Abstract
Parkinson’s disease (PD) is a complex neurodegenerative disorder that is currently incurable. As a consequence of an incomplete understanding of the etiology of the disease, therapeutic strategies mainly focus on symptomatic treatment. Even though the majority of PD cases remain idiopathic (~90%), several genes have been identified to be causative for PD, facilitating the generation of animal models that are a good alternative to study disease pathways and to increase our understanding of the underlying mechanisms of PD. Drosophila melanogaster has proven to be an excellent model in these studies. In this review, we will discuss the different PD models in flies and key findings identified in flies in different affected pathways in PD. Several molecular changes have been identified, of which mitochondrial dysfunction and a defective endo-lysosomal pathway emerge to be the most relevant for PD pathogenesis. Studies in flies have significantly contributed to our knowledge of how disease genes affect and interact in these pathways enabling a better understanding of the disease etiology and providing possible therapeutic targets for the treatment of PD, some of which have already resulted in clinical trials.
1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease and is characterized by bradykinesia, tremor and rigidity [1,2]. These motor signs arise from a lack of dopamine due to a decline of dopaminergic neurons in the substantia nigra [3]. Furthermore, the majority of PD patients present with Lewy bodies, the pathological hallmark of PD that are aggregates of proteins–mostly consisting of alpha-synuclein-, but also containing mitochondria and lipids [4]. In addition to motor symptoms, a wide range of non-motor symptoms occur, including dementia, depression, constipation and sleep problems [5,6,7,8]. These non-motor symptoms may occur years prior to the initial diagnosis [7] and strongly impact the quality of life [9].
PD is a progressive disorder, the prevalence of which increases with age and globally affects 1% of the population above 60 years [10,11,12] imposing a heavy burden on patients, caregivers, and society. An exponential increase in PD prevalence can be expected, in part due to an increasingly aging society [13]. Hence, therapeutic strategies that halt or reverse the disease are invaluable. In the past decades, major advances have been made in our understanding of the disease pathogenesis. However, unfortunately, important gaps remain to be further elucidated and, thus, alleviating signs and symptoms via e.g., dopamine replacement therapy is currently the major therapeutic strategy [14]. To increase our understanding of the underlying pathways resulting in PD, animal models that present with the basic PD-like features, including dopaminergic neuron loss and motor signs are key. Studies in these animal models provide novel insights into disease mechanisms and identify novel therapeutic targets. Unfortunately, an animal that completely represents all signs and symptoms does not exist. Animal models created in different species often reproduce one or several PD-related signs and symptoms, while lacking other essential features. A common and remarkable feature in different genetic mouse models of PD is the lack of dopaminergic neurodegeneration [15,16,17,18], suggestive of a protective mechanism in mice that is absent in humans and pointing to the fact that each animal model has shortcomings when compared to model human diseases.
Nonetheless, Drosophila melanogaster is an excellent model to study the molecular mechanisms in PD. The fruit fly is a relatively simple animal; however, it does contain a complex neuronal circuitry including clusters of dopaminergic neurons [19,20]. D. melanogaster undergoes different stages (egg, larva, pupa, fly) in a short lifecycle of around 10 days (Table 1). Furthermore, many experimental tools exist to genetically manipulate D. melanogaster allowing a relatively rapid creation of genetically modified flies that can serve as a disease model. In this review, we focus on the advantages of using D. melanogaster as animal model to study disease pathways and the key findings in the molecular mechanisms in PD that arose from studies on the fruit fly. First, we will discuss the possibilities of using Drosophila in the study of a variety of human diseases, followed by an overview of the different available PD fly models. Next, we will review in more detail the identification of altered molecular mechanisms in PD fly models with a focus on mitochondrial dysfunction, defective endo-lysosomal pathway and lipids that may constitute a linking factor between these two mechanisms. Finally, the relevance of these findings for PD patients and its consequence for therapeutic strategies will be discussed.

Table 1.
Limitations and advantages of different aspects of using D. melanogaster as animal model to study human diseases.
2. Drosophila melanogaster as Animal Model to Study Human Diseases
Fruit flies are small animals, which facilitates live imaging enabling in vivo recording of biological processes that can further clarify disease-relevant mechanisms, including analyzing dopaminergic neurons that are mainly affected in PD (Table 1). The genome of flies consists of only four chromosomes, which is a markedly lower number than the 23 chromosomes in the human genome. Nonetheless, more than 75% of the human disease-causing genes have an ortholog in flies (Table 1) [21]. Using established methodology to manipulate genes, including the Crispr cas9 system that allows the introduction of specific point mutations [22,23] or the yeast UAS-gal4 system enabling overexpression or knockdown of proteins ubiquitously or in relevant tissue (Table 1) [24,25,26], the proper genetic allele can be created that mimics the genetic disease context to study essential biological processes. For example, overexpression of a specific protein may be more relevant for gain-of-function mutations than knocking down that specific protein. In addition, several collections of mutant flies exist or can be created via toxin-induced mutagenesis. These are especially useful to perform modifier screens, which is one of the benefits of using D. melanogaster in the study of diseases. These modifier screens enable the identification of genes that interact with disease-related mechanisms contributing to the elucidation of novel pathways involved in or providing promising therapeutic targets. Such modifier screens have proven their value in PD research with the identification of vitamin K2, aconitase, sterol regulatory element-binding protein 1 (SREBP1) (see below) [27,28,29]. Furthermore, screens in which specific phenotypes are analyzed that mimic phenotypes of a specific disease model can lead to the identification of new genes that are involved in the disease pathogenesis (Figure 1).
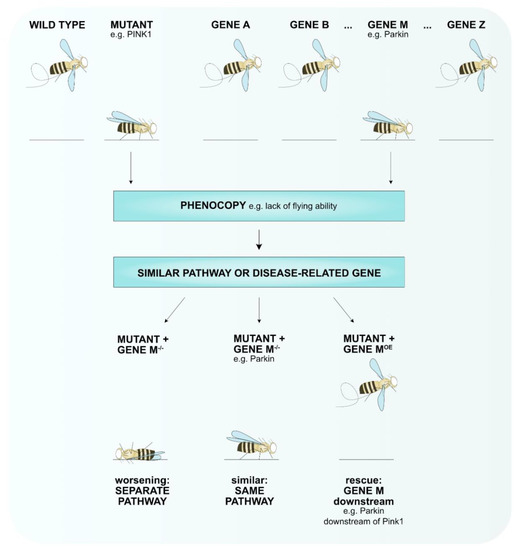
Figure 1.
A phenotypic screen can identify novel genes involved in the disease pathway. A collection of mutant alleles (gene A–gene Z) can be used to identify genes that when mutated result in the same phenotype (phenocopy) e.g., lack of flying ability for pink1-mutant flies that are mimicked by parkin-mutant flies. To test if these genes function in the same pathway, the phenotypes of double-mutant flies can be tested. Loss of gene M worsens the phenotype of the disease mutant, thus, separate pathways play a role that converge into one. Loss of gene M does not enhance or improve the phenotype of the mutant meaning that these genes function in the same pathway (e.g., Pink1-Parkin). Overexpression (OE) of gene M rescues the mutant phenotype hints towards gene M functioning downstream of the mutant (e.g., Parkin functions downstream of Pink1).
The creation of new fly models often emerges from the identification of a disease-causing gene. For example, loss-of-function mutations in Thousand and one amino acid kinase 1 (TOAK1) have been identified to be causative for neurodevelopmental delay and intellectual disability [30]. Knockdown of the Drosophila ortholog Tao shows a delayed development and lethality at the final larval stage accompanying defective brain and neuronal morphology [30] mimicking the disease phenotype. Furthermore, knockdown of Tao presents with altered mitochondrial distribution in axons of motor neurons [30], suggesting mitochondria play a role in the disease pathogenesis, which was confirmed in patient-derived fibroblasts that display defective mitochondrial morphology [30]. Thus, this fly model for neurodevelopmental delay can be used to further investigate the relationship between abnormal mitochondria and brain morphology that results in neurodevelopmental delay. Notably, D. melanogaster can also be used to model diseases that do not occur naturally in flies. Lethal tumor growth and metastasis have not been observed in wild type flies [21]. Nonetheless, genes that play a role in tumor formation in humans, such as those affecting the cell-cycle control have been identified in flies and are being used to study processes of tumor growth [21]. These examples show that D. melanogaster can be employed to study a plethora of human diseases even if the exact disease process does not occur in this animal.
3. Drosophila melanogaster as Animal Model for Parkinson’s Disease
Many animal models exist to study PD. Non-human primate models are the most closely linked to humans and, thus, findings in primate PD models will most likely be resembling those present in PD patients the most. Compared to rodent models, flies are a simple animal model, however, it does represent many of the key features of PD, in contrast to rodent models that often lack loss of dopaminergic neurons [15,16,17]. For a long time, PD was considered to be a sporadic disorder; however, with the identification of SNCA to be causative for PD in the late 90s [31], the importance of genes in the etiology of PD became obvious and has been constantly growing in the past 25 years. Since then, various genes have been identified to be causative for PD (Table 2) [32] and have enabled the creation of genetic animal models of PD leading to a significant increase in our knowledge of its etiology. In addition, many genes have been linked previously to PD; however, were not confirmed or were disproven to be causative for PD [32,33]. Nonetheless, several of these proteins interact with or function in shared mechanisms with the PD-confirmed proteins and thus can provide more information about the affected pathways relevant for PD and, hence, will also be discussed in the relevant sections below.
Table 2.
PD models in flies and their characteristics.
