Abstract
Symptomatic treatments are available for Parkinson’s disease and Alzheimer’s disease. An unmet need is cure or disease modification. This review discusses possible reasons for negative clinical study outcomes on disease modification following promising positive findings from experimental research. It scrutinizes current research paradigms for disease modification with antibodies against pathological protein enrichment, such as α-synuclein, amyloid or tau, based on post mortem findings. Instead a more uniform regenerative and reparative therapeutic approach for chronic neurodegenerative disease entities is proposed with stimulation of an endogenously existing repair system, which acts independent of specific disease mechanisms. The repulsive guidance molecule A pathway is involved in the regulation of peripheral and central neuronal restoration. Therapeutic antagonism of repulsive guidance molecule A reverses neurodegeneration according to experimental outcomes in numerous disease models in rodents and monkeys. Antibodies against repulsive guidance molecule A exist. First clinical studies in neurological conditions with an acute onset are under way. Future clinical trials with these antibodies should initially focus on well characterized uniform cohorts of patients. The efficiency of repulsive guidance molecule A antagonism and associated stimulation of neurogenesis should be demonstrated with objective assessment tools to counteract dilution of therapeutic effects by subjectivity and heterogeneity of chronic disease entities. Such a research concept will hopefully enhance clinical test strategies and improve the future therapeutic armamentarium for chronic neurodegeneration.
1. Introduction
One of the main causes for disability in humans worldwide is onset of neurological disorders, such as stroke and chronic progressive neurodegenerative brain diseases (PND). The most prevalent PNDs are the idiopathic and genetic Parkinson’s disease entity (PD) and the complex of various dementia syndromes, mainly consisting of Alzheimer’s disease (AD), frontotemporal dementia (FTD), mixed dementia (MD), and vascular dementia (VD) [1,2,3]. They are characterized by a common pathophysiologic mechanism, which is aberrant protein aggregation. Well known neuropathological features are β-amyloid and tau-protein enrichment in AD and accumulation of misfolded α-synuclein in PD [4,5]. Incidence of these PNDs will further rise. As an example, estimates of PD prevalence showed a 2.4-fold rise in the last 30 years. Main reasons are an earlier diagnosis associated with better treatment quality and a general rise of human life expectancy [6]. Increased exposure to endogenous and exogenous toxins contributes to a slowly evolving neurodegeneration in the peripheral and central nervous system and accelerates the overall ageing process in a pathological PND related manner. Typical risk factors are pesticides or herbicides, paraquat, rotenone, various metals (i.e., iron, manganese, and lead), gaseous compounds (such as carbon monoxide) and even viruses [7,8]. The rising number of dementia and PD patients will increase the financial burden for health care systems worldwide. To date, it is far from clear, whether the current SARS-CoV-2 outbreak may cause PND like syndromes in the long run, similar to the observed symptomatic PD forms following the 1918 H5N1 influenza pandemic [9]. This review aims to scrutinize current research paradigms for disease modification in PNDs, particularly in PD and dementia syndromes. It scrutinizes current approaches with antibodies against pathological protein enrichment, such as α-synuclein, amyloid, tau, based on post mortem findings. It will finally suggest a more uniform, disease independent therapeutic approach, which aims on neuronal regeneration and repair in the peripheral and central nervous system.
The Current Situation and Unmet Needs
Considerable research activities in the past 60 years have focused on symptomatic therapies for alleviation of PD. A success story was the introduction of the dopamine substitution concept. It alleviates motor and to a considerable extent associated non motor symptoms in PD since the 1960s [10,11]. At that time Levodopa (L-dopa) was initially applied in an appropriate dose. The introduction of L-dopa therapy was based on findings, that high dopamine levels exist in the basal ganglia and that the dopamine precursor L-dopa counteracts reserpine induced dopamine decrease and associated impaired motor behavior (for review: [12]). The subsequent advancement with the launch of dopamine agonists or inhibitors of L-dopa metabolism reflect a consequent further development of this initial therapeutic principle [13]. These drug alternatives aim to spare oral L-dopa dosing. Reasons for this approach were the still ongoing controversy on L-dopa neurotoxicity due to L-dopa induced oxidative stress, L-dopa related impairment of methylation capacity with consecutive acceleration of ageing processes and the onset of mainly oral L-dopa associated motor complications in PD patients [14,15,16,17]. The future availability of subcutaneous L-dopa pump systems with their more continuous L-dopa brain delivery will noticeably reduce oral L-dopa associated fluctuations of motor and associated non motor behavior in PD. This problem is in the foreground of current clinical PD drug development [18,19]. Similar to PD, non-motor symptoms also gained more and more interest in recent years in dementia. A considerable overlap exists between mechanisms of disease progression between PD and dementia syndromes. Thus, the former focus on the dopamine deficiency in PD, respectively the acetylcholine deficit in AD is superseded by a more widespread view. It also considers the individual different decline of other neurotransmitter systems, like serotonin (5-HT) or norepinephrine [20,21] Generally, particularly AD and PD are related to each other, i.e., by signs of microglial activation and neuroinflammation, and even in terms of neuropathological abnormalities [22,23,24] (Table 1). Similar therapeutic approaches are also employed. As an example, acetylcholine esterase inhibiting compounds and glutamate neurotransmission reducing drugs improve cognitive abilities not only in AD, MD, and VD but also in PD plus dementia syndromes [25,26,27].

Table 1.
Interactions of various pathological proteins and disease.
2. Pitfalls of Translational Concepts in Clinical Research
To date, extensive experimental and neuropathological research provided distinct and better insights and understanding of chronic neuronal and associated glial cell death. The predominant responsible and final mechanism cascades are well identified and described in detail [28]. Based on these findings, i.e., antiapoptotic, neuroprotective or oxidative stress reducing compounds, were successfully tested in experimental chronic neurodegenerative and inflammatory disease models [4,29,30,31] (Figure 1).
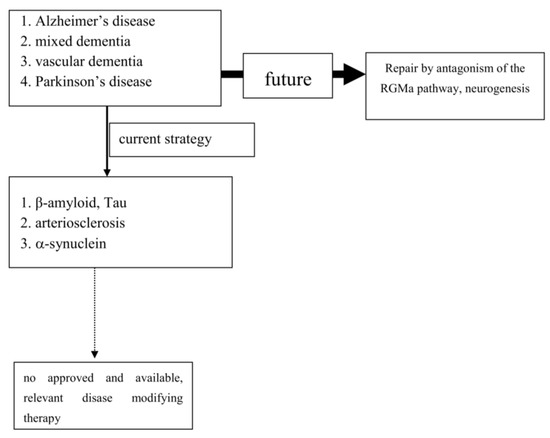
Figure 1.
Past and future concepts for disease modification in progressive neurodegenerative brain diseases (PND).
However, translation into positive clinical study results has failed so far, as trials on cure or disease modification in PNDs were more or less negative. Even transplantation of neurons or administration of neuronal growth factors was negative (as examples: [32,33]). Stimulation of growth factor synthesis, gene modification, and stem cell applications are still discussed as promising tools [34,35,36,37,38,39,40,41,42]. The unmet need for disease modification, respectively repair regeneration for central nervous system disorders, still exists. No therapy has been approved yet. One reason for these failures may be that chronic neurodegenerative processes result from different heterogeneous, but each other complementing metabolic, pathological cascades (as examples for ground breaking research: [43,44,45] for review: [4,5]). All of them end up in neuronal cell death inducing events, such as apoptosis as the suicide programme of the cell [28,29]. However, the processes, which cause chronic neuronal dying, vary. Moreover neuronal death results in an individually pronounced and variable expression of symptoms in patients. Features of personality, socioeconomic factors, such as education, business conditions etc. may interfere with PND related clinical deficits. The “Neuroplasticity” concept has been suggested to be responsible for the compensation of deleterious metabolic processes and the delayed occurrence of symptoms [46]. These aforementioned heterogeneous components interfere with the value of assessment tools in clinical trials and may dilute potential positive effects of a therapeutic intervention. To demonstrate the benefit of disease modification, validated clinical rating scales were mostly used, sometimes even in combination with functional imaging techniques, i.e., visualization of the dopamine neurotransmission in PD (Table 2).
Table 2.
Important trials in Parkinson’s disease, which aimed on modification of progression.
