
Involvement of the Catecholamine Pathway in Glioblastoma Development

 ,
, 

Abstract
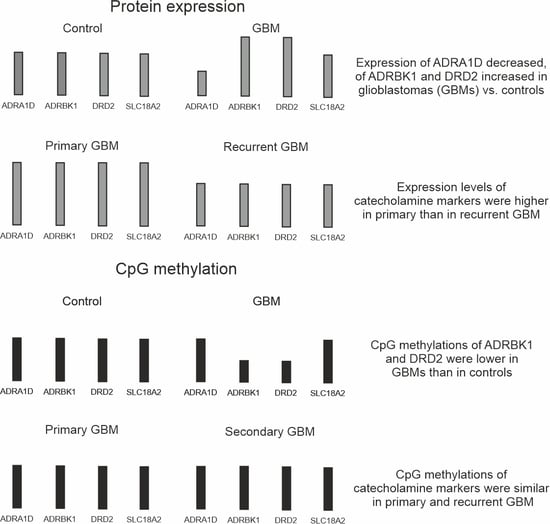
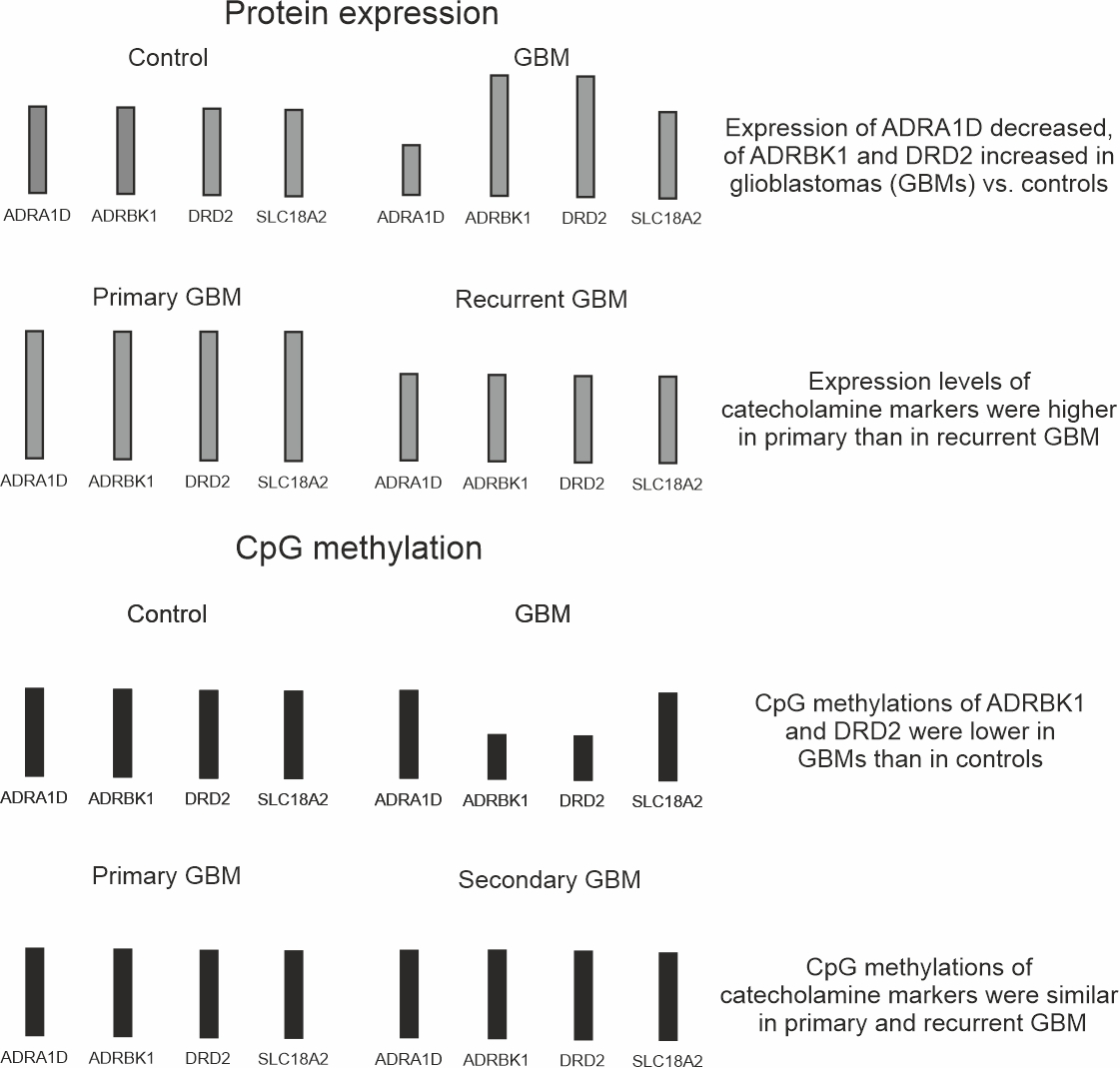{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Subjects of the Study
2.2. DNA Isolation, Library Preparation, CpG Methylation Profiling and Bioinformatic Analyses
2.3. Immunhistochemistry (IHC)
2.4. Statistics and Bioinformatics
3. Results
3.1. Protein Expression Levels of the Four Catecholamine Markers in Paired GBM Samples and Controls
3.2. Promoter and Gene Methylation Profiles of the Four Catecholamine Markers in Individual GBM and Control Samples
3.3. DNA CpG Methylation Levels in Promoter and Gene Regions of Catecholamine Markers in a Database GBM Cohort (Figure 4)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Code Availability
References
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Kraboth, Z.; Galik, B.; Tompa, M.; Kajtar, B.; Urban, P.; Gyenesei, A.; Kalman, B. DNA CpG methylation in sequential glioblastoma specimens. J. Cancer Res. Clin. Oncol. 2020, 146, 2885–2896. [Google Scholar] [CrossRef]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Anderson, D.J. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Stommel, J.M. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef]
- Natsume, A.; Kinjo, S.; Yuki, K.; Kato, T.; Ohno, M.; Motomura, K.; Wakabayashi, T. Glioma-initiating cells and molecular pathology: Implications for therapy. Brain Tumor Pathol. 2011, 28, 1–12. [Google Scholar] [CrossRef]
- Lennarz, W.J.; Lane, M.D. Encyclopedia of Biological Chemistry; Academic Press: Cambridge, MA, USA, 2013; ISBN 978-0-12-378630-2. [Google Scholar]
- Fitzgerald, P.J. Is norepinephrine an etiological factor in some types of cancer? Int. J. Cancer 2009, 124, 257–263. [Google Scholar] [CrossRef]
- Filippi, S.; Parenti, A.; Donnini, S.; Granger, H.J.; Fazzini, A.; Ledda, F. α1D-adrenoceptors cause endothelium-dependent vasodilatation in the rat mesenteric vascular bed. J. Pharmacol. Exp. Ther. 2001, 296, 869–875. [Google Scholar] [PubMed]
- Vinci, M.C.; Bellik, L.; Filippi, S.; Ledda, F.; Parenti, A. Trophic effects induced by α1D-adrenoceptors on endothelial cells are potentiated by hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2140–H2147. [Google Scholar] [CrossRef] [PubMed]
- Underland, L.J.; Mark, E.R.; Katikaneni, R.; Heptulla, R. The impact of dopamine on insulin secretion in healthy controls. Indian J. Crit. Care Med. Peer Rev. Off. Publ. Indian Soc. Crit. Care Med. 2018, 22, 209. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Basu, B.; Chakroborty, D.; Dasgupta, P.S.; Basu, S. The immunoregulatory role of dopamine: An update. Brain Behav. Immun. 2010, 24, 525–528. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Brinkley, D.M.; Morris, D.P.; Smith, M.P.; Louie, R.J.; Schwinn, D.A. Epigenetic regulation of human α1d-adrenergic receptor gene expression: A role for DNA methylation in Spl-dependent regulation. FASEB J. 2007, 21, 1979–1993. [Google Scholar] [CrossRef]
- Calzada, B.C.; De Artiñano, A.A. Alpha-adrenoceptor subtypes. Pharmacol. Res. 2001, 44, 195–208. [Google Scholar] [CrossRef]
- Meyer, J.S.; Quenzer, L.F. Psychopharmacology: Drugs, the Brain, and Behavior; Sinauer Associates: Sunderland, MA, USA, 2005. [Google Scholar]
