
Genetic Approaches Using Zebrafish to Study the Microbiota–Gut–Brain Axis in Neurological Disorders


Abstract
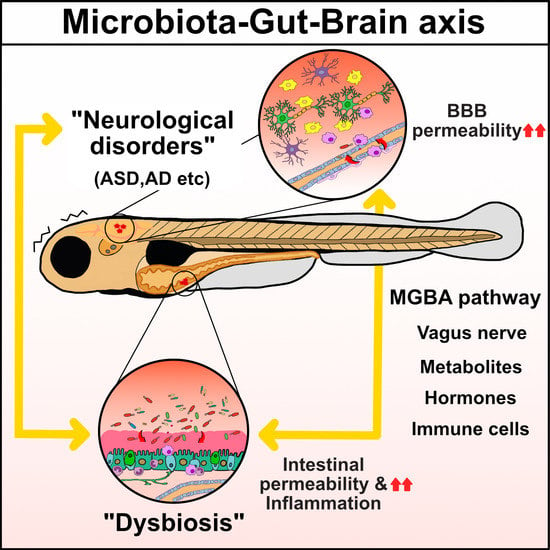
1. Introduction
2. MGBA Pathways
2.1. Regulation of Brain and Intestinal Permeability by the Microbiota
2.2. Neuronal Communication via the Vagus Nerve
2.3. Immune Cell Infiltration into the Brain and Inflammatory Cytokines
2.4. MGBA Metabolites: SCFAs and Tryptophan Derivatives, Including Serotonin
2.4.1. SCFAs
2.4.2. Tryptophan Metabolites and Serotonin
3. MGBA-Associated Neurological Disorders
3.1. Autism. Spectrum Disorder: A Neurodevelopmental Disorder
3.2. Alzheimer’s Disease: A Neurodegeneration Problem
4. Zebrafish as a Model System for MGBA Studies
4.1. The Zebrafish GIT and Associated Cell Types
4.2. Genetic Approaches to Loss-of-Function and Gain-of-Function Studies with Zebrafish Transgenic Lines
4.2.1. Loss-of-Function Approaches
4.2.2. Gain-of-Function Approaches
4.2.3. Zebrafish Transgenic Lines and In Vivo Imaging of Host-Bacterial Interactions
4.3. The Gut Microbiota of Zebrafish in MGBA Studies
4.4. Zebrafish Models for the Neurological Diseases ASD and AD
4.4.1. The Zebrafish ASD Model Representing a Neurodevelopmental Disorder
4.4.2. The Zebrafish AD Model Representing a Neurodegenerative Disease
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT | 5-hydroxytryptamine |
| AD | Alzheimer’s disease |
| Aβ42 | amyloid β42 |
| ASD | autism spectrum disorder |
| BBB | blood–brain barrier |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| EECs | enteroendocrine cells |
| GF | germ-free |
| GIT | gastrointestinal tract |
| GABA | gamma-aminobutyric acid |
| IBD | inflammatory bowel disease |
| IFNγ | interferon γ |
| LPS | lipopolysaccharides |
| MAMP | microbe-associated molecular pattern |
| MGBA | microbiota–gut–brain Axis |
| SCFAs | short chain fatty acids |
| TNF | tumor necrosis factor |
References
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Hoban, A.E.; Stilling, R.M.; Ryan, F.J.; Shanahan, F.; Dinan, T.G.; Claesson, M.J.; Clarke, G.; Cryan, J.F. Regulation of prefrontal cortex myelination by the microbiota. Transl. Psychiatry 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.t.; Mazmanian, S.K. The gut microbiota-brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Bjorkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.L.; Dong, J.; Galipeau, H.J.; Jury, J.; McCarville, J.; Huang, X.; Wang, X.Y.; Naidoo, A.; Anbazhagan, A.N.; Libertucci, J.; et al. Commensal microbiota induces colonic barrier structure and functions that contribute to homeostasis. Sci. Rep. 2018, 8, 14184. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.R.; Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G.; Hyland, N.P. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 2015, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Ploger, S.; Stumpff, F.; Penner, G.B.; Schulzke, J.D.; Gabel, G.; Martens, H.; Shen, Z.; Gunzel, D.; Aschenbach, J.R. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann. N. Y. Acad. Sci. 2012, 1258, 52–59. [Google Scholar] [CrossRef]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohorquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361. [Google Scholar] [CrossRef]
- Fani Maleki, A.; Rivest, S. Innate Immune Cells: Monocytes, Monocyte-Derived Macrophages and Microglia as Therapeutic Targets for Alzheimer’s Disease and Multiple Sclerosis. Front. Cell Neurosci. 2019, 13, 355. [Google Scholar] [CrossRef]
- Malm, T.; Koistinaho, M.; Muona, A.; Magga, J.; Koistinaho, J. The role and therapeutic potential of monocytic cells in Alzheimer’s disease. Glia 2010, 58, 889–900. [Google Scholar] [CrossRef]
- Theriault, P.; ElAli, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.P.; Bellavance, M.A.; Prefontaine, P.; Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 2013, 5, 646–653. [Google Scholar] [CrossRef]
- Baik, S.H.; Cha, M.Y.; Hyun, Y.M.; Cho, H.; Hamza, B.; Kim, D.K.; Han, S.H.; Choi, H.; Kim, K.H.; Moon, M.; et al. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 1286–1292. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Paouri, E.; Georgopoulos, S. Systemic and CNS Inflammation Crosstalk: Implications for Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 559–574. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Lobionda, S.; Sittipo, P.; Kwon, H.Y.; Lee, Y.K. The Role of Gut Microbiota in Intestinal Inflammation with Respect to Diet and Extrinsic Stressors. Microorganisms 2019, 7, 271. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Wu, S.C.; Cao, Z.S.; Chang, K.M.; Juang, J.L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 24. [Google Scholar] [CrossRef]
- Sharon, G.; Garg, N.; Debelius, J.; Knight, R.; Dorrestein, P.C.; Mazmanian, S.K. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014, 20, 719–730. [Google Scholar] [CrossRef]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 1, 461–478. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Pieters, H.J.; Jonkers, D.M. Short-chain fatty acids activate AMP-activated protein kinase and ameliorate ethanol-induced intestinal barrier dysfunction in Caco-2 cell monolayers. J. Nutr. 2013, 143, 1872–1881. [Google Scholar] [CrossRef]