Another critical issue is the therapeutic mode of action, which is utilized for disease modification or cure. As an example, antibodies against pathological misfolded proteins were developed based on neuropathological findings. Enrichment of these altered proteins, i.e., in Lewy bodies (LB) or plaques, are looked upon as the main responsible and important, pathological phenomenon in chronic neurodegenerative brain disorders, such as AD or PD [58]. Failures within physiologic activities of protein metabolism may cause protein degradation and misfolding. However, it is far from clear, whether these abnormalities represent a specific process, which is responsible for disease onset and progression [59]. This pathologic protein accumulation may also be the result of an unspecific side reaction of the metabolic cascade during chronic neurodegenerative processes. It may hypothetically only represent well wrapped protein garbage as consequence of physiologic defence mechanisms [59]. The extent of compensatory capacity, the triggering causes and the moment of initiation of these misfolded protein enrichments during the disease process are not known in detail. However, there is consensus that an essential clinical precondition for disease modifying therapeutic concepts is an early diagnosis, when the disease caused damage is low. Accordingly, biomarkers and/or identification of a genetic predisposition may be excellent tools to screen for PND or PND-at risk-individuals. Their availability may theoretically enable a prodromal diagnosis before the onset of motor symptoms (PD) or cognitive decline (AD). To date, PD and AD are mostly diagnosed relatively late in the disease process due to the compensatory “neuroplasticity” phenomenon in the brain. A treatment allocation following earlier prodromal diagnostic screening will also probably reduce the current abundant missing motivation of PND-at-risk individuals for a testing procedure [60]. A positive test outcome without a causal therapeutic approach may cause a heavy burden for further life. Therefore one may scrutinize, whether the subsequently described clinical research pattern for disease modification is appropriate in chronic neurodegeneration, such as dementia or PD.
3. Dementia Syndromes
Post mortem neuropathological brain investigations describe an accumulation of plaques and tangles with β-amyloid- and/or tau protein pathology in AD. Dystrophic neuritis, astrogliosis, neuropil threads, and microglial activation with neuroinflammation has also been reported [22,23,24,61]. These changes result in an acetylcholine deficit, which is mainly looked upon as responsible for the cognitive impairment. Morphological and functional imaging techniques, such as magnetic resonance imaging tomography or positron emission tomography (PET) with various radiotracers, were developed for visualization of brain function and neuroinflammation [49,62]. Particularly, PET shall help to interrogate the biological mechanisms of disease initiation, progression, and assessment of successful, potential future disease modifying therapies. It also serves as a diagnostic tool. In AD trials, PET is used for a better patient cohort characterization. The enrichment of the radiotracer [18F]-AV-45 is employed for the determination of β-amyloid plaque density [63,64,65]. This approach visualizes “pure” AD forms, particularly in combination with screening for genetic risk factors. The most known one is the ApoE polymorphism. There are three major isoforms, ApoE2, ApoE3, and ApoE4. They impact the risks for developing AD. Carriers of the homozygous ApoE4 allele have the highest AD risk, ApoE3 is considered as normal and ApoE2 is looked upon as protective [66,67]. Accordingly, preclinical researchers developed genetically predisposed AD models. Novel AD drugs were screened and tested in these uniform AD models, designed on the basis of a specific genetic predisposition or acetylcholine deficiency. As an example, antibodies against β-amyloid aggregation or tau pathologies were first successfully investigated in AD animals [68,69]. These experimental investigations proved their efficacy. However compounds failed in clinical trials (for review: [68,69]). This is no surprise for clinicians involved in the real world maintenance of dementia patients. The variability of symptoms, observed in the clinical maintenance of AD patients, was not considered. The most common dementia syndrome represents a mixture between vascular dementia and AD on top of additional manifestations of further comorbidities, i.e., diabetes, hypertension, metabolic syndrome, etc. All of them predispose for dementia and acceleration of ageing. Out of this spectrum of dementia forms, AD is looked upon as the most popular form of dementia. AD allows a specific therapeutic approach based on one pathophysiologic mechanism [5,27]. The strict inclusion criteria of clinical trials mostly try to select “pure” AD patients with nearly no concomitant disorder. However, even this study population represents a broader spectrum of clinical symptoms with individual differing rate of progression. Efficacy of tested compounds is mainly determined with repeat performance of standardised neuropsychological cognitive batteries and clinical rating scales. Both of them were suitable to demonstrate symptomatic effects in trials on the cognition enhancing effects of acetylcholinesterase inhibitors [25,26,68]. However, performance training, which is often executed during dementia trials by caregivers and patients, may dilute their value. As example, the Mini Mental State Examination is often used as selection criterion in dementia trials, but its scores have a considerable bias by the educational level of the patient [70,71,72,73]. Instruments, like the neuropsychiatric inventory, lack of assessment of psychopathological phenomena in relation to exogenous influencing factors, i.e., well-being of caregivers, etc. Does this laboratory study condition within a clinical trial reflect real life of health care in AD? Clinicians point out that non cognition related signs, i.e., motor signs, are further essential quality of life limiting factors and influence progression [74]. To date, the suitability of these assessments for verification of disease modification was scrutinized only to a certain extent. However, use of these clinical rating scales may be responsible for the failed translation of therapeutic interventions on tau- and β-amyloid metabolism from successful preclinical experiments into positive clinical studies in AD. These considerations may be additional reasons, why the Phase III study programs with the two anti-Aβ monoclonal antibodies, bapineuzumab, and solanezumab, were negative [25,26,68]. The discussion on a possible approval of aducanumab, which showed mild effects in faster progressing AD patients, is still ongoing. In contrast to these aforementioned approaches in diagnosed patients, another promising preventive concept is vaccination. Aggregated β1-42-amyloid was employed in animal models for active immunization. Clinical vaccination trials were stopped due to onset of meningoencephalitis in up to 6% of the participants [75,76,77,78]. As a next step, a shift from active to passive immunization is now under way in AD models [75]. It is regarded as more safe, to be better controlled and may also be efficacious [79,80,81,82]. However, will vaccination against aggregated β-amyloid plaques be beneficial for other pathological processes associated with AD, like the tau-protein pathology?
Pragmatism of Clincians
Currently the only way to counteract AD onset or to modify the course of AD is prevention of the major AD risk factors. Arteriosclerosis disease, type II diabetes, midlife hypertension, midlife obesity, metabolic syndrome, smoking, and physical inactivity are looked upon the most common ones. Reduction of these life style associated predisposing risk factors with preventive, more educational approaches is suggested for dementia-at risk-individuals [83,84].