- Weselek, G.; Keiner, S.; Fauser, M.; Wagenführ, L.; Müller, J.; Kaltschmidt, B.; Storch, A. Norepinephrine is a negative regulator of the adult periventricular neural stem cell niche. Stem Cells 2020. [Google Scholar] [CrossRef] [PubMed]
- Entschladen, F.; Drell Iv, T.L.; Lang, K.; Joseph, J.; Zaenker, K.S. Tumour-cell migration, invasion, and metastasis: Navigation by neurotransmitters. Lancet Oncol. 2004, 5, 254–258. [Google Scholar] [CrossRef]
- Schuller, H.M. Neurotransmitter receptor-mediated signaling pathways as modulators of carcinogenesis. In Neuronal Activity in Tumor Tissue; Karger Publishers: Berlin, Germany, 2007; Volume 39, pp. 45–63. [Google Scholar] [CrossRef]
- Adissu, H.A.; Schuller, H.M. Antagonistic growth regulation of cell lines derived from human lung adenocarcinomas of Clara cell and aveolar type II cell lineage: Implications for chemoprevention. Int. J. Oncol. 2004, 24, 1467–1472. [Google Scholar] [CrossRef]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and functional basis of dopamine receptors and their role in neurogenesis: Possible implication for Parkinson’s disease. J. Exp. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.A.; Womeldorff, M.R.; Jensen, R. Meningioma: Signaling pathways and tumor growth. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 169, pp. 137–150. [Google Scholar] [CrossRef]
- Quintana, C.; Beaulieu, J.M. A fresh look at cortical dopamine D2 receptor expressing neurons. Pharmacol. Res. 2019, 139, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Armando, I.; Villar, V.A.M.; Jose, P.A. Dopamine and renal function and blood pressure regulation. Compr. Physiol. 2011, 1, 1075–1117. [Google Scholar] [CrossRef]
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. Midbrain dopaminergic neurons: A review of the molecular circuitry that regulates their development. Dev. Biol. 2013, 379, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Alcaro, A.; Huber, R.; Panksepp, J. Behavioral functions of the mesolimbic dopaminergic system: An affective neuroethological perspective. Brain Res. Rev. 2007, 56, 283–321. [Google Scholar] [CrossRef] [PubMed]
- Berridge, K.C. The debate over dopamine’s role in reward: The case for incentive salience. Psychopharmacology 2007, 191, 391–431. [Google Scholar] [CrossRef]
- Ramanathan, S.; Al-Diwani, A.; Waters, P.; Irani, S.R. The autoantibody-mediated encephalitides: From clinical observations to molecular pathogenesis. J. Neurol. 2019, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Takamura, N.; Nakagawa, S.; Masuda, T.; Boku, S.; Kato, A.; Song, N.; Kusumi, I. The effect of dopamine on adult hippocampal neurogenesis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 50, 116–124. [Google Scholar] [CrossRef]
- Ohira, K. Dopamine as a growth differentiation factor in the mammalian brain. Neural Regen. Res. 2020, 15, 390. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, S.; Kozono, D.; Ng, K.; Futalan, D.; Shen, Y.; Carter, B.S. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget 2014, 5, 882. [Google Scholar] [CrossRef]