- Lewis, K.; Lutgendorff, F.; Phan, V.; Soderholm, J.D.; Sherman, P.M.; McKay, D.M. Enhanced translocation of bacteria across metabolically stressed epithelia is reduced by butyrate. Inflamm. Bowel Dis. 2010, 16, 1138–1148. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, M.; Wang, Y.; Dorfman, R.G.; Liu, H.; Yu, T.; Chen, X.; Tang, D.; Xu, L.; Yin, Y.; et al. Faecalibacterium prausnitzii Produces Butyrate to Maintain Th17/Treg Balance and to Ameliorate Colorectal Colitis by Inhibiting Histone Deacetylase 1. Inflamm. Bowel Dis. 2018, 24, 1926–1940. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Covasa, M.; Stephens, R.W.; Toderean, R.; Cobuz, C. Intestinal Sensing by Gut Microbiota: Targeting Gut Peptides. Front. Endocrinol. 2019, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Katsurada, K.; Yada, T. Neural effects of gut- and brain-derived glucagon-like peptide-1 and its receptor agonist. J. Diabetes Investig. 2016, 7, 64–69. [Google Scholar] [CrossRef]
- Kim, S.W.; Hooker, J.M.; Otto, N.; Win, K.; Muench, L.; Shea, C.; Carter, P.; King, P.; Reid, A.E.; Volkow, N.D.; et al. Whole-body pharmacokinetics of HDAC inhibitor drugs, butyric acid, valproic acid and 4-phenylbutyric acid measured with carbon-11 labeled analogs by PET. Nucl. Med. Biol. 2013, 40, 912–918. [Google Scholar] [CrossRef]
- Vijay, N.; Morris, M.E. Role of monocarboxylate transporters in drug delivery to the brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wang, Q.; Wu, Q.; Mao-Draayer, Y.; Kim, C.H. Bidirectional regulatory potentials of short-chain fatty acids and their G-protein-coupled receptors in autoimmune neuroinflammation. Sci. Rep. 2019, 9, 8831. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef]
- Gershon, M.D.; Tack, J. The serotonin signaling system: From basic understanding to drug development for functional GI disorders. Gastroenterology 2007, 132, 397–414. [Google Scholar] [CrossRef]
- Sikander, A.; Rana, S.V.; Prasad, K.K. Role of serotonin in gastrointestinal motility and irritable bowel syndrome. Clin. Chim. Acta 2009, 403, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Shajib, M.S.; Baranov, A.; Khan, W.I. Diverse Effects of Gut-Derived Serotonin in Intestinal Inflammation. ACS Chem. Neurosci. 2017, 8, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., 3rd; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef]
- Keszthelyi, D.; Troost, F.J.; Jonkers, D.M.; van Eijk, H.M.; Lindsey, P.J.; Dekker, J.; Buurman, W.A.; Masclee, A.A. Serotonergic reinforcement of intestinal barrier function is impaired in irritable bowel syndrome. Aliment. Pharmacol. Ther. 2014, 40, 392–402. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Hoglund, E.; Overli, O.; Winberg, S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Kaszaki, J.; Erces, D.; Varga, G.; Szabo, A.; Vecsei, L.; Boros, M. Kynurenines and intestinal neurotransmission: The role of N-methyl-D-aspartate receptors. J. Neural Transm. 2012, 119, 211–223. [Google Scholar] [CrossRef]
- Lillesaar, C. The serotonergic system in fish. J. Chem. Neuroanat 2011, 41, 294–308. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.L. Gut Microbiota and Dysbiosis in Alzheimer’s Disease: Implications for Pathogenesis and Treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.F.; Frye, R.E.; Gillberg, C.; Casanova, E.L. Editorial: Comorbidity and Autism Spectrum Disorder. Front. Psychiatry 2020, 11. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Yuen, R.K.; Merico, D.; Bookman, M.; Howe, J.L.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Deflaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, A.; Van de Water, J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology 2017, 42, 284–298. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Yenkoyan, K.; Grigoryan, A.; Fereshetyan, K.; Yepremyan, D. Advances in understanding the pathophysiology of autism spectrum disorders. Behav. Brain Res. 2017, 331, 92–101. [Google Scholar] [CrossRef]
- Hashem, S.; Nisar, S.; Bhat, A.A.; Yadav, S.K.; Azeem, M.W.; Bagga, P.; Fakhro, K.; Reddy, R.; Frenneaux, M.P.; Haris, M. Genetics of structural and functional brain changes in autism spectrum disorder. Transl. Psychiatry 2020, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Gladysz, D.; Krzywdzinska, A.; Hozyasz, K.K. Immune Abnormalities in Autism Spectrum Disorder-Could They Hold Promise for Causative Treatment? Mol. Neurobiol. 2018, 55, 6387–6435. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Schultz, S.; Brigida, A.L.; Antonucci, N. Inflammation and Neuro-Immune Dysregulations in Autism Spectrum Disorders. Pharmaceuticals 2018, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Padua, D.; Tillisch, K. Altered brain-gut axis in autism: Comorbidity or causative mechanisms? Bioessays 2014, 36, 933–939. [Google Scholar] [CrossRef]
- Samsam, M.; Ahangari, R.; Naser, S.A. Pathophysiology of autism spectrum disorders: Revisiting gastrointestinal involvement and immune imbalance. World J. Gastroenterol. 2014, 20, 9942–9951. [Google Scholar] [CrossRef] [PubMed]
- Vuong, H.E.; Hsiao, E.Y. Emerging Roles for the Gut Microbiome in Autism Spectrum Disorder. Biol. Psychiatry 2017, 81, 411–423. [Google Scholar] [CrossRef]