4. Parkinson’s Disease
Currently it seems that these misconceptions of AD research will be repeated in PD. PD is mostly diagnosed, when approximately 60% of dopaminergic axons and 30% of nigral dopaminergic neurons are already gone. At that stage motor symptoms appear [85]. Accordingly one may scrutinize, whether testing of a disease modifying therapy for the remaining 70% dopamine synthesizing neurons in the most affected nigrostriatal area in PD may provide clinical relevant changes for the future disease course at all. The still surviving neurons have already lost most of their axons. Therefore they are primed for cell death. Better awareness for non-motor signs and initiatives for earlier detection of PD were initiated. They shall characterize the so-called “prodromal” or “premotor” interval. Heterogeneity of symptoms and the individually differing progression complicates the inauguration of a clinical, reliable classification concept of early PD signs. Moreover various subtypes of the disease entity PD exist [86,87]. No validated, reliable, specific, easy to apply biomarker for an earlier detection of PD or a PD screening was found to date. Still, the LB accumulation with misfolded α-synuclein protein aggregation is looked upon as the most important neuropathologic PD hallmark. Related syndromes, such as Parkinson’ disease dementia (PDD) and dementia with Lewy bodies (DLB), also show these neuronal inclusions. A widely cited model of the dopaminergic neuron degeneration, the Braak model, even considers PD as α-synucleinopathy and sets LB pathology as the essential lime light [88,89,90]. This process of slow neuronal dying follows a certain pattern, for instance starting in the gastrointestinal tract, rising over the brainstem with further spreading all over the brain. This hypothesis is matter of controversy [90,91,92]. No correlations between LB pathology and loss of dopamine generating cells or clinical features of PD have been found yet. No correlations between Braak staging and dopaminergic neuron density in the substantia nigra appeared [93]. These negative results may hypothetically suggest that LB pathology is not essential for nigral degeneration in PD. Post mortem investigations in Leucine-rich repeat kinase 2 (LRRK2)- and Parkin-related PD patients or in PD models with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced nigrostriatal neuronal loss and dopamine deficit show no LB occurrence [94]. Currently employed PD models mainly focus on the loss of dopamine synthesizing nigral neurons. However, chronic neuronal dying also takes place in other neurotransmitter systems in PD, both in the periphery and in the brain [91]. Various neuronal cell types degenerate in an individually different and pronounced manner in various brain regions. This is mirrored by a heterogeneous compilation of motor and non-motor symptoms in PD patients [95,96,97]. Employed in vitro and in vivo PD models mimic the chronic progressive, slow sometimes relapse like progression of neuronal dying to a limited extent. Toxin models with, e.g., 6-OH-dopamine, rotenone, or MPTP application, induce nigrostriatal dopamine deficiency [98]. They only reflect the motor impairment in PD and often result from an acute event after a singular toxin administration. Accordingly, these models mirror the various therapeutic effects of dopamine substitution even in combination with PET, which employs radiotracers for the nigrostriatal dopamine neurotransmission. These functional imaging techniques are also employed in the clinical routine, i.e., as instrument for the confirmation of diagnosis. They are able to mirror PD progression to a certain extent. Effective disease modification was shown in long term studies with PD patients. They showed a lower advance rate with dopamine agonist treatment alone compared with L-dopa/dopadecarboxylase inhibitor (DDC-I) administration as monotherapy (as an example: [99]). This technique was additionally employed for the comparison of disease progression during treatment with two L-dopa/DDC-I dosages against placebo. Progression was faster in the 600 mg L-dopa/carbidopa arm after nine months compared with 300 mg and placebo [100]. No difference appeared, when the effects of dopamine agonist monotherapy versus placebo treatment were investigated in a clinical long term trial with a delayed start design with the same imaging technique as tool for determination of PD progression [48]. The combination of the employed assessment techniques in these aforementioned trials showed that performance of disease modification is possible in chronic neurodegeneration. Avoidance of L-dopa, respectively low L-dopa dosing, is beneficial in the long run in PD, which is still matter of debate [14,15,16,17]. Clinical evidence for a disease-modification/neuroprotective action was also investigated with the MAO-B inhibitors selegiline and rasagiline with various clinical defined endpoints only without the use of expensive functional imaging techniques. The subsequent description of these clinical trials will mirror the complexity of these trials and the interpretation of their outcomes.
4.1. Excurs: Clinical Research on Disease Modification in PD with MAO-B Inhibition
DATATOP (Deprenyl and tocopherol antioxidative therapy of parkinsonism) was a prospective, randomized, double-blind, placebo-controlled trial with 800 PD patients. Treatment naïve PD patients were randomised to either selegiline or α-tocopherol (vitamin E), a combination of both or placebo. Then they were followed until clinical deterioration asked to initiate an additional symptomatic L-dopa therapy. This was interpreted as primary endpoint. Selegiline, but not α-tocopherol, delayed L-dopa treatment compared with placebo after 15 months. The trial was stopped, when 24.3% of participants taking selegiline compared with 43.9% of those without selegiline reached the primary endpoint. Then a change of protocol was performed. Therapy was withdrawn from 367 participants, who did not meet the primary endpoint. These PD patients were evaluated again after one and two months. From baseline to the end of the washout, patients under selegiline therapy showed a slower disease progression according to their Unified Parkinson’s Disease Rating Scale (UPDRS) outcomes, compared with those without selegiline. In comparison to patients without selegiline therapy, the ones on selegiline were slightly better during the washout period in UPDRS part III (motor examination). In the subgroup of DATATOP patients who were not on L-dopa yet (n = 310), no benefit of early initiation of selegiline therapy was found. Those patients with selegiline therapy since trial initiation reached the endpoint of necessary L-dopa therapy sooner than those who had switched from placebo to selegiline. A total of 368 of the DATATOP participants on the L-dopa/carbidopa plus selegiline regimen were further randomised to continue with selegiline or to switch to placebo. The patients on selegiline deteriorated slower compared with those who were randomized to placebo. These results only suggest a beneficial effect of selegiline on disease progression once the patients were on supplemental L-dopa therapy. No beneficial effect was found with vitamin E therapy. The main limitation of this study was a potential confounding symptomatic effect of selegiline on outcomes in combination with the observed placebo effects [56]. SIN-DEP-PAR (Sinemet-Deprenyl-Parlodel) was a prospective, randomized, double-blind study. It randomized 101 patients with mild to moderate PD to either selegiline (10 mg/day) or placebo. Three days later either L-dopa/carbidopa or bromocriptine was supplemented and dosed based on clinical response. Patients on bromocriptine were allowed to add L-dopa/carbidopa if necessary after an escalation of bromocriptine to 20 mg/day or the maximally tolerated dose. Both cohorts were matched and remained in this study for 12 months. Then, a two-month lasting washout period followed. L-dopa and bromocriptine were stopped one week before the final scoring at month 14. At the final evaluation, selegiline treated patients showed less worsened disability compared with the group, which was not on selegiline (0.4 versus 5.8 points on total UPDRS score; p < 0.001). This effect was particularly observed in L-dopa/carbidopa treated patients. Patients on selegiline (p < 0.01) deteriorated less (total UPDRS score: −1.7) compared with those on placebo (+4.8). A total of 23 patients were again rated 2 weeks after stop of L-dopa/carbidopa and bromocriptine again. Selegiline treated patients had less change in their UPDRS scores from baseline compared the ones on placebo [57]. The SELEDO (from selegiline plus L-dopa) trial was a prospective, double-blind study over an interval of five years, which investigated the efficacy of selegiline addition to L-dopa. An optimum titration of L-dopa to individual requirements was done at study start. Then the 116 PD patients were put on either selegiline or placebo. The subsequent necessary L-dopa dose escalation depended on the clinical features. The primary endpoint was defined as the moment when the needed L-dopa dose to control symptoms was 150% or more of the initial dose. Median time to reach this endpoint was 4.9 years in the selegiline group in comparison with 2.6 years in the controls. After five years of treatment, only 50.4% of the selegiline treated patients reached this endpoint in comparison with 74.1% of the placebo treated participants. Both outcomes were significant [101]. These aforementioned selegiline long term trials showed that combination of selegiline and L-dopa provides a greater clinical benefit and less progression than L-dopa monotherapy, when the need for L-dopa is looked upon as an indirect clinical biomarker for disease severity. A certain beneficial effect on the course of the disease was shown with the 1 mg dose in the ADAGIO trial but not with the 2 mg daily dose of rasagiline employing a delayed start design. There are a lot of hypotheses on the failure of the 2 mg arm, which is not explained to date. This controversial outcome only occurred in the ADAGIO (A Randomized Placebo Controlled Study to Show That Rasagiline May Slow Disease Progression for Parkinson’s Disease)—but not in the earlier performed TEMPO (A controlled trial of rasagiline in early Parkinson disease) study. TEMPO showed beneficial effects of rasagiline on disease progression. However, the baseline UPDRS values and durations of disease were lower in the ADAGIO-((UPDRS): 20.4(8.5) (median(SD)); (duration of disease): 4.5(4.6) (months)) than in the TEMPO-participants (25.0(10.84); 12.1(13.2)) [51,52,102]. To date, a certain disease modifying effect of these MAO-B inhibitors is still under discussion and has not been approved as disease modifying effect (Table 2). Duration of trials, sensitivity of applied rating instruments and inconsistency of outcomes are important arguments.