Several fly models to study the underlying mechanisms of PD exist to mimic the human condition, including genetic models (overexpression, knockdown and endogenous) and toxin-induced models. A well-established overexpression model is the overexpression of human alpha-synuclein. Interestingly, SNCA is the only PD gene that does not have a fly ortholog. Nonetheless, the fly models that have been created overexpress wild type-and disease-mutant human SNCA using the UAS-Gal4 system [34] and develop PD-like symptoms, suggesting that the underlying disease mechanisms are evolutionarily conserved. Furthermore, mutations in SNCA causing PD are gain-of-function mutations [35], making the overexpression model a relevant one. In addition to these causative genes, risk factors have been identified that increase the susceptibility for PD (Table 2). While risk factors are not disease-causing, studies that investigate the functional pathways of these genes are useful to identify mechanisms that are linked to PD. The best-studied risk factor for PD is glucocerebrosidase (GCase) encoded by GBA [36,37]. Remarkably, flies have two GBA orthologs (gba1a and gba1b) [38]. To study the function of GCase and how it increases the risk for PD, loss-of-function alleles of both fly genes are being used [38,39] as well as overexpression of human GCase-carrying mutations that are known to increase the risk for PD [40]. This demonstrates that studying the effect of different gene dosages is valuable in the investigation of protein function or involved pathways. Many genes and risk factors are being used in studies to dissect the PD pathogenesis; however, the vast majority of PD cases are currently considered idiopathic. Nonetheless, studies using genetic fly models are useful for various reasons in this context, as well: 1. PD-causing genes have been shown to be altered in sporadic PD [20,41,42,43], suggesting they are functioning as risk factors in these forms of PD; 2. Idiopathic forms of PD show a similar disease progress compared to some of the genetic forms of the disease [44,45], suggesting similar underlying mechanisms to play a role; 3. Mitochondrial defects have first been identified in a subset of idiopathic patients [46] and were later confirmed in genetic forms of PD [47,48] and in animal models [49,50,51,52] to play a role in at least a subset of PD patients. Many of the existing PD fly models reflect PD-related signs and symptoms and their relevance for the disease has been validated numerously. For example, overexpression of human wild type PINK1 in pink1-mutant flies is able to rescue the phenotypes, while overexpression of human mutant PINK1 is not [52,53], suggesting that the function of PINK1 is evolutionarily conserved and that flies are a valid animal model to investigate the function of PINK1 in relation to PD.
Mitochondrial toxins, like 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [54,55] or pesticides such as rotenone [56,57,58] induce parkinsonian-like symptoms that resemble idiopathic forms of PD and can be used to model idiopathic PD in flies [59]. MPTP specifically affects dopaminergic neurons [60], while rotenone is a systemic mitochondrial drug without any preference towards a specific type of neuron or organ that also results in the loss of dopaminergic neurons [59,61]. 6-hydroxydopamine (6-OHDA) is a drug that selectively destroys dopaminergic neurons and is often used to model PD. 6-OHDA cannot pass the blood-brain barrier and therefore, has to be injected into the brain [62]. This mode of application of the drug makes it an unfavorable drug to apply in the fruit fly. Nonetheless, it needs to be considered that application of these drugs to the fly medium is often done in an acute, high-dose fashion, which possibly activates additional mechanisms. In contrast, patients are typically chronically exposed to these drugs for years at a low dose before symptoms arise.
4. The Mitochondrial-PD Connection in Drosophila melanogaster
The first observation that mitochondria are involved in PD came from the observation in the 80s that drug abusers using drugs contaminated with MPTP developed parkinsonian-like symptoms [63]. MPTP and rotenone specifically inhibit complex I of the electron transport chain (ETC) resulting in disrupted energy production [64,65,66]. Later, defective complex I activity was observed in post mortem brains of idiopathic PD patients [67] and PD-causing genes were identified that are linked to mitochondrial function, including PINK1 [47], Parkin [68] and DJ-1 [48] that cause autosomal recessive PD (Table 2). Here, we will discuss the advances that studies using pink1- and parkin-mutant flies have contributed to our knowledge of their shared functions, including mitochondrial remodeling and mitophagy, as well as their independent functions, and the role of DJ-1 in oxidative stress (Figure 2).
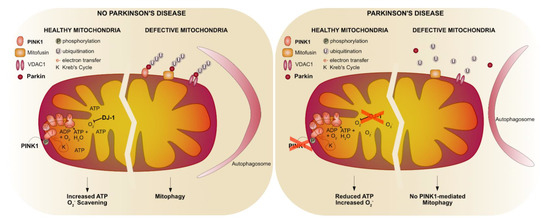
Figure 2.
PD-related proteins in mitochondrial function. In healthy mitochondria, complex I of the electron transport chain (ETC) is phosphorylated in a Pink1-dependent fashion resulting in an efficient electron transfer through the different ETC complexes to finally use oxygen and ADP to form ATP. Oxidative stress (O2.) created by the ETC is scavenged by DJ-1. Upon a defective mitochondrial membrane potential Pink1 remains at the outer mitochondrial membrane that provides a signal for the translocation of Parkin to the mitochondria to ubiquitinate mitochondrial proteins to label these defective mitochondria for autophagosomal degradation. In genetic forms of PD, a lack of PINK1 prevents the phosphorylation of complex I that leads to an inefficient electron transfer and thus a decrease in ATP production. Furthermore, due to loss of PINK1, Parkin is not translocated to the mitochondria resulting in a lack of ubiquitination of mitochondrial proteins to label mitochondria for degradation. Loss of DJ-1 results in increased oxidative stress that results in dysfunctional mitochondria.
4.1. The Pink1-Parkin Pathway
Patients suffering from PINK1 and Parkin-related PD both present with early-onset forms of PD, show a similar disease progression [2] and respond comparably well to therapy. In addition, flies with loss of Pink1 and Parkin function share phenotypic similarities, including locomotion defects, mitochondrial dysfunction and abnormal mitochondrial morphology [49,50]. These observations support the notion that these genes function in the same pathway. Overexpression of Parkin in pink1-mutant flies rescued the pathological phenotypes in the Pink1-deficient flies, whereas overexpression of Pink1 did not induce a rescue in parkin-mutant phenotypes [49,50]. These studies in flies were the first to provide experimental evidence that PINK1 and Parkin function in the same pathway with Parkin acting downstream of PINK1 to maintain mitochondrial integrity. These findings were later confirmed in cellular models (see Section 4.1.2) [69,70].
4.1.1. Mitochondrial Fusion and Fission
The swollen, enlarged mitochondria hint toward a function in mitochondrial remodeling that consists of regulated fusion and fission of mitochondria. Several evolutionarily conserved genes that express GTPase proteins have been identified to be involved in these fusion and fission events. Mitofusins 1 (Mfn1) and 2 (Mfn2) are located at the outer mitochondrial membrane and promote fusion at the outer mitochondrial membrane, optic atrophy 1 (OPA1), located at the intermembrane space provokes fusion of the inner mitochondrial membranes [71,72]. Dynamin-related protein 1 (Drp1) is a cytosolic GTPase that is important for mitochondrial fission [71,73]. Studies that apply genetic alterations of these genes in pink1- or parkin-mutant flies have confirmed the contribution of the Pink1-Parkin pathway in mitochondrial morphology, i.e., stimulation of fission or inhibition of fusion improve the phenotypes, while promotion of fusion or preventing fission events results in the lethality of Pink1- and Parkin-deficient flies [74,75]. Remarkably, recent findings in flies show that PINK1-dependent phosphorylation of Drp1 regulates mitochondrial dynamics that are independent of Parkin [76]. While these data show that loss of Pink1 and Parkin impede mitochondrial remodeling, loss of Drp1 results in distinct mitochondrial phenotypes [77] compared to loss of Pink1 or Parkin [49,50,74,75,78], suggesting that under healthy conditions, Pink1 and Parkin are not part of the key factors of drp1-mediated fission, but are rather activated under pathological conditions.
4.1.2. Mitophagy
The mitochondrial fusion-fission machinery is an important tool in the facilitation of clearing damaged mitochondria [79,80,81], as it selects out defective mitochondria and reduces the mitochondrial size to enable mitochondrial degradation, a process termed mitophagy. Interestingly, Parkin, a ubiquitin ligase [82], ubiquitinates target proteins (that may be located on various organelles such as mitochondria) to serve as a signal for these ubiquitinated proteins and organelles to be degraded [83,84]. Follow-up studies in cellular models have provided further clarification of PINK1-Parkin-related mitophagy. Under healthy conditions, PINK1 is immediately cleaved and imported into mitochondria via the TIM-TOM complex. In defective mitochondria that lead to depolarization of mitochondria, PINK1 remains uncleaved at the outer mitochondrial membrane followed by (auto)phosphorylation of PINK1, a kinase, and Parkin, providing a signal for Parkin to translocate to the mitochondria resulting in mitochondrial degradation [69,85]. These findings were also confirmed in a Drosophila cellular model [86]. Furthermore, studies in these cellular fly models provided evidence for the ubiquitination of Mfn by Parkin to be the critical labeling step for mitochondrial degradation [85,86,87]. F-box domain only protein 7 (Fbxo7) functions in mitophagy via direct interaction with Pink1 and Parkin [88]. Wild type Fbxo7 facilitates mitophagy, while mutant Fbxo7 impedes mitophagy [89]. Mutations in FBXO7 cause atypical parkinsonism and flies do not have a Fbxo7 protein, but overexpression of the human wild type protein rescues Parkin-deficient phenotypes in flies [88], further supporting the role of Fbxo7 in mitophagy. Remarkably, overexpression of wild type Fbxo7 in flies induces age-dependent locomotion defects, dopaminergic neuron loss and swollen mitochondrial [89], mimicking PD-like symptoms.