- Desbonnet, L.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Microbiota is essential for social development in the mouse. Mol. Psychiatry 2014, 19, 146–148. [Google Scholar] [CrossRef]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Fattorusso, A.; Di Genova, L.; Dell’Isola, G.B.; Mencaroni, E.; Esposito, S. Autism Spectrum Disorders and the Gut Microbiota. Nutrients 2019, 11, 521. [Google Scholar] [CrossRef]
- Xu, M.; Xu, X.; Li, J.; Li, F. Association Between Gut Microbiota and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2019, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Bolte, E.R. Autism and Clostridium tetani. Med. Hypotheses 1998, 51, 133–144. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.-W.; Gandal, M.J.; Wang, B.; Kim, Y.-M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 2019, 177, 1600–1618. [Google Scholar] [CrossRef] [PubMed]
- Sandler, R.H.; Finegold, S.M.; Bolte, E.R.; Buchanan, C.P.; Maxwell, A.P.; Vaisanen, M.L.; Nelson, M.N.; Wexler, H.M. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J. Child. Neurol. 2000, 15, 429–435. [Google Scholar] [CrossRef]
- Li, Q.; Han, Y.; Dy, A.B.C.; Hagerman, R.J. The Gut Microbiota and Autism Spectrum Disorders. Front. Cell Neurosci. 2017, 11, 120. [Google Scholar] [CrossRef]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef]
- Israelyan, N.; Margolis, K.G. Serotonin as a link between the gut-brain-microbiome axis in autism spectrum disorders. Pharmacol. Res. 2018, 132, 1–6. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshinaga, N.; Tanabe, S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J. Biol. Chem. 2011, 286, 31263–31271. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, M.; Sapone, A.; Senger, S.; Camhi, S.S.; Kadzielski, S.M.; Buie, T.M.; Kelly, D.L.; Cascella, N.; Fasano, A. Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism. 2016, 7, 49. [Google Scholar] [CrossRef]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef]
- Saurman, V.; Margolis, K.G.; Luna, R.A. Autism Spectrum Disorder as a Brain-Gut-Microbiome Axis Disorder. Dig. Dis. Sci. 2020, 65, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Sgritta, M.; Dooling, S.W.; Buffington, S.A.; Momin, E.N.; Francis, M.B.; Britton, R.A.; Costa-Mattioli, M. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 2019, 101, 246–259.e6. [Google Scholar] [CrossRef]
- Napoli, E.; Duenas, N.; Giulivi, C. Potential therapeutic use of the ketogenic diet in autism spectrum disorders. Front. Pediatrics 2014, 2, 69. [Google Scholar] [CrossRef]
- Newell, C.; Bomhof, M.R.; Reimer, R.A.; Hittel, D.S.; Rho, J.M.; Shearer, J. Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder. Mol. Autism. 2016, 7, 37. [Google Scholar] [CrossRef]
- Ristori, M.V.; Quagliariello, A.; Reddel, S.; Ianiro, G.; Vicari, S.; Gasbarrini, A.; Putignani, L. Autism, Gastrointestinal Symptoms and Modulation of Gut Microbiota by Nutritional Interventions. Nutrients 2019, 11, 2812. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Yu, J.T.; Tan, L.; Wang, Y.L.; Sun, L.; Tan, L. Nutrition and the risk of Alzheimer’s disease. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. The metabolic syndrome and Alzheimer disease. Arch. Neurol. 2007, 64, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Neuner, S.M.; Tcw, J.; Goate, A.M. Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 2020, 143. [Google Scholar] [CrossRef]
- DeJong, E.N.; Surette, M.G.; Bowdish, D.M.E. The Gut Microbiota and Unhealthy Aging: Disentangling Cause from Consequence. Cell Host Microbe 2020, 28, 180–189. [Google Scholar] [CrossRef]
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Investig. 2019, 129, 4050–4057. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, D.; Zhou, G.; Li, C. Dietary Pattern, Gut Microbiota, and Alzheimer’s Disease. J. Agric. Food Chem. 2020, 68, 12800–12809. [Google Scholar] [CrossRef]
- Gale, S.C.; Gao, L.; Mikacenic, C.; Coyle, S.M.; Rafaels, N.; Murray Dudenkov, T.; Madenspacher, J.H.; Draper, D.W.; Ge, W.; Aloor, J.J.; et al. APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 2014, 134, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.T.; Corsini, S.; Kellingray, L.; Hegarty, C.; Le Gall, G.; Narbad, A.; Muller, M.; Tejera, N.; O’Toole, P.W.; Minihane, A.M.; et al. APOE genotype influences the gut microbiome structure and function in humans and mice: Relevance for Alzheimer’s disease pathophysiology. FASEB J. 2019, 33, 8221–8231. [Google Scholar] [CrossRef] [PubMed]
- Correale, C.; Genua, M.; Vetrano, S.; Mazzini, E.; Martinoli, C.; Spinelli, A.; Arena, V.; Peyrin-Biroulet, L.; Caprioli, F.; Passini, N.; et al. Bacterial sensor triggering receptor expressed on myeloid cells-2 regulates the mucosal inflammatory response. Gastroenterology 2013, 144, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.; Iyer, S.; Kerr, W.G. Role of SHIP1 in cancer and mucosal inflammation. Ann. N. Y. Acad. Sci. 2013, 1280, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Lubbers, J.; Rodriguez, E.; van Kooyk, Y. Modulation of Immune Tolerance via Siglec-Sialic Acid Interactions. Front. Immunol. 2018, 9, 2807. [Google Scholar] [CrossRef]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.O.; Holtzman, D.M. Gut Microbiota: From the Forgotten Organ to a Potential Key Player in the Pathology of Alzheimer’s Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef] [PubMed]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.; Frisoni, G.; Neher, J.; Fåk, F.; Jucker, M.; Lasser, T. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 2013, 9, 25–34. [Google Scholar] [CrossRef]