4.2. Current Ongoing Clinical Research Strategies on Disease Modification in PD
These aforementioned studies included patients with sporadic PD [58]. Thus, environmental epigenetic influences, chronic toxin exposure or other still unknown for instance viral or bacterial infections may be main hypothetical causes for PD onset. In the past, these sporadic PD patient cohorts were also employed for performance of transplantation trials with fetal dopamine synthesizing neurons or inhibition of progression with possible disease course modifying therapies (as examples: [33,40]). They were negative, similar to the aforementioned neuroprotection trials in PD patients with selegiline, rasagiline and free radical scavengers, like coenzyme Q10 or tocopherol [47,52,56,103] (Table 2). Currently new attempts are under way with less heterogeneous patient cohorts based on genetic vulnerability factors for PD. Generally, there is also no doubt, that a certain genetic impact is present in some forms of PD. Experimental research demonstrated the corresponding pathological pathways following the initial description of α-synuclein mutations. More than 20 so-called “PD genes” with a different extent of penetration are known nowadays and are allocated to be responsible for sporadic PD. Genetic alterations and mutations in familial PD forms, such as SNCA, Parkin, LRRK2, DJ-1, PINK-1, and UCHL-1, only account for approximately 10% of idiopathic PD patients. Age of onset and clinical symptoms are variable, as example convincingly demonstrated in Glucocerebrosidase (GBA) mutation carriers [104,105,106,107,108]. Neuroprotection trials are again under way in these GBA mutation carrying PD patients. Currently there are also new research initiatives under way in PD with antibodies against α-synuclein based on the corresponding neuropathological findings of protein misfolding in Lewy bodies [109,110]. Various drug mechanisms aim to reduce misfolded α-synuclein and thus disease progression. They focus on boosting of autophagic/lysosomal clearance, reduction of α-synuclein mRNA by modulating histone deacetylase or RNA interference with decreased expression of α-synuclein [58]. Further therapeutic concepts focus on the impeding of the α-synuclein multimerization with heat shock proteins, dissociation of existing misfolded α-synuclein aggregates with small molecules, blocking of α-synuclein entry through receptor blocking, prevention of α-synuclein transport from cell to cell and immunotherapy with neutralization of α-synuclein extracellularly or intracellularly with nanobodies [111]. There is one but essential disadvantage. The basis for this therapeutic concept is always a more or less singular molecular pathology drug approach based on a neuropathological post mortem finding. Can one really assume that this will be sufficient to slow down progression of sporadic PD? Its onset and advance are characterized by various triggers [95]. They have a multifactorial, possibly multigenetic origin. Further different pathological factors for disease progression exist. All of them are responsible for a heterogeneous compilation of clinical symptoms and their progression. Similar to AD, vaccination as preventive approach is also discussed, but outcomes of a vaccination trial in PD are not reported yet.
5. Conclusions
Cure or modification of progression in dementia, particularly AD and PD, is an important unmet need. Clinical trials, which aimed to translate promising experimental research outcomes into relevant positive results, were negative. No therapy has yet been approved despite well identified final main neuronal and related glial cell death mechanisms mostly on the cellular level. Multifactorial origins, heterogeneity of clinical symptoms, variability of each other complementing disease mechanisms and progression are the mostly likely reasons.
6. Outlook
Experimental research convincingly described a number of cellular pathways leading to chronic neuronal degeneration and death in PNDs. Examples are mitochondriopathy, dysfunction of the ubiquitin/proteasome system, oxidative and nitrosative stress, dysregulation of heat shock response, altered iron metabolism and vesicular transport systems, apoptosis, necrosis, autophagy, microglial activation combined with neuroinflammation [23,62,112]. Therapeutic concepts were, and are, developed based on findings, such as that the pro-inflammatory TNF-alpha cytokine is able to modify neuronal plasticity, maturation, and function of human cholinergic neurons also by epigenetic mechanisms [61]. All of these disease related alterations and their possible therapies will consequently change neurotransmission pathways [113]. However to date, preventive or PND progression delaying therapeutic strategies failed following translation into clinical study programs (as an example: [47]). Even the current clinical testing of specific antibodies against certain proteins, which accumulate in the various sporadic PNDs subtypes, may probably fail as shown in AD. This may suggest that the enrichment of these proteins is not specific. Their accumulation overlaps between various clinical PND pictures. This protein enrichment in LB may hypothetically only represent a defence mechanisms against the disease process itself, but do not cause it (Table 1; [114]). To date all clinical studies, which aimed to demonstrate neuroprotection or disease modification, i.e., in PD and AD, showed that research on a specific pathological disease mechanism does not lead to an essential therapeutic innovation in terms of disease course modification (Figure 1). Therefore, the underlying research method is worth to be considered for a modification. As an alternative to this misconception, one may consider the stimulation of an existing repair system in the peripheral and central nervous system as a more promising research paradigm [115,116,117,118,119,120]. Therapeutic strategies, which antagonize the repulsive guidance molecule A (RGMa) pathway, are worth for further development in clinical trials. A RGMa increase in the substantia nigra was found by in situ hybridization and immunohistochemistry in neuromelanin-positive neurons in post-mortem tissue of treated PD patients [116]. It may also be related to L-dopa administration and associated oxidative stress generation to a certain extent [44,121]. Extracellular RGMa inhibits axon regeneration and therefore may accelerate demise of neurons [122,123,124]. However, targeting the RGMa pathway with antibodies or neutralization, respectively, antagonism of the neogenin receptor activity, may start regeneration not only in acute, but also in chronic inflammatory and neurodegenerative disorders [46,125,126,127,128] (Figure 1). It is well known, that considerable metabolic similarities exist both in the peripheral and central nervous system. Therefore, it is hypothesized that other syndromes than PD and AD, may also respond to this approach [118,120,123,129,130]. It may restore neuronal function in the long term as a general concept for repair and may weaken efficiency of toxin exposure [46,116,117,125,126,131,132,133]. Well designed, clinical long term trials with RGMa antagonizing approaches are urgently needed in multiple sclerosis, PD, dementia syndromes, stroke, or spinal cord injury. Neuropathies (NP), diabetic retinopathy, Guillian Barre syndrome and amyotrophic lateral sclerosis are particularly suitable disorders. They allow testing of this approach in rather homogenous, well defined study cohorts with objective assessment tools, such as visual function and visual evoked potentials in retinopathy, or sensory or motor nerve conduction assessment in NP. RGMa antagonism may probably also help to counteract heterogeneous neurological deficits as consequence from severe viral infections, including SARS-CoV-2. Currently two different neutralizing RGMa antibodies (ABT-555; MT-3921) are in phase 2 clinical trials in spinal cord injury. In addition ABT-555 is in phase 2 clinical trials in progressive and relapse-remitting multiple sclerosis and in ischemic stroke. A positive outcome of these clinical trials will support this strategy for regeneration and repair in the damaged human nervous system. A further important pathological mechanism-of-action in PNDs is the potential inhibition of neurogenesis by the RGMa-neogenin pathway also in PD and dementia syndromes [115,118,119,120,134]. Neurogenesis also occurs in the adult human brain, i.e., in the dentate gyrus or the subventricular zone. RGMa blocks neurogenesis in these areas [123,135]. As shown in the hippocampal dentate gyrus, blocking of RGMa promoted formation of new neurons [115]. Targeting RGMa by antibodies may promote neurogenesis in the adult human brain of PND patients. An increased neurogenesis may also improve motor symptoms in PD or cognitive deficits in dementia. This is an alternative to cell replacement and stem cell concepts. Both have a focus on specific cell types only in contrast to the potential of RGMa antagonism in chronic neurodegeneration [34,35,49,136,137,138,139].