For a long time and due to a lack of appropriate tools, it was only possible to study mitophagy events in a cellular system that required the application of mitochondrial drugs to induce mitophagy. The main drawback of this experimental setup was that this did not represent physiological conditions in the disease context. Following the creation of mt-Keima, a pH-sensitive sensor that is targeted to mitochondria [90], flies were created that express mt-Keima creating an in vivo model to study mitophagy [91,92]. Interestingly, studies revealed that loss of Pink1 or Parkin did not display an effect on mitophagy levels in the initial life stages of flies [91,92]; however, basal mitophagy levels were reduced in aging flies [91]. Hence, these data are suggestive of compensatory mechanisms maintaining the basic levels of mitophagy initially. Nonetheless, this compensatory mechanism cannot fulfill the requirements for mitophagy resulting in the age-dependent lowering of mitophagy levels following loss of PINK1 or Parkin.
The relevance of mitophagy in disease pathogenesis remains to be further clarified. Overexpression of Parkin in pink1-mutant flies rescues pathological phenotypes [49,50], independent of the Pink1-dependent translocation of Parkin to the mitochondria, which is absent in Pink1-deficient flies. These observations cannot easily be reconciled with an exclusive function of Pink1 and Parkin in mitophagy as the single underlying disease mechanisms. Furthermore, loss of PINK1 or Parkin results in defects in mitophagy [91] and both PINK1 and Parkin are involved in functions that are independent of one another (see Section 4.1.3 and Section 4.2). In contrast, wild type Fbxo7 that stimulates mitophagy, induces age-dependent PD-like symptoms in flies, challenging the theory that ablation of mitophagy plays a major role in PD pathogenesis. Studies in flies expressing mutant Fbxo7 would aid to clarify this discrepancy. Collectively, these data are indicative of yet to be discovered additional mechanisms to play a role in disease pathogenesis.
4.1.3. Mitochondrial Complex I
Loss of Pink1 results in lower complex I activity [52] that is found to be absent in Parkin-deficient flies [93]. Interestingly, the stimulation of mitochondrial fission that is enabling mitophagy [79,80,81], does not induce a rescue of complex I activity in pink1-mutant flies [93,94]. Bypassing the defective complex I in Pink1-deficient flies via the expression of the yeast complex I, ndi1, rescues the mitochondrial defects in these flies but not in parkin-mutant flies [93]. These data provide evidence that defective complex I plays an important role in PINK1-related PD pathogenesis. Moreover, phosphorylation of ND-42 (the fly ortholog of NdufA10), a complex I subunit that is necessary for proper complex I function, is Pink1-dependent [95], directly linking Pink1 to complex I. These data were confirmed and further validated in patient-derived fibroblasts carrying PINK1 mutations [95], suggesting that the mitochondrial ETC serves as a therapeutic target in PINK1-related PD. Also here, studies in flies have contributed significantly. Application of near-infrared light that boosts ETC complex IV improves the mitochondrial phenotypes in pink1-mutant flies [96] and studies in a mammalian alpha-synuclein model confirmed that this photostimulation is protective for dopaminergic neuron loss [97]. Further studies need to be performed to test the effectiveness of this therapy in PD patients. However, the downside of this therapeutic strategy is the invasiveness to apply the infrared light. Hence, drugs that stimulate the ETC would be the more preferred strategy. Studies in flies identified vitamin k2 as an alternative electron carrier molecule that similar to ubiquinone can stimulate the ETC resulting in a rescue of Pink1-related phenotypes [27], nominating it to be a promising therapeutic strategy in the treatment of PINK1-related PD patients. The application of ubiquinone and vitamin k2 as therapy in patients has been initiated in clinical trials [98,99] and serves as an excellent example of therapeutic targets identified in flies that are now being tested in patients.
One of the first observations in post mortem brains of PD patients was the accumulation of iron in the substantia nigra [100]. However, how iron-mediated toxicity contributes to the PD pathogenesis remains enigmatic. In a genetic modifier screen, mitochondrial aconitase (acon) was identified to improve the phenotypes in pink1-mutant flies [28]. Acon functions in the Krebs cycle [101] and harbors iron-sulfur clusters that are inactivated in loss of Pink1 resulting in the release of iron and reactive oxygen species in the form of hydroxyl peroxide to induce mitochondrial dysfunction [28]. More recent studies showed that increasing the bioavailability of these iron-sulfur clusters in the mitochondrial matrix promotes mitochondrial respiration [102], directly linking iron, oxidative stress and mitochondrial respiration.
4.2. Parkin-Specific Functions
E3 ubiquitin ligases, like Parkin, interact with specific ubiquitin activating (E1) and ubiquitin-conjugating (E2) enzymes to label proteins or organelles for the ubiquitination degrading pathway [103]. Studies in flies and mammalian cells have revealed Rad6, a protein involved in mental retardation, to be one of the E2 conjugating enzymes that bind to Parkin. Lack of Rad6 prevents the translocation of Parkin to the mitochondria and therefore the ubiquitination of mitochondrial proteins [104]. Deubiquitinases oppose that function by removing the ubiquitin from ubiquitin-labeled proteins [105]. USP15, a deubiquitinase specifically counteracts the parkin-mediated ubiquitination of mitochondria and thus prevents mitophagy, while USP15 knockdown stimulates mitochondrial ubiquitination and age-dependent mitophagy defects in Parkin-deficient muscles and dopaminergic neurons [91,106]. In addition, Mask, an anchoring protein also known as ANKHD1 (Ankyrin repeats and KH domain containing protein 1) has been identified in flies to genetically interact with Parkin to inhibit mitophagy [107]. One of the mitochondrial ubiquitination substrates for Parkin is voltage-dependent anion-selective channel 1 (VDAC1) [70,108] and polyubiquitination of VDAC1 [109] triggers mitophagy via the recruitment of p62 and LC3 to the mitochondria [108]. VDAC1 is a component of the mitochondrial transition pore and controls the opening of this pore in the regulation of apoptosis [110]. Interestingly, the first phenotypic characterization of Parkin-deficient flies shows apoptotic cell death in flight muscle [78], providing experimental evidence for Parkin to function in apoptosis. Furthermore, monoubiquitination of VDAC1 is important in the regulation of apoptosis [109] and lack of monoubiquitination of VDAC1 phenocopies parkinsonism, including locomotion defects and loss of dopaminergic neurons [109], suggesting that regulation of apoptosis plays a role in disease pathogenesis. Parkin is a cytosolic protein and thus non-mitochondrial ubiquitination is to be expected. Research in mammalian Parkin-mutant models shows Parkin-dependent PARIS (ZNF746) ubiquitination to regulate mitochondrial biogenesis via peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1a) [111]. Follow-up studies in flies elaborate on this role in mitochondrial biogenesis and reveal that both Pink1 and Parkin promote mitochondrial health via the PARIS-dependent PGC-1a regulatory function on mitochondrial biogenesis [112], suggesting regulators of mitochondrial biogenesis can lead to new therapeutic avenues.
4.3. The Role of Oxidative Stress in PD
DJ-1 is a mitochondrial-linked protein and flies carry two DJ-1 genes, dj-1α (predominantly expressed in testes) and dj-1β (ubiquitously expressed) (Table 2). Homozygous loss of dj-1β results in motor defects that deteriorate with oxidative stress [113,114,115] confirming its function in controlling intracellular reactive oxygen species [116,117]. Double knock-out flies for dj-1α and dj-1β results in a variety of mitochondrial phenotypes that phenocopy those observed in Pink1- and Parkin-deficient flies [49,50,78,118], suggesting a genetic link between these three proteins. Upregulation of DJ-1 improves pink1-mutant phenotypes, however does not rescue parkin-mutant flies [118], suggesting a specific interaction between PINK1 and DJ-1, independent of Parkin. RNA sequencing analyses in dj-1β-mutant flies show decreased levels of NADP+ -dependent isocitrate dehydrogenase (IDH) inducing hypersensitivity to oxidative stress followed by age-dependent mitochondrial defects [119]. IDH uses the reducing potency of NADPH to suppress intracellular and mitochondrial ROS responsible for the mitochondrial phenotypes [119]. The ablation of reducing power is a feature common to that observed upon loss of Pink1 with Acon (see Section 4.1.3) [28] providing more experimental support for the importance of oxidative stress in PD.
5. Endo-Lysosomal PD Connection in Drosophila melanogaster
The implication of mitochondrial dysfunction was the earliest mechanistic insight into PD pathogenesis, however, an increasing amount of evidence is indicative of an important role of the endo-lysosomal pathway in PD [120,121,122]. The endo-lysosomal pathway is important for protein homeostasis and consists of endocytosis, retromer and autolysosome [123,124,125] that we will separately discuss. PD-related proteins that are linked to one or more of these processes are alpha-synuclein, VPS35 and LRRK2 that cause autosomal dominant forms of PD (Figure 3). Interestingly, low-penetrance variants of alpha-synuclein and LRRK2 have been found in idiopathic forms of PD, supporting the importance of this pathway in PD.
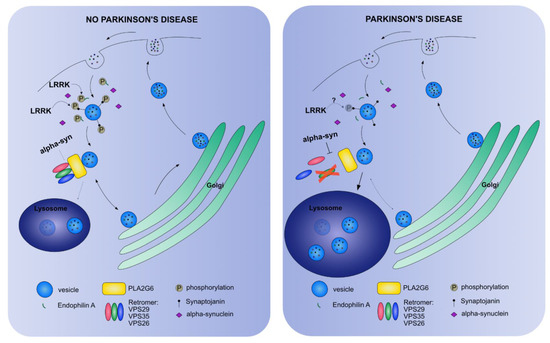
Figure 3.
