad biosynthesis inhibitor apo866 has potent antitumor activity against hematologic malignancies. Blood 2009, 113, 3276–3286. [Google Scholar] [CrossRef] [PubMed]
- Pittelli, M.; Formentini, L.; Faraco, G.; Lapucci, A.; Rapizzi, E.; Cialdai, F.; Romano, G.; Moneti, G.; Moroni, F.; Chiarugi, A. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondriall insensitive nad pool. J. Biol. Chem. 2010, 285, 34106–34114. [Google Scholar] [CrossRef]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. Sirt1 regulates hepatocyte lipid metabolism through activating amp-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. Sirt1 modulation of the acetylation status, cytosolic localization, and activity of lkb1. Possible role in amp-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. Amp-activated protein kinase mediates mitochondriall fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondriall recruitment of drp1 during mitochondriall fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, mff, mid49, and mid51 mediate drp1 recruitment in mitochondriall fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Johanns, M.; Lai, Y.C.; Hsu, M.F.; Jacobs, R.; Vertommen, D.; Van Sande, J.; Dumont, J.E.; Woods, A.; Carling, D.; Hue, L.; et al. Ampk antagonizes hepatic glucagon-stimulated cyclic amp signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4b. Nat. Commun. 2016, 7, 10856. [Google Scholar] [CrossRef]
- Essayan, D.M. Cyclic nucleotide phosphodiesterases. J. Allergy Clin. Immunol. 2001, 108, 671–680. [Google Scholar] [CrossRef]
- Parsons, W.J.; Ramkumar, V.; Stiles, G.L. Isobutylmethylxanthine stimulates adenylate cyclase by blocking the inhibitory regulatory protein, gi. Mol. Pharmacol. 1988, 34, 37–41. [Google Scholar]
- Means, A.R. Calcium, calmodulin and cell cycle regulation. FEBS Lett. 1994, 347, 1–4. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin a and fkbp-fk506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through sirt1-dependent mitochondriall autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Lovy, A.; Ahumada-Castro, U.; Bustos, G.; Farias, P.; Gonzalez-Billault, C.; Molgo, J.; Cardenas, C. Concerted action of ampk and sirtuin-1 induces mitochondriall fragmentation upon inhibition of ca(2+) transfer to mitochondrial. Front. Cell Dev. Biol. 2020, 8, 378. [Google Scholar] [CrossRef]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive insp3 receptor ca2+ transfer to mitochondrial. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Muller, M.; McNeal, A.; Lovy, A.; Jana, F.; Bustos, G.; Urra, F.; Smith, N.; Molgo, J.; Diehl, J.A.; et al. Selective vulnerability of cancer cells by inhibition of ca(2+) transfer from endoplasmic reticulum to mitochondrial. Cell Rep. 2016, 15, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The influence of nicotinamide on health and disease in the central nervous system. Int. J. Tryptophan Res. 2018, 11, 1178646918776658. [Google Scholar] [CrossRef]
- Frieden, M.; James, D.; Castelbou, C.; Danckaert, A.; Martinou, J.C.; Demaurex, N. Ca(2+) homeostasis during mitochondriall fragmentation and perinuclear clustering induced by hfis1. J. Biol. Chem. 2004, 279, 22704–22714. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr. Capacitative calcium entry revisited. Cell Calcium 1990, 11, 611–624. [Google Scholar] [CrossRef]
- Satrustegui, J.; Pardo, B.; Del Arco, A. Mitochondriall transporters as novel targets for intracellular calcium signaling. Physiol. Rev. 2007, 87, 29–67. [Google Scholar] [CrossRef]
- Gellerich, F.N.; Gizatullina, Z.; Arandarcikaite, O.; Jerzembek, D.; Vielhaber, S.; Seppet, E.; Striggow, F. Extramitochondriall ca2+ in the nanomolar range regulates glutamate-dependent oxidative phosphorylation on demand. PLoS ONE 2009, 4, e8181. [Google Scholar] [CrossRef]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of thr231-phosphotau. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef] [PubMed]
- Hathorn, T.; Snyder-Keller, A.; Messer, A. Nicotinamide improves motor deficits and upregulates pgc-1alpha and bdnf gene expression in a mouse model of huntington’s disease. Neurobiol. Dis. 2011, 41, 43–50. [Google Scholar] [CrossRef]
- Anderson, D.W.; Bradbury, K.A.; Schneider, J.S. Broad neuroprotective profile of nicotinamide in different mouse models of mptp-induced parkinsonism. Eur. J. Neurosci. 2008, 28, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Mokudai, T.; Ayoub, I.A.; Sakakibara, Y.; Lee, E.J.; Ogilvy, C.S.; Maynard, K.I. Delayed treatment with nicotinamide (vitamin b(3)) improves neurological outcome and reduces infarct volume after transient focal cerebral ischemia in wistar rats. Stroke 2000, 31, 1679–1685. [Google Scholar] [CrossRef]
- Bold, J.M.; Gardner, C.R.; Walker, R.J. Central effects of nicotinamide and inosine which are not mediated through benzodiazepine receptors. Br. J. Pharmacol. 1985, 84, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Prousky, J. Vitamin b3 for depression: Case report and review of the literature. J. Orthomol. Med. 2010, 25, 137–147. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.B.; Park, J.S.; Jang, S.Y.; Hwang, E.S. Nicotinamide Treatment Facilitates Mitochondrial Fission through Drp1 Activation Mediated by SIRT1-Induced Changes in Cellular Levels of cAMP and Ca2+. Cells 2021, 10, 612. https://doi.org/10.3390/cells10030612
Song SB, Park JS, Jang SY, Hwang ES. Nicotinamide Treatment Facilitates Mitochondrial Fission through Drp1 Activation Mediated by SIRT1-Induced Changes in Cellular Levels of cAMP and Ca2+. Cells. 2021; 10(3):612. https://doi.org/10.3390/cells10030612
Chicago/Turabian StyleSong, Seon Beom, Jin Sung Park, So Young Jang, and Eun Seong Hwang. 2021. "Nicotinamide Treatment Facilitates Mitochondrial Fission through Drp1 Activation Mediated by SIRT1-Induced Changes in Cellular Levels of cAMP and Ca2+" Cells 10, no. 3: 612. https://doi.org/10.3390/cells10030612
APA StyleSong, S. B., Park, J. S., Jang, S. Y., & Hwang, E. S. (2021). Nicotinamide Treatment Facilitates Mitochondrial Fission through Drp1 Activation Mediated by SIRT1-Induced Changes in Cellular Levels of cAMP and Ca2+. Cells, 10(3), 612. https://doi.org/10.3390/cells10030612