- Caragher, S.P.; Shireman, J.M.; Huang, M.; Miska, J.; Atashi, F.; Baisiwala, S.; Lesniak, M.S. Activation of dopamine receptor 2 prompts transcriptomic and metabolic plasticity in glioblastoma. J. Neurosci. 2019, 39, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Hodny, Z. Dopamine signaling: Target in glioblastoma. Oncotarget 2014, 5, 1116. [Google Scholar] [CrossRef] [PubMed]
- Métayé, T.; Gibelin, H.; Perdrisot, R.; Kraimps, J.L. Pathophysiological roles of G-protein-coupled receptor kinases. Cell. Signal. 2005, 17, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Komolov, K.E.; Benovic, J.L. G protein-coupled receptor kinases: Past, present and future. Cell. Signal. 2018, 41, 17–24. [Google Scholar] [CrossRef]
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar] [PubMed]
- Ribas, C.; Penela, P.; Murga, C.; Salcedo, A.; García-Hoz, C.; Jurado-Pueyo, M.; Mayor, F., Jr. The G protein-coupled receptor kinase (GRK) interactome: Role of GRKs in GPCR regulation and signaling. Biochim. Biophys. Acta Biomembr. 2007, 1768, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Murga, C.; Arcones, A.C.; Cruces-Sande, M.; Briones, A.M.; Salaices, M.; Mayor, F., Jr. G protein-coupled receptor kinase 2 (GRK2) as a potential therapeutic target in cardiovascular and metabolic diseases. Front. Pharmacol. 2019, 10, 112. [Google Scholar] [CrossRef]
- Evron, T.; Daigle, T.L.; Caron, M.G. GRK2: Multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 2012, 33, 154–164. [Google Scholar] [CrossRef]
- Sun, W.Y.; Wu, J.J.; Peng, W.T.; Sun, J.C.; Wei, W. The role of G protein-coupled receptor kinases in the pathology of malignant tumors. Acta Pharmacol. Sin. 2018, 39, 1699–1705. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Murga, C.; Ribas, C.; Lafarga, V.; Mayor, F., Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br. J. Pharmacol. 2010, 160, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Bathgate, A. Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: The known and the unknown. Pharmacogenomics 2012, 13, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Nogués, L.; Palacios-García, J.; Reglero, C.; Rivas, V.; Neves, M.; Ribas, C.; Mayor, F., Jr. G Protein-Coupled Receptor Kinases (GRKs) in Tumorigenesis and Cancer Progression: GPCR Regulators and Signaling Hubs. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2018; Volume 48, pp. 78–90. [Google Scholar] [CrossRef]
- Woerner, B.M.; Luo, J.; Brown, K.R.; Jackson, E.; Dahiya, S.M.; Mischel, P.; Rubin, J.B. Suppression of G-protein–Coupled Receptor Kinase 3 Expression Is a Feature of Classical GBM That Is Required for Maximal Growth. Mol. Cancer Res. 2012, 10, 156–166. [Google Scholar] [CrossRef]
- Kaur, G.; Kim, J.; Kaur, R.; Tan, I.; Bloch, O.; Sun, M.Z.; Parsa, A.T. G-protein coupled receptor kinase (GRK)-5 regulates proliferation of glioblastoma-derived stem cells. J. Clin. Neurosci. 2013, 20, 1014–1018. [Google Scholar] [CrossRef]
- Yu, S.; Sun, L.; Jiao, Y.; Lee, L.T.O. The role of G protein-coupled receptor kinases in cancer. Int. J. Biol. Sci. 2018, 14, 189. [Google Scholar] [CrossRef]