A simplified scheme highlighting processes in which PD-related proteins function in the endo-lysosomal pathway. Vesicles are recycled between the plasma membrane and the Golgi (and ER, not shown). LRRK-dependent phosphorylation of Endophilin A and Synaptojanin, and alpha-synuclein are important to facilitate in the endocytosis of vesicles. Recycling vesicles pass the retromer in conjunction with PLA2G6 to continue to the Golgi to recycle their cargo. A small number of vesicles will be transported to the lysosome to be degraded. In PD, the recycling of vesicles is disturbed by defective endocytosis or a dysfunctional retromer that is inhibited by alpha-synuclein. The effect of PD-related mutations on LRRK2-dependent phosphorylation remains unclear. The net effect is that the vesicles are not recycled to the Golgi, but are translocated to the lysosomes that expand and result in neurodegeneration.
5.1. Vesicle Trafficking Endocytosis
Using an alpha-synuclein yeast PD model, defects in the trafficking between the endoplasmic reticulum (ER) and Golgi were observed [126]. SNCA encodes alpha-synuclein that contains a mitochondrial targeting signal [127]. In line with this, alpha-synuclein models have shown oxidative and mitochondrial stress [127], providing overlapping features with other forms of PD that are clearly linked to mitochondrial dysfunction. However, mitochondrial dysfunction appears to be a secondary effect rather than the initial defect related to SNCA mutations. Alpha-synuclein models were the first genetic PD model in flies [34], mimicking the typical PD-like symptoms, including locomotor deficits, dopaminergic neuron loss and alpha-synuclein-aggregated inclusion bodies [34,128,129]. Furthermore, expression of alpha-synuclein in flies results in Ser129 phosphorylated alpha-synuclein [130], similar to what was previously observed in PD patients [131,132]. Phosphorylation increases with aging flies [130] and is crucial for alpha-synuclein-induced dopaminergic neuron loss and inclusion formation that exhibit a neuroprotective role [133].
Combinational studies using yeast and Drosophila alpha-synuclein models for the first time linked Rab proteins to alpha-synuclein [134]. Rab proteins are a family of GTPases that are a member of the Ras superfamily of small G proteins [135]. Rab proteins function in membrane trafficking and more than 60 different Rab proteins are identified that differ according to their subcellular localization [135]. Rab1, the endosomal recycling factor Rab11, the late endosomal Rab7 and post Golgi vesicle trafficking Rab8 are neuroprotective or improve locomotion in flies expressing alpha-synuclein [134,136,137,138] providing ample experimental evidence for a function of alpha-synuclein in vesicle trafficking. Another example of a PD-related protein that is linked to Rab proteins is LRRK2 [139]. LRRK2 is a kinase that affects endocytosis via the LRRK2-dependent phosphorylation of Endophilin A [140,141], an endocytotic protein [142] and Synaptojanin [143] that plays an essential role in synaptic vesicle recycling [144] further supporting vesicle trafficking to be a common function in PD-related proteins. Flies carry a single Leucine Rich Repeat Kinase 2 (LRRK2) ortholog, LRRK [128]. Several models have been created in flies to model LRRK2-related PD. One is the knock-out of LRRK that allows the study of the physiological function of LRRK and shows that LRRK is localized at endosomal and lysosomal membranes [139]. In flies, studies report contradictory results in relation to dopaminergic neurons and locomotion in loss of LRRK [145,146,147]. However, the expression of PD-related mutant Drosophila LRRK or human LRRK2 induces dopaminergic neuron loss and its related locomotion defects [128]. Remarkably, mutations in Synaptojanin induce loss of dopaminergic neurons in flies but do not induce defects in synaptic vesicle endocytosis [143] mystifying the role of vesicle trafficking in PD pathogenesis. Nonetheless, loss of LRRK induces enlarged endosomes and accumulation of autophagosomes that can be rescued by the stimulation of retromer-dependent recycling from late endosomes to the Golgi apparatus [146]. Furthermore, the PD-related protein VPS35 interacts with LRRK2 in the Rab-mediated endocytic pathway [148], linking these PD genes and providing evidence for the importance of the Rab-dependent regulation of retromer-mediated endocytosis in the pathogenesis of PD.
5.2. The Retromer Complex
The retromer is a highly conserved, multi-protein complex that is required for the recycling of transmembrane receptors from endosomes to the trans-Golgi network [149,150,151]. Its recruitment to the endosomal membrane is carried out by the sorting nexin dimer component of the complex [152]. The cargo recognition and binding function are executed by the core vacuolar protein sorting trimer, VPS35, VPS29 and VPS26 [153]. Interestingly, Rab7 binds to this trimer facilitating the recruitment of the retromer to endosomes in concert with the sorting nexin dimer [154], stipulating a possible explanation in which LRRK2 and VPS35 are functionally linked with each other.
One of these core proteins, VPS35 causes autosomal dominant PD [153] and null mutants in flies are lethal [148] underlining the importance of VPS35. Studies that investigate the effects of expression of PD-related mutant VPS35 are conflicting. In one study, these flies induce age-dependent loss of dopaminergic neurons and locomotion deficits [155], while another did not observe such parkinsonian-like phenotypes [156]. However, the latter group found that mutant VPS35 functions in a dominant-negative fashion, which is supported by multiple studies in fly or mouse systems [157,158,159,160]. Furthermore, knockdown of VPS35 aggravated locomotor deficits and provoked accumulation of insoluble alpha-synuclein due to defective lysosomal degradation in alpha-synuclein-expressing flies [161], while overexpression of VPS35 rescues locomotion in LRRK2- and Parkin-models [156,159].
iPLA2-VIA, the Drosophila ortholog of PLA2G6 interacts with the retromer by binding to VPS35 and VPS26 to induce protein and lipid recycling [162]. Mutations in the phospholipase gene PLA2G6 cause different neurodegenerative disorders that, next to shared defects, also result in distinct symptoms [163]. Flies deficient of PLA2G6 show defective locomotion, (dopaminergic) neurodegeneration and defective synaptic transmission [162,164,165,166]. Furthermore, mitochondrial dysfunction and membrane abnormalities are induced by loss of iPLA1-VIA [164] that are most likely secondary phenotypes following upstream defects that are related to its function as a phospholipase that hydrolyzes glycerol phospholipids. iPLA2-VIA deficiency in fly brains does not affect the phospholipid composition [162], but results in phospholipids with shorter acyl chain length [165]. The shortened acyl chain causes ER stress and alpha-synuclein aggregation that can be rescued by restoring the brain lipid composition [165]. Furthermore, loss of iPLA-VIA increases ceramide levels and lowering these ceramide levels suppresses neurodegeneration [162]. Ceramide is the basic sphingolipid synthesized in the ER. Following its transport to the Golgi, ceramide is converted to ceramide phosphoethanolamine (CPE) or sphingomyelin (SM) [167]. CPE/SM are recycled via the retromer complex [168,169] that requires binding of VPS35 to the retromer [162]. CPE/SM that is not undergoing the recycling via the retromer will be relocalized to the lysosomes for hydrolytic breakdown to ceramides and sphingolipids [170]. Impaired retromer function, via loss of iPLA2-VIA or PLA2G6, thus mitigates the CPE/SM recycling, inducing increased lysosomal ceramide levels [122,162].
Retromer depletion impairs the maturation of cathepsin D (CTSD), a lysosomal protease that functions in the metabolism of alpha-synuclein leading to lysosomal accumulation of alpha-synuclein [161]. Knockdown of VPS35 aggravates the locomotion deficits in alpha-synuclein-expressing flies [161]. Interestingly, human cells expressing alpha-synuclein show reduced VPS26 and VPS35 protein levels and increased lysosomal stress that is similar to those observed in PLA2G6 deficiency [162] suggesting alpha-synuclein plays a role in sphingolipid metabolism [122]. Mammalian alpha-synuclein cells indeed show increased ceramide levels and myriocin, to inhibit ceramide synthesis, improves alpha-synuclein-induced degeneration in flies [162]. These mechanistic studies link LRRK2, VPS35, PLA2G6 and alpha-synuclein to the retromer providing strong evidence for the retromer, an important regulator of sphingolipid metabolism [170], and therefore also of sphingolipid ceramide, to play a central role in PD pathogenesis. Interestingly, ceramide is an important regulator of autophagy [171].
5.3. The Autolysosome
Autophagy is tightly regulated. Protein aggregates or damaged organelles are targeted and packed in the autophagosome followed by the fusion of the autophagosome with lysosomes to form the autolysosome that enables the degradation of protein aggregates or damaged organelles [172]. The LRRK2-dependent phosphorylation of Endophilin A recruits Atg3 (autophagy-related 3) to promote autophagy at the synapse [140,141]. Furthermore, Synaptojanin is a phosphatase that dephosphorylates PI(3) and PI(3,5)P2, two lipids localized on autophagosomal membranes. Synaptojanin-mutant flies show defective dephosphorylation [143], indicating autophagosomal abnormalities and suggesting an important role for defective autophagy in LRRK2 PD. Autophagy-related or lysosomal proteins are important for the activation, flux and completion of autophagy, including the transmembrane lysosomal ATPase ATP13A2 [172,173,174]. Mutations in ATP13A2 lead to juvenile-onset PD, or Kufor-Rakeb syndrome [175]. A combinational study using mice and flies that are deficient for ATP13A2 show that ATP13A2 is involved in the proper fusion of the autophagosome with the lysosome in a histone deacetylase 6 (HDAC6)-dependent manner [176]. This fusion is an important step for the degradation of proteins or organelles in autophagy [177]. Interestingly, HDAC6 is protective for alpha-synuclein-induced dopaminergic degeneration [178].