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Nichols, E.; Szoeke, C.E.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Deuschl, G.; Beghi, E.; Fazekas, F.; Varga, T.; Christoforidi, K.A.; Sipido, E.; Bassetti, C.L.; Vos, T.; Feigin, V.L. The burden of neurological diseases in Europe: An analysis for the Global Burden of Disease Study 2017. Lancet Public Health 2020, 5, e551–e567. [Google Scholar] [CrossRef]
- Chaplot, K.; Jarvela, T.S.; Lindberg, I. Secreted Chaperones in Neurodegeneration. Front. Aging Neurosci. 2020, 12, 268. [Google Scholar] [CrossRef]
- Gracia, P.; Camino, J.D.; Volpicelli-Daley, L.; Cremades, N. Multiplicity of α-Synuclein Aggregated Species and Their Possible Roles in Disease. Int. J. Mol. Sci. 2020, 21, 8043. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Abushouk, A.I.; Gabr, M.; Negida, A.; Abdel-Daim, M.M. Parkinson’s disease and pesticides: A meta-analysis of disease connection and genetic alterations. Biomed. Pharmacother. 2017, 90, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ma, T.; Qu, B.; Ji, Y.; Liu, Z. Pesticide-Induced Gene Mutations and Parkinson Disease Risk: A Meta-Analysis. Genet. Test. Mol. Biomark. 2013, 17, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Ter Meulen, V. Coronaviruses: A challenge of today and a call for extended human postmortem brain analyses. J. Neural Transm. 2020, 127, 1217–1228. [Google Scholar] [CrossRef]
- Birkmayer, W.; Hornykiewicz, O. The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wien. Klin. Wochenschr. 1961, 73, 787–788. [Google Scholar] [PubMed]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. Modification of Parkinsonism—Chronic treatment with L-dopa. N. Engl. J. Med. 1969, 280, 337–345. [Google Scholar] [CrossRef]
- Carlsson, A. Biochemical and pharmacological aspects of Parkinsonism. Acta Neurol. Scand. Suppl. 1972, 51, 11–42. [Google Scholar] [PubMed]
- Tolosa, E.; Marti, M.J.; Valldeoriola, F.; Molinuevo, J.L. History of levodopa and dopamine agonists in Parkinson’s disease treatment. Neurology 1998, 50 (Suppl. 6), S2–S10. [Google Scholar] [CrossRef]
- De Bie, R.M.A.; Clarke, C.E.; Espay, A.J.; Fox, S.H.; Lang, A.E. Initiation of pharmacological therapy in Parkinson’s disease: When, why, and how. Lancet Neurol. 2020, 19, 452–461. [Google Scholar] [CrossRef]
- Leal, R.M.; Rascol, O.; Ferreira, J.J. The “long and winding road” of the disease-modifying effects of levodopa has not ended yet. Mov. Disord. 2020, 35, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Müller, T. Detoxification and antioxidative therapy for levodopa-induced neurodegeneration in Parkinson’s disease. Expert Rev. Neurother. 2013, 13, 707–718. [Google Scholar] [CrossRef]
- Verschuur, C.V.; Suwijn, S.R.; Boel, J.A.; Post, B.; Bloem, B.R.; van Hilten, J.J.; van Laar, T.; Tissingh, G.; Munts, A.G.; Deuschl, G.; et al. Randomized Delayed-Start Trial of Levodopa in Parkinson’s Disease. N. Engl. J. Med. 2019, 380, 315–324. [Google Scholar] [CrossRef]
- Müller, T. Pharmacokinetics and pharmacodynamics of levodopa/carbidopa cotherapies for Parkinson’s disease. Expert Opin. Drug Metab. Toxicol. 2020, 16, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Ramot, Y.; Nyska, A.; Maronpot, R.R.; Shaltiel-Karyo, R.; Tsarfati, Y.; Manno, R.A.; Sacco, G.; Yacoby-Zeevi, O. Ninety-day Local Tolerability and Toxicity Study of ND0612, a Novel Formulation of Levodopa/Carbidopa, Administered by Subcutaneous Continuous Infusion in Minipigs. Toxicol. Pathol. 2017, 45, 764–773. [Google Scholar] [CrossRef]
- Gannon, M.; Che, P.; Chen, Y.; Jiao, K.; Roberson, E.D.; Wang, Q. Noradrenergic dysfunction in Alzheimer’s disease. Front. Neurosci. 2015, 9, 220. [Google Scholar] [CrossRef]
- Moll, G.; Gsell, W.; Wichart, I.; Jellinger, K.; Riederer, P. Cholinergic and monoaminergic neuromediator systems in DAT. Neuropathological and neurochemical findings. In Alzheimer’s Disease. Epidemiology, Neuropathology, Neurochemistry, and Clinics; Maurer, K., Riederer, P., Beckmann, H., Eds.; Springer: Vienna, Austria, 1990; pp. 235–243. [Google Scholar]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions about Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2020, 108352. [Google Scholar] [CrossRef]
- Koola, M.M. Galantamine-Memantine combination in the treatment of Alzheimer’s disease and beyond. Psychiatry Res. 2020, 293, 113409. [Google Scholar] [CrossRef] [PubMed]
- Petrazzuoli, F.; Vinker, S.; Palmqvist, S.; Midlöv, P.; De Lepeleire, J.; Pirani, A.; Frese, T.; Buono, N.; Ahrensberg, J.; Asenova, R.; et al. Unburdening dementia—A basic social process grounded theory based on a primary care physician survey from 25 countries. Scand. J. Prim. Health Care 2020, 38, 253–264. [Google Scholar] [CrossRef]
- Viel, T.A.; Toricelli, M.; Pereira, A.A.R.; Abrao, G.S.; Malerba, H.N.; Maia, J.; Buck, H.S. Mechanisms of neuroplasticity and brain degeneration: Strategies for protection during the aging process. Neural Regen. Res. 2021, 16, 58–67. [Google Scholar] [CrossRef]
- Boonman, Z.; Isacson, O. Apoptosis in Neuronal Development and Transplantation: Role of Caspases and Trophic Factors. Exp. Neurol. 1999, 156, 1–15. [Google Scholar] [CrossRef]
- Demicheva, E.; Cui, Y.-F.; Bardwell, P.; Barghorn, S.; Kron, M.; Meyer, A.H.; Schmidt, M.; Gerlach, B.; Leddy, M.; Barlow, E.; et al. Targeting Repulsive Guidance Molecule A to Promote Regeneration and Neuroprotection in Multiple Sclerosis. Cell Rep. 2015, 10, 1887–1898. [Google Scholar] [CrossRef]
- Saitoh, Y.; Takahashi, Y. Riluzole for the treatment of amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 2020, 10, 343–355. [Google Scholar] [CrossRef]
- Gross, R.E.; Watts, R.L.; Hauser, R.A.; Bakay, R.A.; Reichmann, H.; von Kummer, R.; Ondo, W.G.; Reissig, E.; Eisner, W.; Steiner-Schulze, H.; et al. Intrastriatal transplantation of microcarrier-bound human retinal pigment epithelial cells versus sham surgery in patients with advanced Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2011, 10, 509–519. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Guarnieri, G.; Sarchielli, E.; Vannelli, G.B.; Morelli, A. Cell-based therapy in Alzheimer’s disease: Can human fetal cholinergic neurons “untangle the skein”? Neural Regen. Res. 2018, 13, 2105–2107. [Google Scholar]
- Liu, Z.; Cheung, H.-H. Stem Cell-Based Therapies for Parkinson Disease. Int. J. Mol. Sci. 2020, 21, 8060. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Sampaio, T.B.; Savall, A.S.; Gutierrez, M.E.Z.; Pinton, S. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: Implications for pathogenesis and therapy. Neural Regen. Res. 2017, 12, 549–557. [Google Scholar] [PubMed]
- Barker, R.A.; Mason, S.L.; Harrower, T.P.; Swain, R.A.; Ho, A.K.; Sahakian, B.J.; Mathur, R.; Elneil, S.; Thornton, S.; Hurrelbrink, C.; et al. The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2013, 84, 657–665. [Google Scholar] [CrossRef]
- Lige, L.; Zengmin, T. Transplantation of neural precursor cells in the treatment for parkinson disease: An efficacy and safety analysis. Turk. Neurosurg. 2015, 26, 378–383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olanow, C.W.; Goetz, C.G.; Kordower, J.H.; Stoessl, A.J.; Sossi, V.; Brin, M.F.; Shannon, K.M.; Nauert, G.M.; Perl, D.P.; Godbold, J.; et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann. Neurol. 2003, 54, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.P.; Ravichandran, J.; Carkaci-Salli, N. Neural regeneration therapies for Alzheimer’s and Parkinson’s disease-related disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165506. [Google Scholar] [CrossRef] [PubMed]