- Bowman, G.L.; Kaye, J.A.; Moore, M.; Waichunas, D.; Carlson, N.E.; Quinn, J.F. Blood-brain barrier impairment in Alzheimer disease: Stability and functional significance. Neurology 2007, 68, 1809–1814. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Zhao, Y.; Cong, L.; Jaber, V.; Lukiw, W.J. Microbiome-Derived Lipopolysaccharide Enriched in the Perinuclear Region of Alzheimer’s Disease Brain. Front. Immunol. 2017, 8, 1064. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Bora, S.H.; Xu, G.; Borchelt, D.R.; Price, D.L.; Koliatsos, V.E. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 2003, 14, 133–145. [Google Scholar] [CrossRef]
- Bulgart, H.R.; Neczypor, E.W.; Wold, L.E.; Mackos, A.R. Microbial involvement in Alzheimer disease development and progression. Mol. Neurodegener. 2020, 15, 42. [Google Scholar] [CrossRef]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflammation 2019, 16, 180. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes alpha-synuclein aggregation and motor impairment in mice. Elife 2020, 9. [Google Scholar] [CrossRef]
- Santoriello, C.; Zon, L.I. Hooked! Modeling human disease in zebrafish. J. Clin. Investig. 2012, 122, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.M.; Nguyen, A.T.; Odem, M.A.; Eisenhoffer, G.T.; Krachler, A.M. The zebrafish as a model for gastrointestinal tract-microbe interactions. Cell Microbiol. 2020, 22. [Google Scholar] [CrossRef]
- Fontana, B.D.; Mezzomo, N.J.; Kalueff, A.V.; Rosemberg, D.B. The developing utility of zebrafish models of neurological and neuropsychiatric disorders: A critical review. Exp. Neurol. 2018, 299, 157–171. [Google Scholar] [CrossRef]
- Sakai, C.; Ijaz, S.; Hoffman, E.J. Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future. Front. Mol. Neurosci. 2018, 11, 294. [Google Scholar] [CrossRef]
- Pham, L.N.; Kanther, M.; Semova, I.; Rawls, J.F. Methods for generating and colonizing gnotobiotic zebrafish. Nat. Protoc. 2008, 3, 1862. [Google Scholar] [CrossRef]
- Melancon, E.; Gomez De La Torre Canny, S.; Sichel, S.; Kelly, M.; Wiles, T.J.; Rawls, J.F.; Eisen, J.S.; Guillemin, K. Best practices for germ-free derivation and gnotobiotic zebrafish husbandry. Methods Cell Biol. 2017, 138, 61–100. [Google Scholar] [CrossRef]
- Stephens, W.Z.; Burns, A.R.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J. The composition of the zebrafish intestinal microbial community varies across development. ISME J. 2016, 10, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Gaulke, C.A.; Beaver, L.M.; Armour, C.R.; Humphreys, I.R.; Barton, C.L.; Tanguay, R.L.; Ho, E.; Sharpton, T.J. An integrated gene catalog of the zebrafish gut microbiome reveals significant homology with mammalian microbiomes. BioRxiv 2020. [Google Scholar] [CrossRef]
- Orger, M.B.; de Polavieja, G.G. Zebrafish Behavior: Opportunities and Challenges. Annu. Rev. Neurosci. 2017, 40, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Basnet, R.M.; Zizioli, D.; Taweedet, S.; Finazzi, D.; Memo, M. Zebrafish larvae as a behavioral model in neuropharmacology. Biomedicines 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Gebhardt, M.; Stewart, A.M.; Cachat, J.M.; Brimmer, M.; Chawla, J.S.; Craddock, C.; Kyzar, E.J.; Roth, A.; Landsman, S.; et al. Towards a comprehensive catalog of zebrafish behavior 1.0 and beyond. Zebrafish 2013, 10, 70–86. [Google Scholar] [CrossRef]
- Vaz, R.; Hofmeister, W.; Lindstrand, A. Zebrafish models of neurodevelopmental disorders: Limitations and benefits of current tools and techniques. Int. J. Mol. Sci. 2019, 20, 1296. [Google Scholar] [CrossRef]
- Wallace, K.N.; Akhter, S.; Smith, E.M.; Lorent, K.; Pack, M. Intestinal growth and differentiation in zebrafish. Mech. Dev. 2005, 122, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Du, J.; Lam, S.H.; Mathavan, S.; Matsudaira, P.; Gong, Z. Morphological and molecular evidence for functional organization along the rostrocaudal axis of the adult zebrafish intestine. BMC Genom. 2010, 11, 392. [Google Scholar] [CrossRef] [PubMed]
- Brugman, S. The zebrafish as a model to study intestinal inflammation. Dev. Comp. Immunol. 2016, 64, 82–92. [Google Scholar] [CrossRef]
- Wallace, K.N.; Pack, M. Unique and conserved aspects of gut development in zebrafish. Dev. Biol. 2003, 255, 12–29. [Google Scholar] [CrossRef]
- Moran-Ramos, S.; Tovar, A.R.; Torres, N. Diet: Friend or foe of enteroendocrine cells--how it interacts with enteroendocrine cells. Adv. Nutr. 2012, 3, 8–20. [Google Scholar] [CrossRef]
- Ye, L.; Mueller, O.; Bagwell, J.; Bagnat, M.; Liddle, R.A.; Rawls, J.F. High fat diet induces microbiota-dependent silencing of enteroendocrine cells. Elife 2019, 8. [Google Scholar] [CrossRef]
- Ye, L.; Bae, M.; Cassilly, C.D.; Jabba, S.V.; Thorpe, D.W.; Martin, A.M.; Lu, H.Y.; Wang, J.; Thompson, J.D.; Lickwar, C.R.; et al. Enteroendocrine cells sense bacterial tryptophan catabolites to activate enteric and vagal neuronal pathways. Cell Host Microbe 2020. [Google Scholar] [CrossRef]