6. Lipids as Connecting Factor between Mitochondrial Dysfunction and a Defective Endo-Lysosomal Pathway in Drosophila melanogaster
As discussed above, research revealed two seemingly distinct molecular pathways in PD. However, how these are connected to converge to the same signs and symptoms remains mostly enigmatic. Recently, emerging evidence highlight the importance of lipids in PD. Lipids are important for proper mitochondrial function and are key in the endo-lysosomal pathway and may constitute the missing link between mitochondria and endo-lysosomes in relation to PD. Numerous studies report lipid alterations in PD patients [4,179] and PD-related models [162,180,181]. A genome-wide association study identified sterol regulatory element-binding protein 1 (SREBP-1) as a risk locus for PD [182]. Furthermore, a screen to identify genes that promote mitophagy in Drosophila S2 cells revealed SREBP-1 to regulate mitophagy [29]. SREBP-1 is a regulator of lipogenesis and these data support distorted regulation of mitophagy to employ a risk factor for the development of PD. Furthermore, a screen to identify modifiers of Pink1, showed that partial genetic or pharmacological inhibition of fatty acid synthase 2 (FASN2) improves Pink1-deficient phenotypes that are evolutionarily conserved [181]. Lower FASN2 is indirectly linked to increased cardiolipin levels [181]. Cardiolipin is a mitochondrial-specific lipid residing at the inner mitochondrial membrane [183,184,185] that is decreased in pink1-mutant flies and supplementation of cardiolipin specifically improves the inefficient electron transfer between complex I and ubiquinone [181]. Furthermore, cardiolipin can be redistributed to the outer mitochondrial membrane to serve as a signal for dysfunctional mitochondria to undergo mitophagy [185]. Thus, cardiolipin is a lipid that serves as a link between mitochondrial function and the endo-lysosomal pathway, two mechanisms involved in PD and hence, providing evidence for lipids to connect both mechanisms in PD. Furthermore, several PD-related proteins are involved in lipid metabolism, including GCase.
6.1. Glucorerebrosidase
GBA encodes Glucocerebrosidase (GCase), a lysosomal enzyme that is key in the sphingolipid homeostasis by hydrolyzing glucosylceramide to form ceramide [186,187]. Homozygous mutations in GBA cause the lysosomal storage disorder Gaucher’s disease [188] and heterozygously mutated alleles are the most common risk factor in PD [189,190]. D. melanogaster Gba1a is predominantly expressed in the adult fly gut and Gba1b is expressed in the adult brain and adult fat body [191]. Loss of Gba1b results in locomotion defects and neurodegeneration [39]. Furthermore, changes in lipid metabolism and accumulation of glucosylceramide, its substrate, are observed [192]. Interestingly, numerous studies show GCase to interact with alpha-synuclein [189,193]. However, the deleterious effect of loss of Gba in alpha-synuclein expressing flies remains unclear [39,194]. Nonetheless, expression of the human PD-related-mutant GBA in flies induces parkinsonian phenotypes, including age-dependent locomotion defects, dopaminergic neurodegeneration and alpha-synuclein aggregation [38,40,195]. Furthermore, co-expression of mutant human GBA and alpha-synuclein worsens these symptoms compared to the expression of GBA or alpha-synuclein alone [40] providing further proof for mutant GBA to serve as a risk factor and for the importance of sphingolipids in PD pathogenesis.
6.2. Identification of Underlying Mechanisms in Non-Motor Symptoms in PD
The origin of the non-motor symptoms is poorly understood, and non-motor symptoms that pose a strong burden on the quality of life are dementia and sleep problems with the accompanying deregulation of the circadian rhythm [5,7,8]. Dementia or memory and learning difficulties have been observed in Pink1- and Parkin-deficient flies [196] and in alpha-synuclein overexpressing flies [197]; however, mechanistic insights in the development of these phenotypes have yet to be discovered. Furthermore, aged flies overexpressing the human alpha-synuclein A30P develop fragmentation of sleep that could be improved by treatment with sleep-promoting substances such as Kamikihito and Unkei-to [198]. In addition to identifying potential novel drugs to relieve sleep defects, studies in flies have also contributed to understanding the underlying mechanisms that cause these sleep defects. Using a combination of genetic tools available in the fruit fly, researchers uncovered the mechanisms underlying the deregulation of the circadian rhythm in a pink1- and parkin-mutant fly model. They identified increased ER-mitochondrial contact sites that were previously linked to PD [199], resulting in abnormal lipid trafficking followed by the depletion of the lipid phosphatidylserine at the ER. The lack of phosphatidylserine disrupts the ER-lipid balance at these ER-mitochondrial contact sites followed by the prevention of vesicle formation in neuropeptidergic neurons [200]. Interestingly, the increase of these ER-mitochondrial contact sites is sufficient to provoke sleep defects in flies [200] and the supplementation of phosphatidylserine improved these sleep defects in the PD fly models. These findings were confirmed in patient-derived fibroblasts [200], hinting towards the use of phosphatidylserine as a therapeutic agent in the alleviation of sleep problems in PD patients.
7. Validation in a Mammalian System
As discussed above, the use of D. melanogaster is valuable in the research of the underlying mechanisms in PD. One such study, identifying vitamin K2 as a modifier of the pink1-mutant phenotypes that is currently being tested as a therapeutic strategy in PD patients carrying PINK1 mutations [99]. Nonetheless, research in fly models also raises caveats that one needs to be aware of. The short life span of flies (~80 days) can be used as an advantage; however, in an age-dependent disorder such as PD, this life span may be too short to observe signs and symptoms that only arise at later stages of the disease that are not represented in flies. Furthermore, while flies do contain complex neuronal connections, these connections are not per se evolutionarily conserved to the human brain. Hence, biological processes that have only been developed and evolved in a vertebrate system will not be identified in the invertebrate fruit fly. One such example is the recent finding that the mitochondrial phenotypes following loss of Parkin or PINK1 activate the STING-dependent inflammatory response. Consequently, ablation of STING prevented the inflammatory response and the accompanying neurodegenerative phenotypes [201]. D. melanogaster carries a fly ortholog of STING (Sting) [202]; however, loss of Drosophila Sting fails to suppress the behavioral and mitochondrial defects in pink1- and parkin-mutant flies [203]. Hence, flies can be successfully used for general mechanisms, but studies in mammalian/human systems are necessary to validate the relevance in the disease context and to test specific human functions.
8. Conclusions
PD research in flies has resulted in many different findings that are relevant for patients and that have led to the identification of new therapeutic targets. Studies in flies have significantly contributed to the identification of genetic interactions between PD-related proteins affecting similar pathways that finally converge on similar PD signs and symptoms. In addition, many of the PD-related proteins exert distinct, independent functions providing an explanation for the difference in clinical presentation and suggesting stratification of therapeutic strategies in combination with a therapy that targets the main, shared PD pathogenesis.
Author Contributions
Conceptualization, M.V. and C.K.; writing—original draft preparation, M.V.; writing—review and editing, M.V. and C.K. Both authors have read and agreed to the published version of the manuscript.
Funding
Support was provided by an Else-Kröner-Fresenius-Stiftung grant (2017_A56) to M. Vos. C. Klein is supported by the DFG (FOR2488).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank Marija Dulovic-Mahlow for helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fahn, S.; Jankovic, J. Principles and Practice of Movement Disorders; Churchill Livingstone Elsevier: Philadelphia, PA, USA, 2007. [Google Scholar]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.-J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 715–724. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435. [Google Scholar] [CrossRef]
- Lee, H.; Koh, S. Many Faces of Parkinson’s Disease: Non-Motor Symptoms of Parkinson’s Disease. Mov. Disord. 2015, 8, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Marras, C.; Klein, C. Nonmotor Signs in Genetic Forms of Parkinson’s Disease. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 129–178. ISBN 9780128137086. [Google Scholar]
- Hermanowicz, N.; Jones, S.A.; Hauser, R.A. Impact of non-motor symptoms in Parkinson’s disease: A PMDAlliance survey. Neuropsychiatr. Dis. Treat. 2019, 15, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef] [PubMed]
- GBD Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [CrossRef]
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, in press. [Google Scholar]
- You, H.; Mariani, L.; Mangone, G.; Le Febvre de Nailly, D.; Charbonnier-Beaupel, F.; Corvol, J. Molecular basis of dopamine replacement therapy and its side effects in Parkinson’s disease. Cell Tissue Res. Cell Tissue Res. 2018, 373, 111–135. [Google Scholar] [CrossRef]
- Neumann, M.; Kahle, P.J.; Giasson, B.I.; Ozmen, L.; Borroni, E.; Spooren, W.; Müller, V.; Odoy, S.; Fujiwara, H.; Hasegawa, M.; et al. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J. Clin. Investig. 2002, 110, 1429–1439. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.Y. Neuronal [alpha]-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human [alpha]-Synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef]
- Yamamoto, S.; Seto, E. Dopamine dynamics and signaling in Drosophila: An overview of genes, drugs and behavioral paradigms. Exp. Anim. 2014, 63, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Nagoshi, E. Drosophila Models of Sporadic Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 3343. [Google Scholar] [CrossRef] [PubMed]
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005, 6, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, L.; Huang, X. Genome modification by CRISPR/Cas9. FEBS J. 2014, 281, 5186–5193. [Google Scholar] [CrossRef]
- Gratz, S.; Cummings, A.; Nguyen, J.; Hamm, D.; Donohue, L.; Harrison, M.; Wildonger, J.; O’Connor-Giles, K. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Webster, N.; Jin, J.; Green, S.; Hollis, M.; Chambon, P. The yeast UASG is a transcriptional enhancer in human HeLa cells in the presence of the GAL4 trans-activator. Cell 1988, 52, 169–178. [Google Scholar] [CrossRef]
- Kakidani, H.; Ptashne, M. GAL4 activates gene expression in mammalian cells. Cell 1988, 52, 161–167. [Google Scholar] [CrossRef]
- Duffy JB GAL4 system in Drosophila: A fly geneticist’s Swiss army knife. Genes Brain Behav. 2002, 34, 1–15.