- Russ, K.; Flores, J.; Brudek, T.; Doudet, D. Neonatal human retinal pigment epithelial cells secrete limited trophic factors in vitro and in vivo following striatal implantation in parkinsonian rats. J. Neural Transm. 2015, 123, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Jha, N.K.; Jha, S.K.; Ramani, K.; Ambasta, R.K. Tau Phosphorylation, Molecular Chaperones, and Ubiquitin E3 Ligase: Clinical Relevance in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 43, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Liedhegner, E.A.; Steller, K.M.; Mieyal, J.J. Levodopa activates apoptosis signaling kinase 1 (ASK1) and promotes apoptosis in a neuronal model: Implications for the treatment of Parkinson’s disease. Chem. Res. Toxicol. 2011, 24, 1644–1652. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Yi, H.; Inaba, K.; Akao, Y.; Shamoto-Nagai, M. Mitochondria in neurodegenerative disorders: Regulation of the redox state and death signaling leading to neuronal death and survival. J. Neural Transm. 2009, 116, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Mothe, A.J.; Coelho, M.; Huang, L.; Monnier, P.P.; Cui, Y.-F.; Mueller, B.K.; Jacobson, P.B.; Tator, C.H. Delayed administration of the human anti-RGMa monoclonal antibody elezanumab promotes functional recovery including spontaneous voiding after spinal cord injury in rats. Neurobiol. Dis. 2020, 143, 104995. [Google Scholar] [CrossRef]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Schapira, A.H.; McDermott, M.P.; Barone, P.; Comella, C.L.; Albrecht, S.; Hsu, H.H.; Massey, D.H.; Mizuno, Y.; Poewe, W.; Rascol, O.; et al. Pramipexole in patients with early Parkinson’s disease (PROUD): A randomised delayed-start trial. Lancet Neurol. 2013, 12, 747–755. [Google Scholar] [CrossRef]
- Whone, A.L.; Watts, R.L.; Stoessl, A.J.; Davis, M.; Reske, S.; Nahmias, C.; Lang, A.E.; Rascol, O.; Ribeiro, M.J.; Remy, P.; et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Ann Neurol. 2003, 54, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Oertel, W.H.; Wolters, E.; Sampaio, C.; Gimenez-Roldan, S.; Bergamasco, B.; Dujardin, M.; Grosset, D.G.; Arnold, G.; Leenders, K.L.; Hundemer, H.P.; et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov. Disord. 2006, 21, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group. A Controlled, Randomized, Delayed-Start Study of Rasagiline in Early Parkinson Disease. Arch. Neurol. 2004, 61, 561–566. [Google Scholar] [CrossRef]
- Rascol, O.; Fitzer-Attas, C.J.; Hauser, R.; Jankovic, J.; Lang, A.; Langston, J.W.; Melamed, E.; Poewe, W.; Stocchi, F.; Tolosa, E.; et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): Prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol. 2011, 10, 415–423. [Google Scholar] [CrossRef]
- Parkinson Study Group. A controlled trial of rasagiline in early Parkinson disease: The TEMPO Study. Arch. Neurol. 2002, 59, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. JAMA 2000, 284, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Pålhagen, S.; Heinonen, E.; Hägglund, J.; Kaugesaar, T.; Kontants, H.; Mäki-Ikola, O.; Palm, R.; Turunen, J.; Swedish Parkinson Study Group. Selegiline delays the onset of disability in de novo parkinsonian patients. Neurology 1998, 51, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group. Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP patients requiring levodopa. Ann. Neurol. 1996, 39, 37–45. [Google Scholar] [CrossRef]
- Olanow, C.; Hauser, R.; Gauguster, L.; Malapira, T.; Koller, W.; Hubble, J.; Bushenbark, K.; Lilienfeld, D.; Esterlitz, J. The effect of deprenyl and levodopa on the progression of Parkinson’s disease. Ann. Neurol. 1995, 38, 771–777. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef]
- Sian-Hulsmann, J.; Monoranu, C.; Strobel, S.; Riederer, P. Lewy Bodies: A Spectator or Salient Killer? CNS Neurol. Disord. Drug Targets 2015, 14, 947–955. [Google Scholar] [CrossRef]
- Müller, T. Investigational agents for the management of Huntington’s disease. Expert Opin. Investig. Drugs 2016, 26, 175–185. [Google Scholar] [CrossRef]
- Guarnieri, G.; Sarchielli, E.; Comeglio, P.; Herrera-Puerta, E.; Piaceri, I.; Nacmias, B.; Benelli, M.; Kelsey, G.; Maggi, M.; Gallina, P.; et al. Tumor Necrosis Factor α Influences Phenotypic Plasticity and Promotes Epigenetic Changes in Human Basal Forebrain Cholinergic Neuroblasts. Int. J. Mol. Sci. 2020, 21, 6128. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Kim, M.-J.; Coughlin, J.M.; Henter, I.D.; Owen, D.R.; Innis, R.B. PET imaging of neuroinflammation in neurological disorders. Lancet Neurol. 2020, 19, 940–950. [Google Scholar] [CrossRef]
- Avila, J.; Pallas, N.; Bolos, M.; Sayas, C.L.; Hernandez, F. Intracellular and extracelleular microtubule associated protein tau as a therapeutic target in Alzheimer disease and other tauopathies. Expert Opin. Ther. Targets 2015, 20, 653–661. [Google Scholar] [CrossRef]
- Awasthi, M.; Singh, S.; Pandey, V.P.; Dwivedi, U.N. Alzheimer’s disease: An overview of amyloid beta dependent pathogenesis and its therapeutic implications along with in silico approaches emphasizing the role of natural products. J. Neurol. Sci. 2016, 361, 256–271. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, L.; Zhou, T.Y.; Lu, W. Alzheimer’s disease progression model based on integrated biomarkers and clinical measures. Acta Pharmacol. Sin. 2014, 35, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vieitez, E.; Nielsen, H.M. Associations between APOE Variants, Tau and α-Synuclein. Adv. Exp. Med. Biol. 2019, 1184, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Ballard, C.; Aarsland, D.; Cummings, J.; O’Brien, J.; Mills, R.; Molinuevo, J.L.; Fladby, T.; Williams, G.; Doherty, P.; Corbett, A.; et al. Drug repositioning and repurposing for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 661–673. [Google Scholar] [CrossRef]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef]
- Giordani, B.; Boivin, M.; Hall, A.; Foster, N.; Lehtinen, S.; Bluemlein, L.; Berent, S. The utility and generality of Mini-Mental State Examination scores in Alzheimer’s disease. Neurology 1990, 40, 1894–1896. [Google Scholar] [CrossRef] [PubMed]
- Hoops, S.; Nazem, S.; Siderowf, A.D.; Duda, J.E.; Xie, S.X.; Stern, M.B.; Weintraub, D. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 2009, 73, 1738–1745. [Google Scholar] [CrossRef]
- Walter, S.; Dufouil, C.; Gross, A.L.; Jones, R.N.; Mungas, D.; Filshtein, T.J.; Manly, J.J.; Arpawong, T.E.; Glymour, M.M. Neuropsychological Test Performance and MRI Markers of Dementia Risk: Reducing Education Bias. Alzheimer Dis. Assoc. Disord. 2019, 33, 179–185. [Google Scholar] [CrossRef]
- Zhou, A.; Jia, J. The value of the clock drawing test and the mini-mental state examination for identifying vascular cognitive impairment no dementia. Int. J. Geriatr. Psychiatry 2008, 23, 422–426. [Google Scholar] [CrossRef]
- Scarmeas, N.; Albert, M.; Brandt, J.; Blacker, D.; Hadjigeorgiou, G.; Papadimitriou, A.; Dubois, B.; Sarazin, M.; Wegesin, D.; Marder, K.; et al. Motor signs predict poor outcomes in Alzheimer disease. Neurology 2005, 64, 1696–1703. [Google Scholar] [CrossRef]