- Olsson, C.; Holmberg, A.; Holmgren, S. Development of enteric and vagal innervation of the zebrafish (Danio rerio) gut. J. Comp. Neurol. 2008, 508, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Ganz, J.; Bayrer, J.; Becker, L.; Bogunovic, M.; Rao, M. Advances in Enteric Neurobiology: The “Brain” in the Gut in Health and Disease. J. Neurosci. 2018, 38, 9346–9354. [Google Scholar] [CrossRef]
- Shepherd, I.; Eisen, J. Development of the zebrafish enteric nervous system. Methods Cell Biol. 2011, 101, 143–160. [Google Scholar] [CrossRef]
- Ganz, J. Gut feelings: Studying enteric nervous system development, function, and disease in the zebrafish model system. Dev. Dyn. 2018, 247, 268–278. [Google Scholar] [CrossRef]
- Trede, N.S.; Langenau, D.M.; Traver, D.; Look, A.T.; Zon, L. The use of zebrafish to understand immunity. Immunity 2004, 20, 367–379. [Google Scholar] [CrossRef]
- Oosterhof, N.; Boddeke, E.; van Ham, T.J. Immune cell dynamics in the CNS: Learning from the zebrafish. Glia 2015, 63, 719–735. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Robu, M.E.; Larson, J.D.; Nasevicius, A.; Beiraghi, S.; Brenner, C.; Farber, S.A.; Ekker, S.C. p53 activation by knockdown technologies. PLoS Genet. 2007, 3, e78. [Google Scholar] [CrossRef]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef]
- Nüsslein-Volhard, C. The zebrafish issue of development. Development 2012, 139, 4099–4103. [Google Scholar] [CrossRef]
- Sertori, R.; Trengove, M.; Basheer, F.; Ward, A.C.; Liongue, C. Genome editing in zebrafish: A practical overview. Brief. Funct. Genom. 2016, 15, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- Liu, K.; Petree, C.; Requena, T.; Varshney, P.; Varshney, G.K. Expanding the CRISPR Toolbox in Zebrafish for Studying Development and Disease. Front. Cell Dev. Biol. 2019, 7, 13. [Google Scholar] [CrossRef]
- Wierson, W.A.; Welker, J.M.; Almeida, M.P.; Mann, C.M.; Webster, D.A.; Torrie, M.E.; Weiss, T.J.; Kambakam, S.; Vollbrecht, M.K.; Lan, M. Efficient targeted integration directed by short homology in zebrafish and mammalian cells. Elife 2020, 9. [Google Scholar] [CrossRef]
- Cho, H.-J.; Lee, J.-G.; Kim, J.-H.; Kim, S.-Y.; Huh, Y.H.; Kim, H.-J.; Lee, K.-S.; Yu, K.; Lee, J.-S. Vascular defects of DYRK1A knockouts are ameliorated by modulating calcium signaling in zebrafish. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.M.; Fujimoto, E.; Grabher, C.; Mangum, B.D.; Hardy, M.E.; Campbell, D.S.; Parant, J.M.; Yost, H.J.; Kanki, J.P.; Chien, C.B. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007, 236, 3088–3099. [Google Scholar] [CrossRef] [PubMed]
- Scheer, N.; Campos-Ortega, J.A. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mech. Dev. 1999, 80, 153–158. [Google Scholar] [CrossRef]
- Goll, M.G.; Anderson, R.; Stainier, D.Y.; Spradling, A.C.; Halpern, M.E. Transcriptional silencing and reactivation in transgenic zebrafish. Genetics 2009, 182, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ouyang, J.; Qie, J.; Zhang, G.; Liu, L.; Yang, P. Optimization of the Gal4/UAS transgenic tools in zebrafish. Appl. Microbiol. Biotechnol. 2019, 103, 1789–1799. [Google Scholar] [CrossRef]
- Boniface, E.J.; Lu, J.; Victoroff, T.; Zhu, M.; Chen, W. FlEx-based transgenic reporter lines for visualization of Cre and Flp activity in live zebrafish. Genesis 2009, 47, 484–491. [Google Scholar] [CrossRef]
- Subedi, A.; Macurak, M.; Gee, S.T.; Monge, E.; Goll, M.G.; Potter, C.J.; Parsons, M.J.; Halpern, M.E. Adoption of the Q transcriptional regulatory system for zebrafish transgenesis. Methods 2014, 66, 433–440. [Google Scholar] [CrossRef]
- Carney, T.J.; Mosimann, C. Switch and Trace: Recombinase Genetics in Zebrafish. Trends Genet. 2018, 34, 362–378. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.M.; Akitake, C.M.; Goll, M.G.; Rhee, J.M.; Gosse, N.; Baier, H.; Halpern, M.E.; Leach, S.D.; Parsons, M.J. Transactivation from Gal4-VP16 transgenic insertions for tissue-specific cell labeling and ablation in zebrafish. Dev. Biol. 2007, 304, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zeng, J.; Jing, M.; Zhou, J.; Feng, J.; Owen, S.F.; Luo, Y.; Li, F.; Wang, H.; Yamaguchi, T.; et al. A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice. Cell 2018, 174, 481–496.e419. [Google Scholar] [CrossRef]
- Villani, A.; Benjaminsen, J.; Moritz, C.; Henke, K.; Hartmann, J.; Norlin, N.; Richter, K.; Schieber, N.L.; Franke, T.; Schwab, Y.; et al. Clearance by Microglia Depends on Packaging of Phagosomes into a Unique Cellular Compartment. Dev. Cell 2019, 49, 77–88.e7. [Google Scholar] [CrossRef]
- Wang, Z.; Lin, L.; Chen, W.; Zheng, X.; Zhang, Y.; Liu, Q.; Yang, D. Neutrophil plays critical role during Edwardsiella piscicida immersion infection in zebrafish larvae. Fish. Shellfish Immunol. 2019, 87, 565–572. [Google Scholar] [CrossRef]
- Rosowski, E.E.; Deng, Q.; Keller, N.P.; Huttenlocher, A. Rac2 Functions in Both Neutrophils and Macrophages To Mediate Motility and Host Defense in Larval Zebrafish. J. Immunol. 2016, 197, 4780–4790. [Google Scholar] [CrossRef]
- Thakur, P.C.; Davison, J.M.; Stuckenholz, C.; Lu, L.; Bahary, N. Dysregulated phosphatidylinositol signaling promotes endoplasmic-reticulum-stress-mediated intestinal mucosal injury and inflammation in zebrafish. Dis. Model. Mech. 2014, 7, 93–106. [Google Scholar] [CrossRef]
- Walton, E.M.; Cronan, M.R.; Beerman, R.W.; Tobin, D.M. The Macrophage-Specific Promoter mfap4 Allows Live, Long-Term Analysis of Macrophage Behavior during Mycobacterial Infection in Zebrafish. PLoS ONE 2015, 10, e138949. [Google Scholar] [CrossRef] [PubMed]