- Hu, C.; Tao, L.; Cao, X.; Chen, L. The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian J. Pharm. Sci. 2020, 15, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, K.G. Presynaptic regulation of dopamine release: Role of the DAT and VMAT2 transporters. Neurochem. Int. 2019, 122, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Omote, H.; Miyaji, T.; Hiasa, M.; Juge, N.; Moriyama, Y. Structure, function, and drug interactions of neurotransmitter transporters in the postgenomic era. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Lawal, H.O.; Krantz, D.E. SLC18: Vesicular neurotransmitter transporters for monoamines and acetylcholine. Mol. Asp. Med. 2013, 34, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.H.; Nowosielski, M. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018, 24, 1611–1624. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Molecular mechanisms of RNA-triggered gene silencing machineries. Acc. Chem. Res. 2012, 45, 1122–1131. [Google Scholar] [CrossRef]
- Moarii, M.; Boeva, V.; Vert, J.P.; Reyal, F. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genom. 2015, 16, 1–14. [Google Scholar] [CrossRef]
- Kobayashi, K. Role of catecholamine signaling in brain and nervous system functions: New insights from mouse molecular genetic study. J. Investig. Dermatol. Symp. Proc. 2001, 6, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Westmoreland, S.V.; Bahn, M.E.; Chen, G.L.; Yang, H.; Vallender, E.J.; Miller, G.M. Rhesus monkey trace amine-associated receptor 1 signaling: Enhancement by monoamine transporters and attenuation by the D2 autoreceptor in vitro. J. Pharmacol. Exp. Ther. 2007, 321, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Stupp, R.; Homicsko, K. Standards of care and novel approaches in the management of glioblastoma multiforme. Chin. J. Cancer 2014, 33, 32. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Xu, J.; Hu, J.; Guo, F.; Wan, L.; Cheng, M.; Pang, L. Single-cell RNA-seq reveals the invasive trajectory and molecular cascades underlying glioblastoma progression. Mol. Oncol. 2019, 13, 2588–2603. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Chakroborty, D.; Chowdhury, U.R.; Dasgupta, P.S.; Basu, S. Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin. Cancer Res. 2008, 14, 2502–2510. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Wang, C.; Chen, X.; Duan, C.; Zhang, X.; Zhang, J.; Yang, J. Dopamine induces growth inhibition and vascular normalization through reprogramming M2-polarized macrophages in rat C6 glioma. Toxicol. Appl. Pharmacol. 2015, 286, 112–123. [Google Scholar] [CrossRef]
- Marisetty, A.L.; Lu, L.; Veo, B.L.; Liu, B.; Coarfa, C.; Kamal, M.M.; Majumder, S. REST-DRD2 mechanism impacts glioblastoma stem cell–mediated tumorigenesis. Neuro-oncology 2019, 21, 775–785. [Google Scholar] [CrossRef]
- Pathania, A.S.; Ren, X.; Mahdi, M.Y.; Shackleford, G.M.; Erdreich-Epstein, A. GRK2 promotes growth of medulloblastoma cells and protects them from chemotherapy-induced apoptosis. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mundell, S.J.; Kelly, E. The effect of inhibitors of receptor internalization on the desensitization and resensitization of three Gs-coupled receptor responses. Br. J. Pharmacol. 1998, 125, 1594–1600. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Kugimiya, T.; Miyazaki, T. Substance P receptor in U373 MG human astrocytoma cells activates mitogen-activated protein kinases ERK1/2 through Src. Brain Tumor Pathol. 2005, 22, 1–8. [Google Scholar] [CrossRef]