- Pohanka, M. Vaccination to Alzheimer Disease. Is it a Promising Tool or a Blind Way? Curr. Med. Chem. 2016, 23, 1432–1441. [Google Scholar] [CrossRef]
- Song, G.; Yang, H.; Shen, N.; Pham, P.; Brown, B.; Lin, X.; Hong, Y.; Sinu, P.; Cai, J.; Li, X.; et al. An Immunomodulatory Therapeutic Vaccine Targeting Oligomeric Amyloid-beta. J. Alzheimers Dis. 2020, 77, 1639–1653. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Zhou, H.D.; Zhou, X.F. Modified immunotherapies against Alzheimer’s disease: Toward safer and effective amyloid clearance. J. Alzheimers Dis. 2010, 21, 1065–1075. [Google Scholar] [CrossRef]
- Yang, C.; Xiao, S. New developments of clinical trial in immunotherapy for Alzheimer’s disease. Curr. Pharm. Biotechnol. 2015, 16, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Joly-Amado, A.; Davtyan, H.; Serraneau, K.; Jules, P.; Zitnyar, A.; Pressman, E.; Zagorski, K.; Antonyan, T.; Hovakimyan, A.; Paek, H.; et al. Active immunization with tau epitope in a mouse model of tauopathy induced strong antibody response together with improvement in short memory and pSer396-tau pathology. Neurobiol. Dis. 2020, 134, 104636. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Horowitz, A.; Lesch, K.-P.; Dandekar, T. Delaying memory decline: Different options and emerging solutions. Transl. Psychiatry 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, Z.; Xing, Z.; Zuo, Z.; Yuan, L.; Wu, Y.; Jiang, M.; Qi, F.; Yao, Z. Influenza vaccination in early Alzheimer’s disease rescues amyloidosis and ameliorates cognitive deficits in APP/PS1 mice by inhibiting regulatory T cells. J. Neuroinflamm. 2020, 17, 65. [Google Scholar] [CrossRef]
- Liang, Z.; Zhao, Y.; Ruan, L.; Zhu, L.; Jin, K.; Zhuge, Q.; Su, D.-M.; Zhao, Y. Impact of aging immune system on neurodegeneration and potential immunotherapies. Prog. Neurobiol. 2017, 157, 2–28. [Google Scholar] [CrossRef]
- Lisko, I.; Kulmala, J.; Annetorp, M.; Ngandu, T.; Mangialasche, F.; Kivipelto, M. How can dementia and disability be prevented in older adults: Where are we today and where are we going? J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Nugent, S.; Potvin, O.; Cunnane, S.C.; Chen, T.-H.; Duchesne, S. Associating Type 2 Diabetes Risk Factor Genes and FDG-PET Brain Metabolism in Normal Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 580633. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Berg, D.; Godau, J.; Seppi, K.; Behnke, S.; Liepelt-Scarfone, I.; Lerche, S.; Stockner, H.; Gaenslen, A.; Mahlknecht, P.; Huber, H.; et al. The PRIPS study: Screening battery for subjects at risk for Parkinson’s disease. Eur. J. Neurol. 2013, 20, 102–108. [Google Scholar] [CrossRef]
- Mahlknecht, P.; Seppi, K.; Poewe, W. The Concept of Prodromal Parkinson’s Disease. J. Parkinson’s Dis. 2015, 5, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rub, U.; Gai, W.P.; Del, T.K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.; McCann, H.; Shepherd, C. Evaluation of the Braak hypothesis: How far can it explain the pathogenesis of Parkinson’s disease? Expert Rev. Neurother. 2012, 12, 673–686. [Google Scholar] [CrossRef]
- Kingsbury, A.E.; Bandopadhyay, R.; Silveira-Moriyama, L.; Ayling, H.; Kallis, C.; Sterlacci, W.; Maeir, H.; Poewe, W.; Lees, A.J. Brain stem pathology in Parkinson’s disease: An evaluation of the Braak staging model. Mov. Disord. 2010, 25, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural Transm. 2019, 126, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J. Examining Braak’s hypothesis by imaging Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S83–S88. [Google Scholar] [CrossRef]
- Patterson, L.; Rushton, S.P.; Attems, J.; Thomas, A.J.; Morris, C.M. Degeneration of dopaminergic circuitry influences depressive symptoms in Lewy body disorders. Brain Pathol. 2018, 29, 544–557. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.-N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical Correlations With Lewy Body Pathology inLRRK2-Related Parkinson Disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef]
- Weiner, W.J. There Is No Parkinson Disease. Arch. Neurol. 2008, 65, 705–708. [Google Scholar] [CrossRef]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 (Suppl. 1), 14–20. [Google Scholar] [CrossRef]
- Przuntek, H.; Riederer, P. Diagnostic staging of Parkinson?s disease: Conceptual aspects. J. Neural Transm. 2004, 111, 201–216. [Google Scholar] [CrossRef]
- Shabir, O.; Moll, T.A.; Matuszyk, M.M.; Eyre, B.; Dake, M.D.; Berwick, J.; Francis, S.E. Preclinical models of disease and multimorbidity with focus upon cardiovascular disease and dementia. Mech. Ageing Dev. 2020, 192, 111361. [Google Scholar] [CrossRef]
- Parkinson Study Group. Dopamine Transporter Brain Imaging to Assess the Effects of Pramipexole vs Levodopa on Parkinson Disease Progression. JAMA 2002, 287, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Schifitto, G.; Zhao, H.; et al. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar]
- Przuntek, H.; Conrad, B.; Dichgans, J.; Kraus, P.; Krauseneck, P.; Pergande, G.; Rinne, U.; Schimrigk, K.; Schnitker, J.; Vogel, H. SELEDO: A 5-year long-term trial on the effect of selegiline in early parkinsonian patients treated with levodopa. Eur. J. Neurol. 1999, 6, 141–150. [Google Scholar] [CrossRef]
- Olanow, C.W.; Hauser, R.A.; Jankovic, J.; Langston, W.; Lang, A.; Poewe, W.; Tolosa, E.; Stocchi, F.; Melamed, E.; Eyal, E.; et al. A randomized, double-blind, placebo-controlled, delayed start study to assess rasagiline as a disease modifying therapy in Parkinson’s disease (the ADAGIO study): Rationale, design, and baseline characteristics. Mov. Disord. 2008, 23, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Shoulson, I.; Oakes, D.; Fahn, S.; Lang, A.; Langston, J.W.; LeWitt, P.; Olanow, C.W.; Penney, J.B.; Tanner, C.; Kieburtz, K.; et al. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: A randomized placebo-controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann. Neurol. 2002, 51, 604–612. [Google Scholar] [CrossRef]
- Balestrino, R.; Tunesi, S.; Tesei, S.; Lopiano, L.; Zecchinelli, A.L.; Goldwurm, S. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord. 2020, 35, 2111–2114. [Google Scholar] [CrossRef]
- Greuel, A.; Trezzi, J.P.; Glaab, E.; Ruppert, M.C.; Maier, F.; Jäger, C.; Hodak, Z.; Lohmann, K.; Ma, Y.; Eidelberg, D.; et al. GBA Variants in Parkinson’s Disease: Clinical, Metabolomic, and Multimodal Neuroimaging Phenotypes. Mov. Disord. 2020, 35, 2201–2210. [Google Scholar] [CrossRef]
- Mullin, S.; Stokholm, M.G.; Hughes, D.; Mehta, A.; Parbo, P.; Hinz, R.; Pavese, N.; Brooks, D.J.; Schapira, A.H. Brain Microglial Activation Increased in Glucocerebrosidase (GBA) Mutation Carriers without Parkinson’s disease. Mov. Disord. 2021, 36, 774–779. [Google Scholar] [CrossRef]
- Straniero, L.; Asselta, R.; Bonvegna, S.; Rimoldi, V.; Melistaccio, G.; Soldà, G.; Aureli, M.; Della Porta, M.; Lucca, U.; Di Fonzo, A.; et al. The SPID-GBA study: Sex distribution, Penetrance, Incidence, and Dementia in GBA-PD. Neurol. Genet. 2020, 6, e523. [Google Scholar] [CrossRef]