- Dee, C.T.; Nagaraju, R.T.; Athanasiadis, E.I.; Gray, C.; Fernandez Del Ama, L.; Johnston, S.A.; Secombes, C.J.; Cvejic, A.; Hurlstone, A.F. CD4-Transgenic Zebrafish Reveal Tissue-Resident Th2- and Regulatory T Cell-like Populations and Diverse Mononuclear Phagocytes. J. Immunol. 2016, 197, 3520–3530. [Google Scholar] [CrossRef]
- Rossi, F.; Casano, A.M.; Henke, K.; Richter, K.; Peri, F. The SLC7A7 Transporter Identifies Microglial Precursors prior to Entry into the Brain. Cell Rep. 2015, 11, 1008–1017. [Google Scholar] [CrossRef]
- Sanderson, L.E.; Chien, A.T.; Astin, J.W.; Crosier, K.E.; Crosier, P.S.; Hall, C.J. An inducible transgene reports activation of macrophages in live zebrafish larvae. Dev. Comp. Immunol. 2015, 53, 63–69. [Google Scholar] [CrossRef]
- Peri, F.; Nusslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 2008, 133, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Sieger, D.; Moritz, C.; Ziegenhals, T.; Prykhozhij, S.; Peri, F. Long-range Ca2+ waves transmit brain-damage signals to microglia. Dev. Cell 2012, 22, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Page, D.M.; Wittamer, V.; Bertrand, J.Y.; Lewis, K.L.; Pratt, D.N.; Delgado, N.; Schale, S.E.; McGue, C.; Jacobsen, B.H.; Doty, A.; et al. An evolutionarily conserved program of B-cell development and activation in zebrafish. Blood 2013, 122, 1–11. [Google Scholar] [CrossRef]
- Alvers, A.L.; Ryan, S.; Scherz, P.J.; Huisken, J.; Bagnat, M. Single continuous lumen formation in the zebrafish gut is mediated by smoothened-dependent tissue remodeling. Development 2014, 141, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.C.; Espenschied, S.T.; Matty, M.A.; Mueller, O.; Tobin, D.M.; Rawls, J.F. Intestinal Serum amyloid A suppresses systemic neutrophil activation and bactericidal activity in response to microbiota colonization. PLoS Pathog. 2019, 15, e1007381. [Google Scholar] [CrossRef]
- Troll, J.V.; Hamilton, M.K.; Abel, M.L.; Ganz, J.; Bates, J.M.; Stephens, W.Z.; Melancon, E.; van der Vaart, M.; Meijer, A.H.; Distel, M.; et al. Microbiota promote secretory cell determination in the intestinal epithelium by modulating host Notch signaling. Development 2018, 145. [Google Scholar] [CrossRef]
- Hall, C.; Flores, M.V.; Chien, A.; Davidson, A.; Crosier, K.; Crosier, P. Transgenic zebrafish reporter lines reveal conserved Toll-like receptor signaling potential in embryonic myeloid leukocytes and adult immune cell lineages. J. Leukoc Biol. 2009, 85, 751–765. [Google Scholar] [CrossRef]
- Kanther, M.; Sun, X.; Muhlbauer, M.; Mackey, L.C.; Flynn, E.J., 3rd; Bagnat, M.; Jobin, C.; Rawls, J.F. Microbial colonization induces dynamic temporal and spatial patterns of NF-kappaB activation in the zebrafish digestive tract. Gastroenterology 2011, 141, 197–207. [Google Scholar] [CrossRef]
- Ogryzko, N.V.; Lewis, A.; Wilson, H.L.; Meijer, A.H.; Renshaw, S.A.; Elks, P.M. Hif-1alpha-Induced Expression of Il-1beta Protects against Mycobacterial Infection in Zebrafish. J. Immunol. 2019, 202, 494–502. [Google Scholar] [CrossRef]
- Tsarouchas, T.M.; Wehner, D.; Cavone, L.; Munir, T.; Keatinge, M.; Lambertus, M.; Underhill, A.; Barrett, T.; Kassapis, E.; Ogryzko, N.; et al. Dynamic control of proinflammatory cytokines Il-1beta and Tnf-alpha by macrophages in zebrafish spinal cord regeneration. Nat. Commun. 2018, 9, 4670. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, C.; Lischinsky, J.E.; Jing, M.; Zhou, J.; Wang, H.; Zhang, Y.; Dong, A.; Wu, Z.; Wu, H.; et al. A Genetically Encoded Fluorescent Sensor for Rapid and Specific In Vivo Detection of Norepinephrine. Neuron 2019, 102, 745–761. [Google Scholar] [CrossRef]
- Rawls, J.F.; Mahowald, M.A.; Ley, R.E.; Gordon, J.I. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 2006, 127, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [CrossRef]
- Kostic, A.D.; Howitt, M.R.; Garrett, W.S. Exploring host-microbiota interactions in animal models and humans. Genes Dev. 2013, 27, 701–718. [Google Scholar] [CrossRef]
- de Abreu, M.S.; Giacomini, A.; Sysoev, M.; Demin, K.A.; Alekseeva, P.A.; Spagnoli, S.T.; Kalueff, A.V. Modeling gut-brain interactions in zebrafish. Brain Res. Bull. 2019, 148, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Rawls, J.F.; Samuel, B.S.; Gordon, J.I. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc. Natl. Acad. Sci. USA 2004, 101, 4596–4601. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.M.; Mittge, E.; Kuhlman, J.; Baden, K.N.; Cheesman, S.E.; Guillemin, K. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev. Biol. 2006, 297, 374–386. [Google Scholar] [CrossRef]
- Bates, J.M.; Akerlund, J.; Mittge, E.; Guillemin, K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2007, 2, 371–382. [Google Scholar] [CrossRef]
- Semova, I.; Carten, J.D.; Stombaugh, J.; Mackey, L.C.; Knight, R.; Farber, S.A.; Rawls, J.F. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 2012, 12, 277–288. [Google Scholar] [CrossRef]
- Falcinelli, S.; Rodiles, A.; Unniappan, S.; Picchietti, S.; Gioacchini, G.; Merrifield, D.L.; Carnevali, O. Probiotic treatment reduces appetite and glucose level in the zebrafish model. Sci. Rep. 2016, 6, 18061. [Google Scholar] [CrossRef]