- Vos, M.; Esposito, G.; Edirisinghe, J.N.; Vilain, S.; Haddad, D.M.; Slabbaert, J.R.; Van Meensel, S.; Schaap, O.; De Strooper, B.; Meganathan, R.; et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 2012, 336, 1306–1310. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Vos, M.; Vilain, S.; Swerts, J.; De Sousa Valadas, J.; Van Meensel, S.; Schaap, O.; Verstreken, P. Van Aconitase Causes Iron Toxicity in Drosophila pink1 Mutants. PLoS Genet. 2013, 9, e1003478. [Google Scholar] [CrossRef]
- Ivatt, R.; Sanchez-Martinez, A.; Godena, V.; Brown, S.; Ziviani, E.; Whitworth, A. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499. [Google Scholar] [CrossRef]
- Dulovic-Mahlow, M.; Trinh, J.; Kandaswamy, K.K.; Braathen, G.J.; Di Donato, N.; Rahikkala, E.; Beblo, S.; Werber, M.; Krajka, V.; Busk, Ø.L.; et al. De Novo Variants in TAOK1 Cause Neurodevelopmental Disorders. Am. J. Hum. Genet. 2019, 105, 213–220. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Marras, C.; Lang, A.; van de Warrenburg, B.; Sue, C.; Tabrizi, S.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Lohmann, K.; Lang, A.; Klein, C. Fixing the broken system of genetic locus symbols: Parkinson disease and dystonia as examples. Neurology 2012, 78, 1016–1024. [Google Scholar] [CrossRef]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson’s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Mazzulli, J. Is Parkinson’s disease a lysosomal disorder? Brain 2018, 141, 2255–2262. [Google Scholar] [CrossRef]
- Maor, G.; Rencus-Lazar, S.; Filocamo, M.; Steller, H.; Segal, D.; Horowitz, M. Unfolded protein response in Gaucher disease: From human to Drosophila. Orphanet J. Rare Dis. 2013, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Trinh, K.; Thomas, R.; Yu, S.; Germanos, A.; Whitley, B.; Sardi, S.; Montine, T.; Pallanck, L. Glucocerebrosidase Deficiency in Drosophila Results in α-Synuclein-Independent Protein Aggregation and Neurodegeneration. PLoS Genet. 2016, 12, e1005944. [Google Scholar] [CrossRef]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef]
- Chai, C.; Lim, K.-L. Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr. Genomics 2013, 14, 486–501. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Role of Mendelian genes in “sporadic” Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, S66–S70. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Markopoulou, K.; Putzke, J.; Whaley, N.; Farrer, M.; Wszolek, Z.; Uitti, R. Phenotypic commonalities in familial and sporadic Parkinson disease. Arch. Neurol. 2006, 63, 579–583. [Google Scholar] [CrossRef]
- Papapetropoulos, S.; Adi, N.; Ellul, J.; Argyriou, A.; Chroni, E. A prospective study of familial versus sporadic Parkinson’s disease. Neurodegener. Dis. 2007, 4, 424–427. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Qian, L.; Xiong, H.; Liu, J.; Neckameyer, W.S.; Oldham, S.; Xia, K.; Wang, J.; Bodmer, R.; Zhang, Z. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 13520–13525. [Google Scholar] [CrossRef]
- Tetrud, J.W.; Langston, J.W.; Garbe, P.L.; Ruttenber, A.J. Mild parkinsonism in persons exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurology 1989, 39, 1483–1487. [Google Scholar] [CrossRef]
- Tetrud, J.W.; Langston, J.W. MPTP-induced parkinsonism as a model for Parkinson’s disease. Acta Neurol. Scand. Suppl. 1989, 126, 35–40. [Google Scholar] [CrossRef]
- Alam, M.; Schmidt, W.J. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav. Brain Res. 2002, 136, 317–324. [Google Scholar] [CrossRef]
- Inden, M.; Kitamura, Y.; Takeuchi, H.; Yanagida, T.; Takata, K.; Kobayashi, Y.; Taniguchi, T.; Yoshimoto, K.; Kaneko, M.; Okuma, Y.; et al. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J. Neurochem. 2007, 101, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Coulom, H.; Birman, S. Chronic Exposure to Rotenone Models Sporadic Parkinson’s Disease in Drosophila melanogaster. J. Neurosci. 2004, 24, 10993–10998. [Google Scholar] [CrossRef]
- Trevor, A.J.; Castagnoli, N.; Singer, T.P. The formation of reactive intermediates in the MAO-catalyzed oxidation of the nigrostriatal toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 513–519. [Google Scholar] [CrossRef]
- Esteve-Rudd, J.; Fernandez-Sanchez, L.; Lax, P.; De Juan, E.; Martin-Nieto, J.; Cuenca, N. Rotenone induces degeneration of photoreceptors and impairs the dopaminergic system in the rat retina. Neurobiol. Dis. 2011, 44, 102–115. [Google Scholar] [CrossRef]
- Simola, N.; Morelli, M.; Carta, A. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 1, 1269. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V. Mechanisms of MPTP toxicity. Mov. Disord. 1998, 13, 35–38. [Google Scholar]
- Xiong, N.; Long, X.; Xiong, J.; Jia, M.; Chen, C.; Huang, J.; Ghoorah, D.; Kong, X.; Lin, Z.; Wang, T. Mitochondrial complex I inhibitor rotenone-induced toxicity and its potential mechanisms in Parkinson’s disease models. Crit. Rev. Toxicol. 2012, 42, 613–632. [Google Scholar] [CrossRef]
- Manneschi, L.; Dotti, M.T.; Battisti, C.; de Stefano, N.; Federico, A. Muscle respiratory chain enzyme activities in parkinson’s disease and in multisystem extrapyramidal disorders with parkinsonism as the main clinical feature. Arch. Gerontol. Geriatr. 1994, 19, 155–161. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 2009, 5, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Rube, D.; van der Bliek, A. Mitochondrial morphology is dynamic and varied. Mol. Cell. Biochem. 2004, 256, 331–339. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar] [CrossRef]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK1 phosphorylates Drp1S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef]
- Verstreken, P.; Ly, C.V.; Venken, K.J.T.; Koh, T.-W.; Zhou, Y.; Bellen, H.J. Synaptic Mitochondria Are Critical for Mobilization of Reserve Pool Vesicles at Drosophila Neuromuscular Junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.; Mohamed, H.; Wikstrom, J.; Walzer, G.; Stiles, L.; Haigh, S.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Scarffe, L.; Stevens, D.; Dawson, V.; Dawson, T. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef]
- Winklhofer, K. Parkin and mitochondrial quality control: Toward assembling the puzzle. Trends Cell Biol. 2014, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y. PINK1-Parkin signaling in Parkinson’s disease: Lessons from Drosophila. Neurosci. Res. 2020, 159, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [PubMed]
- Ziviani, E.; Whitworth, A.J. How could Parkin-mediated ubiquitination of mitofusin promote mitophagy? Autophagy 2010, 6, 660–662. [Google Scholar] [CrossRef]
- Burchell, V.; Nelson, D.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.; Pogson, J.; Randle, S.; Wray, S.; Lewis, P.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Zhou, Z.; Lee, J.; Tan, E. Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef]
- Cornelissen, T.; Vilain, S.; Vints, K.; Gounko, N.; Verstreken, P.; Vandenberghe, W. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila. Elife 2018, 7, e35878. [Google Scholar] [CrossRef]
- Lee, J.J.; Sanchez-Martinez, A.; Zarate, A.M.; Benincá, C.; Mayor, U.; Clague, M.J.; Whitworth, A.J. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 2018, 217, 1613–1622. [Google Scholar] [CrossRef]
- Vilain, S.; Esposito, G.; Haddad, D.; Schaap, O.; Dobreva, M.P.; Vos, M.; Van Meensel, S.; Morais, V.A.; De Strooper, B.; Verstreken, P. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet. 2012, 8, e1002456. [Google Scholar] [CrossRef]
- Liu, W.; Acín-Peréz, R.; Geghman, K.D.; Manfredi, G.; Lu, B.; Li, C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. USA 2011, 108, 12920–12924. [Google Scholar] [CrossRef]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Vos, M.; Lovisa, B.; Geens, A.; Morais, V.A.; Wagnières, G.; Van Den Bergh, H.; Ginggen, A.; De Strooper, B.; Tardy, Y.; Verstreken, P. Near-infrared 808 nm light boosts complex IV-dependent respiration and rescues a Parkinson-related pink1 model. PLoS ONE 2013, 8, e78562. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Lovisa, B.; Perrin, J.; Wagnières, G.; van den Bergh, H.; Tardy, Y.; Lashuel, H. Photobiomodulation Suppresses Alpha-Synuclein-Induced Toxicity in an AAV-Based Rat Genetic Model of Parkinson’s Disease. PLoS ONE 2015, 10, e0140880. [Google Scholar]
- Prasuhn, J.; Brüggemann, N.; Hessler, N.; Berg, D.; Gasser, T.; Brockmann, K.; Olbrich, D.; Ziegler, A.; König, I.; Klein, C.; et al. An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: Concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol. Res. Pract. 2019, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Kasten, M.; Vos, M.; König, I.R.; Schmid, S.M.; Wilms, B.; Klein, C.; Brüggemann, N. The Use of Vitamin K2 in Patients With Parkinson’s Disease and Mitochondrial Dysfunction (PD-K2): A Theranostic Pilot Study in a Placebo-Controlled Parallel Group Design. Front. Neurol. 2021, 11, 592104. [Google Scholar] [CrossRef]