- Thaler, A.; Shenhar-Tsarfaty, S.; Shaked, Y.; Gurevich, T.; Omer, N.; Bar-Shira, A.; Gana-Weisz, M.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; et al. Metabolic syndrome does not influence the phenotype of LRRK2 and GBA related Parkinson’s disease. Sci. Rep. 2020, 10, 9329. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, A.-M.; Gravel, C.; Lévesque, M. Treating Parkinson’s Disease with Antibodies: Previous Studies and Future Directions. J. Parkinson’s Dis. 2021, 11, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Jamal, F. Immunotherapies Targeting α-Synuclein in Parkinson Disease. Fed. Pract. 2020, 37, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Kuhn, W.; Muller, T.; Nastos, I.; Poehlau, D. The neuroimmune hypothesis in Parkinson’s disease. Rev. Neurosci. 1997, 8, 29–34. [Google Scholar] [CrossRef]
- Sian-Hulsmann, J.; Riederer, P. The role of alpha-synuclein as ferrireductase in neurodegeneration associated with Parkinson’s disease. J. Neural Transm. 2020, 127, 749–754. [Google Scholar] [CrossRef]
- Jellinger, K.A. Interaction between α-Synuclein and Other Proteins in Neurodegenerative Disorders. Sci. World J. 2011, 11, 1893–1907. [Google Scholar] [CrossRef]
- Isaksen, T.J.; Yamashita, T. Repulsive Guidance Molecule A Regulates Adult Neurogenesis via the Neogenin Receptor. Neurosci. Insights 2020, 15. [Google Scholar] [CrossRef]
- Korecka, J.A.; Moloney, E.B.; Eggers, R.; Hobo, B.; Scheffer, S.; Ras-Verloop, N.; Pasterkamp, R.J.; Swaab, D.F.; Smit, A.B.; van Kesteren, R.E.; et al. Repulsive Guidance Molecule a (RGMa) Induces Neuropathological and Behavioral Changes That Closely Resemble Parkinson’s Disease. J. Neurosci. 2017, 37, 9361–9379. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Barghorn, S.; Lütge, S.; Haas, T.; Mueller, R.; Gerlach, B.; Öhm, G.; Eilert, K.; Trommer, I.; Mueller, B.K. Decreased levels of repulsive guidance molecule A in association with beneficial effects of repeated intrathecal triamcinolone acetonide application in progressive multiple sclerosis patients. J. Neural Transm. 2014, 122, 841–848. [Google Scholar] [CrossRef]
- Oda, W.; Fujita, Y.; Baba, K.; Mochizuki, H.; Niwa, H.; Yamashita, T. Inhibition of repulsive guidance molecule-a protects dopaminergic neurons in a mouse model of Parkinson’s disease. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Robinson, R.A.; Griffiths, S.C.; van de Haar, L.L.; Malinauskas, T.; van Battum, E.Y.; Zelina, P.; Schwab, R.A.; Karia, D.; Malinauskaite, L.; Brignani, S.; et al. Simultaneous binding of Guidance Cues NET1 and RGM blocks extracellular NEO1 signaling. Cell 2021. [Google Scholar] [CrossRef]
- Satoh, J.; Tabunoki, H.; Ishida, T.; Saito, Y.; Arima, K. Accumulation of a repulsive axonal guidance molecule RGMa in amyloid plaques: A possible hallmark of regenerative failure in Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 2013, 39, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Trommer, I.; Muhlack, S.; Mueller, B.K. Levodopa increases oxidative stress and repulsive guidance molecule A levels: A pilot study in patients with Parkinson’s disease. J. Neural Transm. 2016, 123, 401–406. [Google Scholar] [CrossRef]
- Babitt, J.L.; Zhang, Y.; Samad, T.A.; Xia, Y.; Tang, J.; Campagna, J.A.; Schneyer, A.L.; Woolf, C.J.; Lin, H.Y. Repulsive Guidance Molecule (RGMa), a DRAGON Homologue, Is a Bone Morphogenetic Protein Co-receptor. J. Biol. Chem. 2005, 280, 29820–29827. [Google Scholar] [CrossRef] [PubMed]
- Key, B.; Lah, G.J. Repulsive guidance molecule A (RGMa): A molecule for all seasons. Cell Adh. Migr. 2012, 6, 85–90. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Malinauskas, T.; Peer, T.V.; Bishop, B.; Mueller, T.D.; Siebold, C. Repulsive guidance molecules lock growth differentiation factor 5 in an inhibitory complex. Proc. Natl. Acad. Sci. USA 2020, 117, 15620–15631. [Google Scholar] [CrossRef]
- Kubo, T.; Tokita, S.; Yamashita, T. Repulsive Guidance Molecule-a and Demyelination: Implications for Multiple Sclerosis. J. Neuroimmune Pharmacol. 2011, 7, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Mothe, A.J.; Tassew, N.G.; Shabanzadeh, A.P.; Penheiro, R.; Vigouroux, R.J.; Huang, L.; Grinnell, C.; Cui, Y.-F.; Fung, E.; Monnier, P.P.; et al. RGMa inhibition with human monoclonal antibodies promotes regeneration, plasticity and repair, and attenuates neuropathic pain after spinal cord injury. Sci. Rep. 2017, 7, 10529. [Google Scholar] [CrossRef]
- Charish, J.; Shabanzadeh, A.P.; Chen, D.; Mehlen, P.; Sethuramanujam, S.; Harada, H.; Bonilha, V.L.; Awatramani, G.; Bremner, R.; Monnier, P.P. Neogenin neutralization prevents photoreceptor loss in inherited retinal degeneration. J. Clin. Investig. 2020, 130, 2054–2068. [Google Scholar] [CrossRef]
- Shabanzadeh, A.P.; Tassew, N.G.; Szydlowska, K.; Tymianski, M.; Banerjee, P.; Vigouroux, R.J.; Eubanks, J.H.; Huang, L.; Geraerts, M.; Koeberle, P.D.; et al. Uncoupling Neogenin association with lipid rafts promotes neuronal survival and functional recovery after stroke. Cell Death Dis. 2015, 6, e1744. [Google Scholar] [CrossRef]
- Song, M.; Tian, F.; Xia, H.; Xie, Y. Repulsive guidance molecule a suppresses seizures and mossy fiber sprouting via the FAK‑p120RasGAP‑Ras signaling pathway. Mol. Med. Rep. 2019, 19, 3255–3262. [Google Scholar] [CrossRef]
- Tanabe, S.; Yamashita, T. Repulsive Guidance Molecule-a Is Involved in Th17-Cell-Induced Neurodegeneration in Autoimmune Encephalomyelitis. Cell Rep. 2014, 9, 1459–1470. [Google Scholar] [CrossRef]
- Chen, J.; Shifman, M.I. Inhibition of neogenin promotes neuronal survival and improved behavior recovery after spinal cord injury. Neuroscience 2019, 408, 430–447. [Google Scholar] [CrossRef]
- Nakagawa, H.; Ninomiya, T.; Yamashita, T.; Takada, M. Treatment with the Neutralizing Antibody against Repulsive Guidance Molecule-a Promotes Recovery from Impaired Manual Dexterity in a Primate Model of Spinal Cord Injury. Cereb. Cortex 2018, 29, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Sun, P. Promoting functions of microRNA-29a/199B in neurological recovery in rats with spinal cord injury through inhibition of the RGMA/STAT3 axis. J. Orthop. Surg. Res. 2020, 15, 427. [Google Scholar] [CrossRef]
- Isaksen, T.J.; Fujita, Y.; Yamashita, T. Repulsive Guidance Molecule A Suppresses Adult Neurogenesis. Stem Cell Rep. 2020, 14, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Shi, H.; Xiong, S.; Hu, F.; Xiong, W.-C.; Liu, J. The neogenin/DCC homolog UNC-40 promotes BMP signaling via the RGM protein DRAG-1 in C. elegans. Development 2013, 140, 4070–4080. [Google Scholar] [CrossRef] [PubMed]
- Schweyer, K.; Rüschoff-Steiner, C.; Arias-Carrión, O.; Oertel, W.H.; Rösler, T.W.; Höglinger, G.U. Neuronal precursor cells with dopaminergic commitment in the rostral migratory stream of the mouse. Sci. Rep. 2019, 9, 13359. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Crews, L.; Pathel, P.; Morvinski-Friedmann, D.; Kosberg, K.; Roberts, S.; Patrick, C.; Winner, B.; Winkler, J.; et al. α-Synuclein Induces Alterations in Adult Neurogenesis in Parkinson Disease Models via p53-mediated Repression of Notch1*. J. Biol. Chem. 2012, 287, 31691–31702. [Google Scholar] [CrossRef]
- Winner, B.; Regensburger, M.; Schreglmann, S.; Boyer, L.; Prots, I.; Rockenstein, E.; Mante, M.; Zhao, C.; Winkler, J.; Masliah, E.; et al. Role of α-Synuclein in Adult Neurogenesis and Neuronal Maturation in the Dentate Gyrus. J. Neurosci. 2012, 32, 16906–16916. [Google Scholar] [CrossRef]
- Winner, B.; Marchetto, M.C.; Winkler, J.; Gage, F.H. Human-induced pluripotent stem cells pave the road for a better understanding of motor neuron disease. Hum. Mol. Genet. 2014, 23, R27–R34. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).