- Eiden, L.E.; Weihe, E. VMAT2: A dynamic regulator of brain monoaminergic neuronal function interacting with drugs of abuse. Ann. N. Y. Acad. Sci. 2011, 1216, 86. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Krantz, D.E. Vesicular Neurotransmitter Transporters. In Handbook of Neurochemistry and Molecular Neurobiology; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Lohr, K.M.; Chen, M.; Hoffman, C.A.; McDaniel, M.J.; Stout, K.A.; Dunn, A.R.; Miller, G.W. Vesicular monoamine transporter 2 (VMAT2) level regulates MPTP vulnerability and clearance of excess dopamine in mouse striatal terminals. Toxicol. Sci. 2016, 153, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Sanacora, G.; Anand, A.; Roach, L.M.; Fasula, M.K.; Finkelstein, C.O.; Charney, D.S. Monoamine depletion in unmedicated depressed subjects. Biol. Psychiatry 2002, 51, 469–473. [Google Scholar] [CrossRef]
- Schweimer, J.; Saft, S.; Hauber, W. Involvement of catecholamine neurotransmission in the rat anterior cingulate in effort-related decision making. Behav. Neurosci. 2005, 119, 1687. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Curschmann, J. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, X.; Gao, L.; Liu, X.; Li, J.; Huang, X.; Zeng, T. Synergistic suppression of glioblastoma cell growth by combined application of temozolomide and dopamine D2 receptor antagonists. World Neurosurg. 2019, 128, e468–e477. [Google Scholar] [CrossRef] [PubMed]
- Nogués, L.; Reglero, C.; Rivas, V.; Salcedo, A.; Lafarga, V.; Neves, M.; Zhou, X.Z. G protein-coupled receptor kinase 2 (GRK2) promotes breast tumorigenesis through a HDAC6-Pin1 axis. EBioMedicine 2016, 13, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Al-Shanti, N.; Stewart, C. C2 skeletal myoblast survival, death, proliferation and differentiation: Regulation by Adra1d. Cell. Physiol. Biochem. 2010, 25, 253–262. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Horstmann, H. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Woo, P.J. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Suvà, M. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Shim, J.K.; Yoon, S.J.; Kim, S.H.; Chang, J.H.; Kang, S.G. Transcriptome profiling-based identification of prognostic subtypes and multi-omics signatures of glioblastoma. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Levitin, H.M.; Frattini, V.; Bush, E.C.; Boyett, D.M.; Samanamud, J.; Sims, P.A. Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Med. 2018, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraboth, Z.; Kajtár, B.; Gálik, B.; Gyenesei, A.; Miseta, A.; Kalman, B. Involvement of the Catecholamine Pathway in Glioblastoma Development. Cells 2021, 10, 549. https://doi.org/10.3390/cells10030549
Kraboth Z, Kajtár B, Gálik B, Gyenesei A, Miseta A, Kalman B. Involvement of the Catecholamine Pathway in Glioblastoma Development. Cells. 2021; 10(3):549. https://doi.org/10.3390/cells10030549
Chicago/Turabian StyleKraboth, Zoltán, Bela Kajtár, Bence Gálik, Attila Gyenesei, Attila Miseta, and Bernadette Kalman. 2021. "Involvement of the Catecholamine Pathway in Glioblastoma Development" Cells 10, no. 3: 549. https://doi.org/10.3390/cells10030549
APA StyleKraboth, Z., Kajtár, B., Gálik, B., Gyenesei, A., Miseta, A., & Kalman, B. (2021). Involvement of the Catecholamine Pathway in Glioblastoma Development. Cells, 10(3), 549. https://doi.org/10.3390/cells10030549