- Okazaki, F.; Zang, L.; Nakayama, H.; Chen, Z.; Gao, Z.J.; Chiba, H.; Hui, S.P.; Aoki, T.; Nishimura, N.; Shimada, Y. Microbiome Alteration in Type 2 Diabetes Mellitus Model of Zebrafish. Sci. Rep. 2019, 9, 867. [Google Scholar] [CrossRef] [PubMed]
- Bertotto, L.B.; Catron, T.R.; Tal, T. Exploring interactions between xenobiotics, microbiota, and neurotoxicity in zebrafish. Neurotoxicology 2020, 76, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Luczynski, P.; McVey Neufeld, K.A.; Oriach, C.S.; Clarke, G.; Dinan, T.G.; Cryan, J.F. Growing up in a Bubble: Using Germ-Free Animals to Assess the Influence of the Gut Microbiota on Brain and Behavior. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef]
- Davis, D.J.; Bryda, E.C.; Gillespie, C.H.; Ericsson, A.C. Microbial modulation of behavior and stress responses in zebrafish larvae. Behav. Brain Res. 2016, 311, 219–227. [Google Scholar] [CrossRef]
- Phelps, D.; Brinkman, N.E.; Keely, S.P.; Anneken, E.M.; Catron, T.R.; Betancourt, D.; Wood, C.E.; Espenschied, S.T.; Rawls, J.F.; Tal, T. Microbial colonization is required for normal neurobehavioral development in zebrafish. Sci. Rep. 2017, 7, 11244. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.J.; Doerr, H.M.; Grzelak, A.K.; Busi, S.B.; Jasarevic, E.; Ericsson, A.C.; Bryda, E.C. Lactobacillus plantarum attenuates anxiety-related behavior and protects against stress-induced dysbiosis in adult zebrafish. Sci. Rep. 2016, 6, 33726. [Google Scholar] [CrossRef]
- Borrelli, L.; Aceto, S.; Agnisola, C.; De Paolo, S.; Dipineto, L.; Stilling, R.M.; Dinan, T.G.; Cryan, J.F.; Menna, L.F.; Fioretti, A. Probiotic modulation of the microbiota-gut-brain axis and behaviour in zebrafish. Sci. Rep. 2016, 6, 30046. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Meshalkina, D.A.; Kizlyk, M.N.; Kysil, E.V.; Collier, A.D.; Echevarria, D.J.; Abreu, M.S.; Barcellos, L.J.; Song, C.; Warnick, J.E.; Kyzar, E.J. Zebrafish models of autism spectrum disorder. Exp. Neurol. 2018, 299, 207–216. [Google Scholar] [CrossRef]
- Sailer, L.; Duclot, F.; Wang, Z.; Kabbaj, M. Consequences of prenatal exposure to valproic acid in the socially monogamous prairie voles. Sci. Rep. 2019, 9, 2453. [Google Scholar] [CrossRef] [PubMed]
- Maaswinkel, H.; Zhu, L.; Weng, W. Assessing social engagement in heterogeneous groups of zebrafish: A new paradigm for autism-like behavioral responses. PLoS ONE 2013, 8, e75955. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lei, L.; Tian, L.; Hou, F.; Roper, C.; Ge, X.; Zhao, Y.; Chen, Y.; Dong, Q.; Tanguay, R.L. Developmental and behavioral alterations in zebrafish embryonically exposed to valproic acid (VPA): An aquatic model for autism. Neurotoxicol. Teratol. 2018, 66, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Rea, V.; Van Raay, T.J. Using Zebrafish to Model Autism Spectrum Disorder: A Comparison of ASD Risk Genes Between Zebrafish and Their Mammalian Counterparts. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef]
- Kim, O.-H.; Cho, H.-J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.-H.; Hwang, K.-S.; Jeong, Y.-M.; Yang, S.-Y.; Yu, K. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism. 2017, 8, 50. [Google Scholar] [CrossRef]
- Liu, C.X.; Li, C.Y.; Hu, C.C.; Wang, Y.; Lin, J.; Jiang, Y.H.; Li, Q.; Xu, X. CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism. 2018, 9, 23. [Google Scholar] [CrossRef]
- James, D.M.; Kozol, R.A.; Kajiwara, Y.; Wahl, A.L.; Storrs, E.C.; Buxbaum, J.D.; Klein, M.; Moshiree, B.; Dallman, J.E. Intestinal dysmotility in a zebrafish (Danio rerio) shank3a;shank3b mutant model of autism. Mol. Autism. 2019, 10, 3. [Google Scholar] [CrossRef]
- Liu, F.; Li, J.; Wu, F.; Zheng, H.; Peng, Q.; Zhou, H. Altered composition and function of intestinal microbiota in autism spectrum disorders: A systematic review. Transl. Psychiatry 2019, 9, 43. [Google Scholar] [CrossRef]
- Bruckner, J.J.; Stednitz, S.J.; Grice, M.Z.; Larsch, J.; Tallafuss, A.; Washbourne, P.; Eisen, J.S. The microbiota promotes social behavior by neuro-immune modulation of neurite complexity. BioRxiv 2020. [Google Scholar] [CrossRef]
- Nery, L.R.; Eltz, N.S.; Hackman, C.; Fonseca, R.; Altenhofen, S.; Guerra, H.N.; Freitas, V.M.; Bonan, C.D.; Vianna, M.R. Brain intraventricular injection of amyloid-beta in zebrafish embryo impairs cognition and increases tau phosphorylation, effects reversed by lithium. PLoS ONE 2014, 9, e105862. [Google Scholar] [CrossRef] [PubMed]
- Javed, I.; Peng, G.; Xing, Y.; Yu, T.; Zhao, M.; Kakinen, A.; Faridi, A.; Parish, C.L.; Ding, F.; Davis, T.P.; et al. Inhibition of amyloid beta toxicity in zebrafish with a chaperone-gold nanoparticle dual strategy. Nat. Commun. 2019, 10, 3780. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, G.G.; Lim, S.; Leighton, P.; Allison, W.T.; Rihel, J. Sleep is bi-directionally modified by amyloid beta oligomers. Elife 2020, 9, e53995. [Google Scholar] [CrossRef]
- Bhattarai, P.; Thomas, A.K.; Cosacak, M.I.; Papadimitriou, C.; Mashkaryan, V.; Froc, C.; Reinhardt, S.; Kurth, T.; Dahl, A.; Zhang, Y.; et al. IL4/STAT6 Signaling Activates Neural Stem Cell Proliferation and Neurogenesis upon Amyloid-beta42 Aggregation in Adult Zebrafish Brain. Cell Rep. 2016, 17, 941–948. [Google Scholar] [CrossRef]