- Sofic, E.; Paulus, W.; Jellinger, K.; Riederer, P.; Youdim, M.B. Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J. Neurochem. 1991, 56, 978–982. [Google Scholar] [CrossRef]
- Beinert, H.; Kennedy, M.C. Aconitase, a two-faced protein: Enzyme and iron regulatory factor. FASEB J. 1993, 7, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Xu, J.; Huang, Y.; Zhai, Y.; Ma, Z.; Zhou, B.; Cao, Z. Elevating bioavailable iron levels in mitochondria suppresses the defective phenotypes caused by PINK1 loss-of-function in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2020, 532, 285–291. [Google Scholar] [CrossRef]
- Pickart, C.; Eddins, M. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D.M.; Vilain, S.; Vos, M.; Esposito, G.; Matta, S.; Kalscheuer, V.M.; Craessaerts, K.; Leysen, M.; Nascimento, R.; Vianna-Morgante, A.M.; et al. Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin-dependent mitophagy. Mol. Cell 2013, 50, 831–843. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef]
- Zhu, M.; Li, X.; Tian, X.; Wu, C. Mask loss-of-function rescues mitochondrial impairment and muscle degeneration of Drosophila pink1 and parkin mutants. Hum. Mol. Genet. 2015, 24, 3272–3285. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell. Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. USA 2020, 117, 4281–4291. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Shin, J.; Ko, H.; Kang, H.; Lee, Y.; Lee, Y.; Pletinkova, O.; Troconso, J.; Dawson, V.; Dawson, T. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, S.; Yuan, C.; Khan, M.; Karuppagounder, S.; Wang, L.; Xiong, Y.; Kang, S.; Lee, Y.; Dawson, V.; Dawson, T. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol. Neurodegener. 2020, 15, 17. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.; Cha, G.; Lee, S.; Kim, S.; Chung, J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 2005, 361, 133–139. [Google Scholar] [CrossRef]
- Lavara-Culebras, E.; Paricio, N. Drosophila DJ-1 mutants are sensitive to oxidative stress and show reduced lifespan and motor deficits. Gene 2007, 400, 158–165. [Google Scholar] [CrossRef]
- Meulener, M.C.; Xu, K.; Thomson, L.; Ischiropoulos, H.; Bonini, N.M. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc. Natl. Acad. Sci. USA 2006, 103, 12517–12522. [Google Scholar] [CrossRef] [PubMed]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2, e362. [Google Scholar] [CrossRef] [PubMed]
- Martín-Nieto, J.; Uribe, M.; Esteve-Rudd, J.; Herrero, M.; Campello, L. A role for DJ-1 against oxidative stress in the mammalian retina. Neurosci. Lett. 2019, 708, 134361. [Google Scholar] [CrossRef]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef]
- Yang, J.; Kim, M.; Yoon, W.; Kim, E.; Kim, H.; Lee, Y.; Min, B.; Kang, K.; Son, J.; Park, H.; et al. Isocitrate protects DJ-1 null dopaminergic cells from oxidative stress through NADP+-dependent isocitrate dehydrogenase (IDH). PLoS Genet. 2017, 13, e1006975. [Google Scholar] [CrossRef]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.; Maddison, D.; Smith, G.; Peters, O. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Wang, L.; Marcogliese, P.; Bellen, H. Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends Endocrinol. Metab. 2019, 30, 106–117. [Google Scholar] [CrossRef]
- Kuijpers, M.; Azarnia Tehran, D.; Haucke, V.; Soykan, T. The axonal endolysosomal and autophagic systems. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Andres-Alonso, M.; Kreutz, M.; Karpova, A. Autophagy and the endolysosomal system in presynaptic function. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Giovedì, S.; Ravanelli, M.; Parisi, B.; Bettegazzi, B.; Guarnieri, F. Dysfunctional Autophagy and Endolysosomal System in Neurodegenerative Diseases: Relevance and Therapeutic Options. Front. Cell Neurosci. 2020, 14, 602116. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.; Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yu, J. Modeling Parkinson’s Disease in Drosophila: What Have We Learned for Dominant Traits? Front. Neurol. 2018, 9, 228. [Google Scholar] [CrossRef]
- Mohite, G.; Dwivedi, S.; Das, S.; Kumar, R.; Paluri, S.; Mehra, S.; Ruhela, N.; Arunima, S.; Jha, N.; Maji, S. Parkinson’s Disease Associated α-Synuclein Familial Mutants Promote Dopaminergic Neuronal Death in Drosophila melanogaster. ACS Chem. Neurosci. 2018, 9, 2628–2638. [Google Scholar] [CrossRef]
- Takahashi, M.; Kanuka, H.; Fujiwara, H.; Koyama, A.; Hasegawa, M.; Miura, M.; Iwatsubo, T. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 2003, 336, 155–158. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kawashima, A.; Ruberu, N.; Fujiwara, H.; Koyama, S.; Sawabe, M.; Arai, T.; Nagura, H.; Yamanouchi, H.; Hasegawa, M.; et al. Accumulation of phosphorylated alpha-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 2003, 62, 644–654. [Google Scholar] [CrossRef]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H.; Olkkonen, V.M. The Rab GTPase family. Genome Biol. 2001, 2, reviews3007.1. [Google Scholar] [CrossRef]
- Breda, C.; Nugent, M.L.; Estranero, J.G.; Kyriacou, C.P.; Outeiro, T.F.; Steinert, J.R.; Giorgini, F. Rab11 modulates α-synuclein-mediated defects in synaptic transmission and behaviour. Hum. Mol. Genet. 2015, 24, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Dinter, E.; Saridaki, T.; Nippold, M.; Plum, S.; Diederichs, L.; Komnig, D.; Fensky, L.; May, C.; Marcus, K.; Voigt, A.; et al. Rab7 induces clearance of α-synuclein aggregates. J. Neurochem. 2016, 138, 758–774. [Google Scholar] [CrossRef]
- Yin, G.; Lopes da Fonseca, T.; Eisbach, S.; Anduaga, A.; Breda, C.; Orcellet, M.; Szegő, É.; Guerreiro, P.; Lázaro, D.; Braus, G.; et al. α-Synuclein interacts with the switch region of Rab8a in a Ser129 phosphorylation-dependent manner. Neurobiol. Dis. 2014, 70, 149–161. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef]
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.-J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 Controls an EndoA Phosphorylation Cycle in Synaptic Endocytosis. Neuron 2012, 75, 1008–1021. [Google Scholar] [CrossRef]
- Soukup, S.; Kuenen, S.; Vanhauwaert, R.; Manetsberger, J.; Hernández-Díaz, S.; Swerts, J.; Schoovaerts, N.; Vilain, S.; Gounko, N.; Vints, K.; et al. A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 2016, 92, 829–844. [Google Scholar] [CrossRef]
- Huttner, W.; Schmidt, A. Lipids, lipid modification and lipid-protein interaction in membrane budding and fission—Insights from the roles of endophilin A1 and synaptophysin in synaptic vesicle endocytosis. Curr. Opin. Neurol. 2000, 10, 543–551. [Google Scholar] [CrossRef]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC1 domain in synaptojanin is required for~autophagosome maturation at presynaptic~terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef]
- Song, W.; Zinsmaier, K. Endophilin and synaptojanin hook up to promote synaptic vesicle endocytosis. Neuron 2003, 40, 665–667. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, W.; Lee, S.; Chung, J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun. 2007, 358, 534–539. [Google Scholar] [CrossRef]
- Dodson, M.W.; Leung, L.K.; Lone, M.; Lizzio, M.A.; Guo, M. Novel ethyl methanesulfonate (EMS)-induced null alleles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis. Model. Mech. 2014, 7, 1351–1363. [Google Scholar] [CrossRef]
- Wang, D.; Tang, B.; Zhao, G.; Pan, Q.; Xia, K.; Bodmer, R.; Zhang, Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol. Neurodegener. 2008, 3, 3. [Google Scholar] [CrossRef]
- Inoshita, T.; Arano, T.; Hosaka, Y.; Meng, H.; Umezaki, Y.; Kosugi, S.; Morimoto, T.; Koike, M.; Chang, H.; Imai, Y.; et al. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum. Mol. Genet. 2017, 26, 2933–2948. [Google Scholar] [CrossRef]
- Pfeffer, S. Membrane transport: Retromer to the rescue. Curr. Biol. 2001, 11, R109–R111. [Google Scholar] [CrossRef]
- Seaman, M. Recycle your receptors with retromer. Trends Cell. Biol. 2005, 15, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.; Cullen, P. Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perpect. Biol. 2014, 6, a016774. [Google Scholar] [CrossRef]
- Gallon, M.; Cullen, P. Retromer and sorting nexins in endosomal sorting. Biochem. Soc. Trans. 2015, 43, 33–47. [Google Scholar] [CrossRef]