- Bhattarai, P.; Cosacak, M.I.; Mashkaryan, V.; Demir, S.; Popova, S.D.; Govindarajan, N.; Brandt, K.; Zhang, Y.; Chang, W.; Ampatzis, K.; et al. Neuron-glia interaction through Serotonin-BDNF-NGFR axis enables regenerative neurogenesis in Alzheimer’s model of adult zebrafish brain. PLoS Biol. 2020, 18, e3000585. [Google Scholar] [CrossRef] [PubMed]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.M.; Berg, S.; Hellberg, S.; Falting, J.; Distel, M.; Koster, R.W.; Schmid, B.; et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef]
- Lopez, A.; Lee, S.E.; Wojta, K.; Ramos, E.M.; Klein, E.; Chen, J.; Boxer, A.L.; Gorno-Tempini, M.L.; Geschwind, D.H.; Schlotawa, L.; et al. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 2017, 140, 1128–1146. [Google Scholar] [CrossRef]
- Cosacak, M.I.; Bhattarai, P.; Bocova, L.; Dzewas, T.; Mashkaryan, V.; Papadimitriou, C.; Brandt, K.; Hollak, H.; Antos, C.L.; Kizil, C. Human TAU(P301L) overexpression results in TAU hyperphosphorylation without neurofibrillary tangles in adult zebrafish brain. Sci. Rep. 2017, 7, 12959. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Cell Type | Promoter (Gene Name) | Zebrafish Transgenic Line | Features | References |
|---|---|---|---|---|
| Myeloid cell | spi1b/pu.1 (Spi-1 proto-oncogene b) | Tg(spi1b:GAL4,UAS:EGFP) | GFP or RFP expression combined with other UAS lines | [166] |
| Tg(spi1b:GAL4,UAS:TagRFP) | ||||
| Neutrophil | mpx (myeloid-specific peroxidase) | Tg(mpx:GFP) | GFP or mCherry expression | [167] |
| Tg(mpx:mCherry) | [168] | |||
| lyz (lysozyme) | Tg(lyz:EGFP) | GFP expression | [169] | |
| Macrophage | mpeg1.1 (macrophage expressed 1, tandem duplicate 1) | Tg(mpeg1:dendra2) | Dendra2 converted by UV from green to red for cell tracking | [170] |
| Tg(mpeg1: GFP-CAAX) | Expression of membrane-GFP in macrophages | [166] | ||
| mfap (microfibril associated protein 4) | Tg(mfap4:dLanYFP-CAAX) | Expression of membrane-YFP in macrophages | [170] | |
| csf1ra/fms (colony stimulating factor 1 receptor, a) | TgBAC(csf1ra:GFP) | GFP expression | [171] | |
| Tg(fms::nfsB-mCherry) | mCherry expression conditional cell ablation by metronidazole treatment | [172] | ||
| irg1 (immunoresponsive gene 1) | Tg(irg1:EGFP) | GFP expression Expression of activated macrophages | [173] | |
| Microglia (the resident innate immune cells in the brain) | apoeb (apolipoprotein Eb) | Tg(apoeb:lyn-EGFP) | expression of membrane-GFP in microglia | [174] |
| slc7a7 (solute carrier family 7 member 7) | Tg(slc7a7:Kaede) | Microglia precursor expression Kaede converted by UV from green to red for cell tracking | [172] | |
| p2ry12 (purinergic receptor P2Y12) | TgBAC(p2ry12:p2ry12-GFP) | GFP expression | [175] | |
| T cell | lck (LCK proto-oncogene, Src family tyrosine kinase) | Tg(lck:GFP) | GFP expression | [171] |
| B cell | ighm (immunoglobulin heavy constant mu) | Tg(IgM1:eGFP) | GFP expression | [176] |
| nodose ganglia (vagus nerve) | isl1 (ISL LIM Homeobox 1) | Tg(isl1:EGFP) | a subset of isl1+ cells | [141] |
| enterocyte | cldn15la (claudin 15-like a) | TgBAC(cldn15la-GFP); Tg(-0.349cldn15la: mCherry) | absorption | [177,178] |
| enteroendocrine | neurod1 (Neuronal Differentiation 1) | TgBAC(neurod1:EGFP) Tg(neurod1:RFP) | intestinal hormone release | [140] |
| nkx2.2a (NK2 homeobox 2a) | Tg(nkx2.2a:mEGFP) | intestinal hormone release | [179] |
| Immune-Signaling Component | Description | Zebrafish Transgenic Line | References |
|---|---|---|---|
| myd88 (myeloid differentiation response gene 88) | major adaptor protein of Toll-like receptor (TLR) –mediated signaling | Tg(myd88:EGFP) Tg(myd88:DsRED2) | [180] |
| NF-κB (nuclear factor κ-light chain enhancer of activated B cells) | transcription factor; activated by bacteria-derived cytokines | Tg(NF-κB:EGFP) | [181] |
| il1β (interleukin 1, beta) | pro-inflammatory cytokine | TgBAC(il1β:eGFP) | [182] |
| tnf-α (tumor necrosis factor α) | pro-inflammatory cytokine | Tg(tnf-α:GFP) | [183] |
| MGBA Cell Types | Description | Zebrafish Transgenic Line | References |
|---|---|---|---|
| dopamine | in vivo dopamine sensor | Tg(elval3: DA1m) | [165] |
| norepinephrine | in vivo norepinephrine sensor | Tg(HuC:GRABNE1m) | [184] |
| Kaede | UV-photoconvertible sensor | Tg(Gal4-VP16;UAS:EGFP) X Tg(UAS:Kaede) | [164] |
| CaMPARI | UV-photoconvertible calcium sensor | Tg(neurod1:CaMPARI) | [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-G.; Cho, H.-J.; Jeong, Y.-M.; Lee, J.-S. Genetic Approaches Using Zebrafish to Study the Microbiota–Gut–Brain Axis in Neurological Disorders. Cells 2021, 10, 566. https://doi.org/10.3390/cells10030566
Lee J-G, Cho H-J, Jeong Y-M, Lee J-S. Genetic Approaches Using Zebrafish to Study the Microbiota–Gut–Brain Axis in Neurological Disorders. Cells. 2021; 10(3):566. https://doi.org/10.3390/cells10030566
Chicago/Turabian StyleLee, Jae-Geun, Hyun-Ju Cho, Yun-Mi Jeong, and Jeong-Soo Lee. 2021. "Genetic Approaches Using Zebrafish to Study the Microbiota–Gut–Brain Axis in Neurological Disorders" Cells 10, no. 3: 566. https://doi.org/10.3390/cells10030566
APA StyleLee, J.-G., Cho, H.-J., Jeong, Y.-M., & Lee, J.-S. (2021). Genetic Approaches Using Zebrafish to Study the Microbiota–Gut–Brain Axis in Neurological Disorders. Cells, 10(3), 566. https://doi.org/10.3390/cells10030566