- Williams, E.T.; Chen, X.; Moore, D.J. VPS35, the Retromer Complex and Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Rojas, R.; van Vlijmen, T.; Mardones, G.A.; Prabhu, Y.; Rojas, A.L.; Mohammed, S.; Heck, A.J.R.; Raposo, G.; van der Sluijs, P.; Bonifacino, J.S. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 2008, 183, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Toh, J.; Ho, P.; Tio, M.; Zhao, Y.; Tan, E.-K. In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Mol. Brain 2014, 7, 73. [Google Scholar] [CrossRef][Green Version]
- Malik, B.R.; Godena, V.K.; Whitworth, A.J. VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin. Hum. Mol. Genet. 2015, 24, 6106–6117. [Google Scholar] [CrossRef]
- Ishizu, N.; Yui, D.; Hebisawa, A.; Aizawa, H.; Cui, W.; Fujita, Y.; Hashimoto, K.; Ajioka, I.; Mizusawa, H.; Yokota, T.; et al. Impaired striatal dopamine release in homozygous Vps35 D620N knock-in mice. Hum. Mol. Genet. 2016, 25, 4507–4517. [Google Scholar] [CrossRef] [PubMed]
- Munsie, L.N.; Milnerwood, A.J.; Seibler, P.; Beccano-Kelly, D.A.; Tatarnikov, I.; Khinda, J.; Volta, M.; Kadgien, C.; Cao, L.P.; Tapia, L.; et al. Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson’s disease VPS35 mutation p.D620N. Hum. Mol. Genet. 2015, 24, 1691–1703. [Google Scholar] [CrossRef]
- Linhart, R.; Wong, S.A.; Cao, J.; Tran, M.; Huynh, A.; Ardrey, C.; Park, J.M.; Hsu, C.; Taha, S.; Peterson, R.; et al. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of Leucine-Rich Repeat Kinase 2 (LRRK2). Mol. Neurodegener. 2014, 9, 23. [Google Scholar] [CrossRef]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Miura, E.; Hasegawa, T.; Konno, M.; Suzuki, M.; Sugeno, N.; Fujikake, N.; Geisler, S.; Tabuchi, M.; Oshima, R.; Kikuchi, A.; et al. VPS35 dysfunction impairs lysosomal degradation of α-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 2014, 71, 1–13. [Google Scholar] [CrossRef]
- Lin, G.; Lee, P.-T.; Chen, K.; Mao, D.; Tan, K.L.; Zuo, Z.; Lin, W.-W.; Wang, L.; Bellen, H.J. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to α-Synuclein Gain. Cell Metab. 2018, 28, 605–618.e6. [Google Scholar] [CrossRef]
- Guo, Y.-P.; Tang, B.-S.; Guo, J.-F. PLA2G6-Associated Neurodegeneration (PLAN): Review of Clinical Phenotypes and Genotypes. Front. Neurol. 2018, 9, 1100. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.-I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. USA 2019, 116, 20689–20699. [Google Scholar] [CrossRef]
- Iliadi, K.G.; Gluscencova, O.B.; Iliadi, N.; Boulianne, G.L. Mutations in the Drosophila homolog of human PLA2G6 give rise to age-dependent loss of psychomotor activity and neurodegeneration. Sci. Rep. 2018, 8, 2939. [Google Scholar] [CrossRef]
- Panevska, A.; Skočaj, M.; Križaj, I.; Maček, P.; Sepčić, K. Ceramide phosphoethanolamine, an enigmatic cellular membrane sphingolipid. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1284–1292. [Google Scholar] [CrossRef]
- Taniguchi, M.; Okazaki, T. The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration—from cell and animal models to human disorders. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 692–703. [Google Scholar] [CrossRef]
- Choudhury, A.; Dominguez, M.; Puri, V.; Sharma, D.K.; Narita, K.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Investig. 2002, 109, 1541–1550. [Google Scholar] [CrossRef]
- Fröhlich, F.; Petit, C.; Kory, N.; Christiano, R.; Hannibal-Bach, H.-K.; Graham, M.; Liu, X.; Ejsing, C.S.; Farese, R.V.; Walther, T.C. The GARP complex is required for cellular sphingolipid homeostasis. Elife 2015, 4, e08712. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ogretmen, B. Autophagy paradox and ceramide. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 783–792. [Google Scholar] [CrossRef]
- Fujioka, Y.; Noda, N.N. Biomolecular condensates in autophagy regulation. Curr. Opin. Cell Biol. 2021, 69, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.-H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef]
- Dehay, B.; Martinez-Vicente, M.; Ramirez, A.; Perier, C.; Klein, C.; Vila, M.; Bezard, E. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy 2012, 8, 1389–1391. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Hadeed, A.; Al-Din, A.; Wreikat, A.; Lees, A. Kufor Rakeb disease: Autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov. Disord. 2015, 20, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J. Cell Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef]
- Du, G.; Liu, X.; Chen, X.; Song, M.; Yan, Y.; Jiao, R.; Wang, C.-C. Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Mol. Biol. Cell 2010, 21, 2128–2137. [Google Scholar] [CrossRef]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.-K.; Gräber-Sultan, S.; Schleicher, E.; et al. Plasma Ceramide and Glucosylceramide Metabolism Is Altered in Sporadic Parkinson’s Disease and Associated with Cognitive Impairment: A Pilot Study. PLoS ONE 2013, 8, e73094. [Google Scholar]
- Goya, M.E.; Xue, F.; Sampedro-Torres-Quevedo, C.; Arnaouteli, S.; Riquelme-Dominguez, L.; Romanowski, A.; Brydon, J.; Ball, K.L.; Stanley-Wall, N.R.; Doitsidou, M. Probiotic Bacillus subtilis Protects against α-Synuclein Aggregation in C. elegans. Cell Rep. 2020, 30, 367–380. [Google Scholar] [CrossRef]
- Vos, M.; Geens, A.; Böhm, C.; Deaulmerie, L.; Swerts, J.; Rossi, M.; Craessaerts, K.; Leites, E.P.; Seibler, P.; Rakovic, A.; et al. Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J. Cell Biol. 2017, 216, 695–708. [Google Scholar] [CrossRef]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Brody, S.; Hostetler, K. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur. J. Biochem. 1993, 212, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin Affects the Supramolecular Organization of ATP Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Vielhaber, G.; Pfeiffer, S.; Brade, L.; Lindner, B.; Goldmann, T.; Vollmer, E.; Hintze, U.; Wittern, K.; Wepf, R. Localization of ceramide and glucosylceramide in human epidermis by immunogold electron microscopy. J. Investig. Dermatol. 2001, 117, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.; Schapira, A. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef]
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: A comprehensive review. Transl. Neurodegener. 2021, 10, 4. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.; Singleton, A. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Robinson, S.W.; Herzyk, P.; Dow, J.A.T.; Leader, D.P. FlyAtlas: Database of gene expression in the tissues of Drosophila melanogaster. Nucleic Acids Res. 2013, 41, D744–D750. [Google Scholar] [CrossRef]
- Cabasso, O.; Paul, S.; Dorot, O.; Maor, G.; Krivoruk, O.; Pasmanik-Chor, M.; Mirzaian, M.; Ferraz, M.; Aerts, J.; Horowitz, M. Drosophila melanogaster Mutated in its GBA1b Ortholog Recapitulates Neuronopathic Gaucher Disease. J. Clin. Med. 2019, 8, 1420. [Google Scholar] [CrossRef]
- Brockmann, K. GBA-Associated Synucleinopathies: Prime Candidates for Alpha-Synuclein Targeting Compounds. Front. Cell Dev. Biol. 2020, 8, 562522. [Google Scholar] [CrossRef] [PubMed]
- Abul Khair, S.B.; Dhanushkodi, N.R.; Ardah, M.T.; Chen, W.; Yang, Y.; Haque, M.E. Silencing of Glucocerebrosidase Gene in Drosophila Enhances the Aggregation of Parkinson’s Disease Associated α-Synuclein Mutant A53T and Affects Locomotor Activity. Front. Neurosci. 2018, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [CrossRef] [PubMed]
- Julienne, H.; Buhl, E.; Leslie, D.S.; Hodge, J.J.L. Drosophila PINK1 and parkin loss-of-function mutants display a range of non-motor Parkinson’s disease phenotypes. Neurobiol. Dis. 2017, 104, 15–23. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The New Mutation, E46K, of Alpha-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Ito, K.; Kawasaki, H.; Suzuki, T.; Takahara, T.; Ishida, N. Effects of Kamikihito and Unkei-to on Sleep Behavior of Wild Type and Parkinson Model in Drosophila. Front. Psychiatry 2017, 8, 132. [Google Scholar] [CrossRef]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [PubMed]
- Valadas, J.S.; Esposito, G.; Vandekerkhove, D.; Miskiewicz, K.; Deaulmerie, L.; Raitano, S.; Seibler, P.; Klein, C.; Verstreken, P. ER Lipid Defects in Neuropeptidergic Neurons Impair Sleep Patterns in Parkinson’s Disease. Neuron 2018, 98, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Martin, M.; Hiroyasu, A.; Guzman, R.M.; Roberts, S.A.; Goodman, A.G. Analysis of Drosophila STING Reveals an Evolutionarily Conserved Antimicrobial Function. Cell Rep. 2018, 23, 3537–3550.e6. [Google Scholar] [CrossRef]
- Lee, J.J.; Andreazza, S.; Whitworth, A.J. The STING pathway does not contribute to behavioural or mitochondrial phenotypes in Drosophila Pink1/parkin or mtDNA mutator models. Sci. Rep. 2020, 10, 2693. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).