
Iron Deficiency in Pulmonary Arterial Hypertension: A Deep Dive into the Mechanisms

 , and
, and
Abstract
1. Introduction
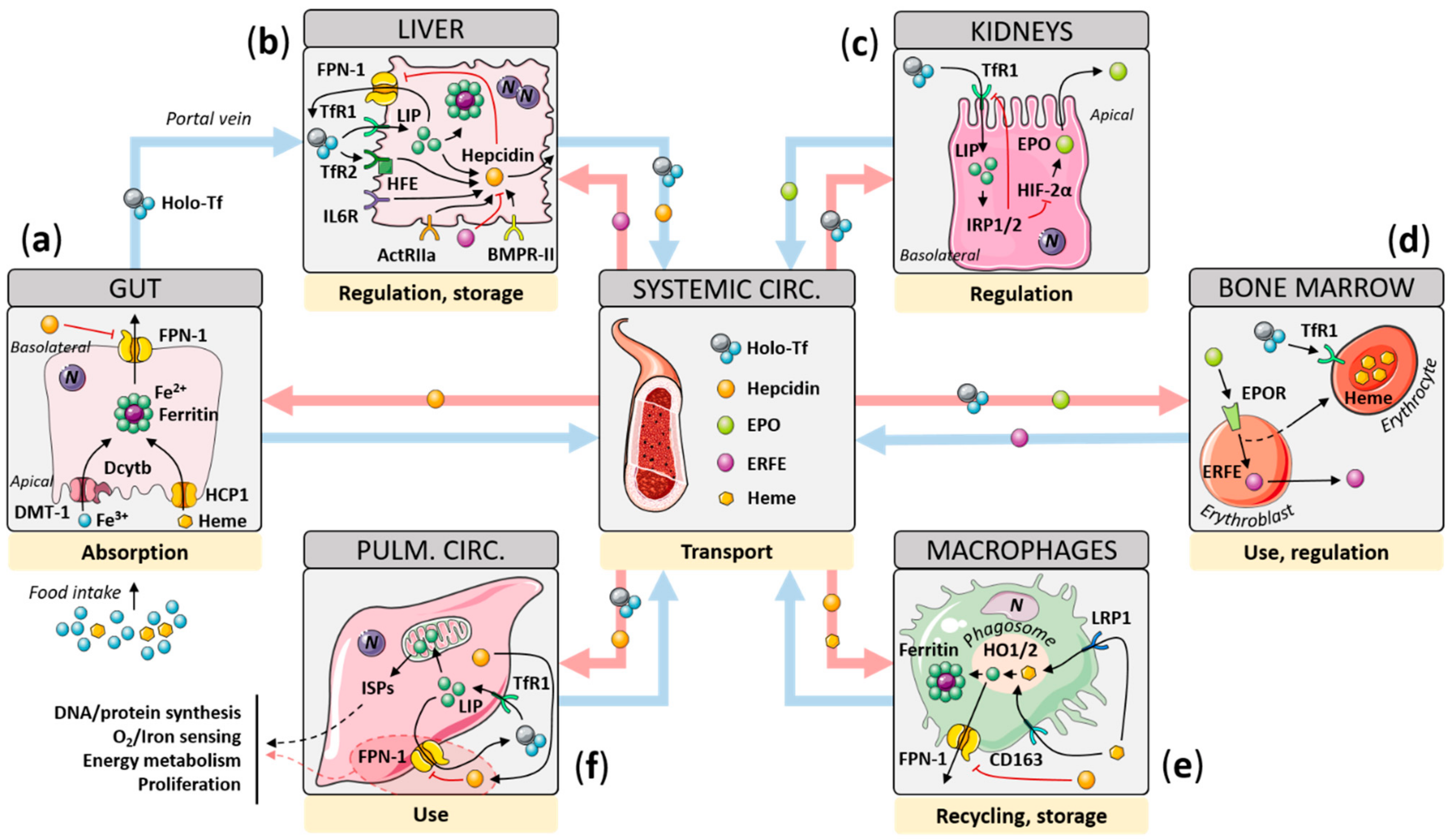
2. Iron Homeostasis: Mechanisms of Regulation
2.1. Gut
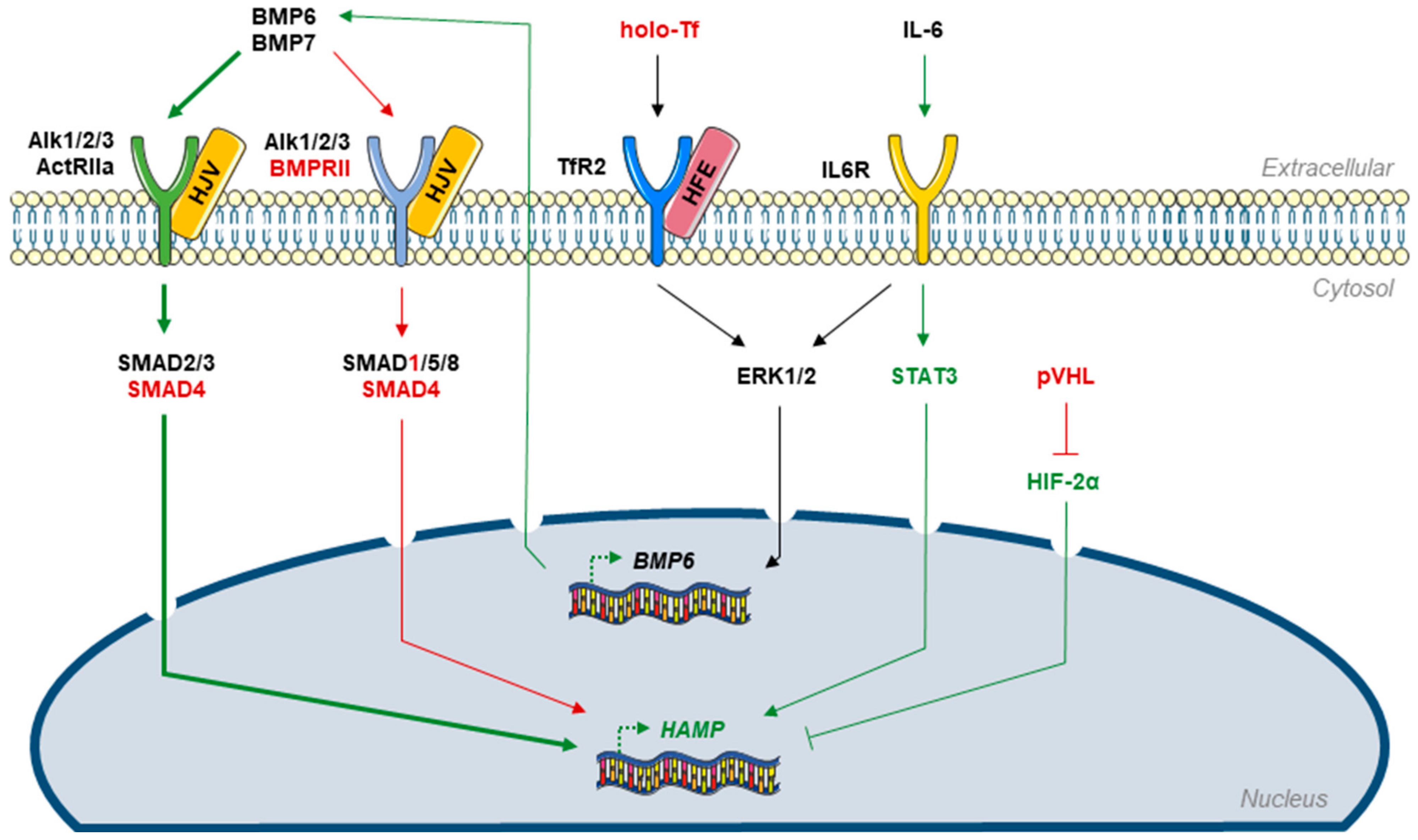
2.2. Liver
2.3. Kidney
2.4. Scavenging Macrophages
2.5. Cellular Iron Sensing, Consumption, and Functions
3. Iron Status in PAH and Experimental PH
3.1. Iron Levels and Manipulation in PAH
3.2. Iron Levels in Experimental PH
3.3. Iron Manipulation in Experimental PH
3.4. Role of Iron in the RV
4. Molecular Links between Iron Deficiency and PAH
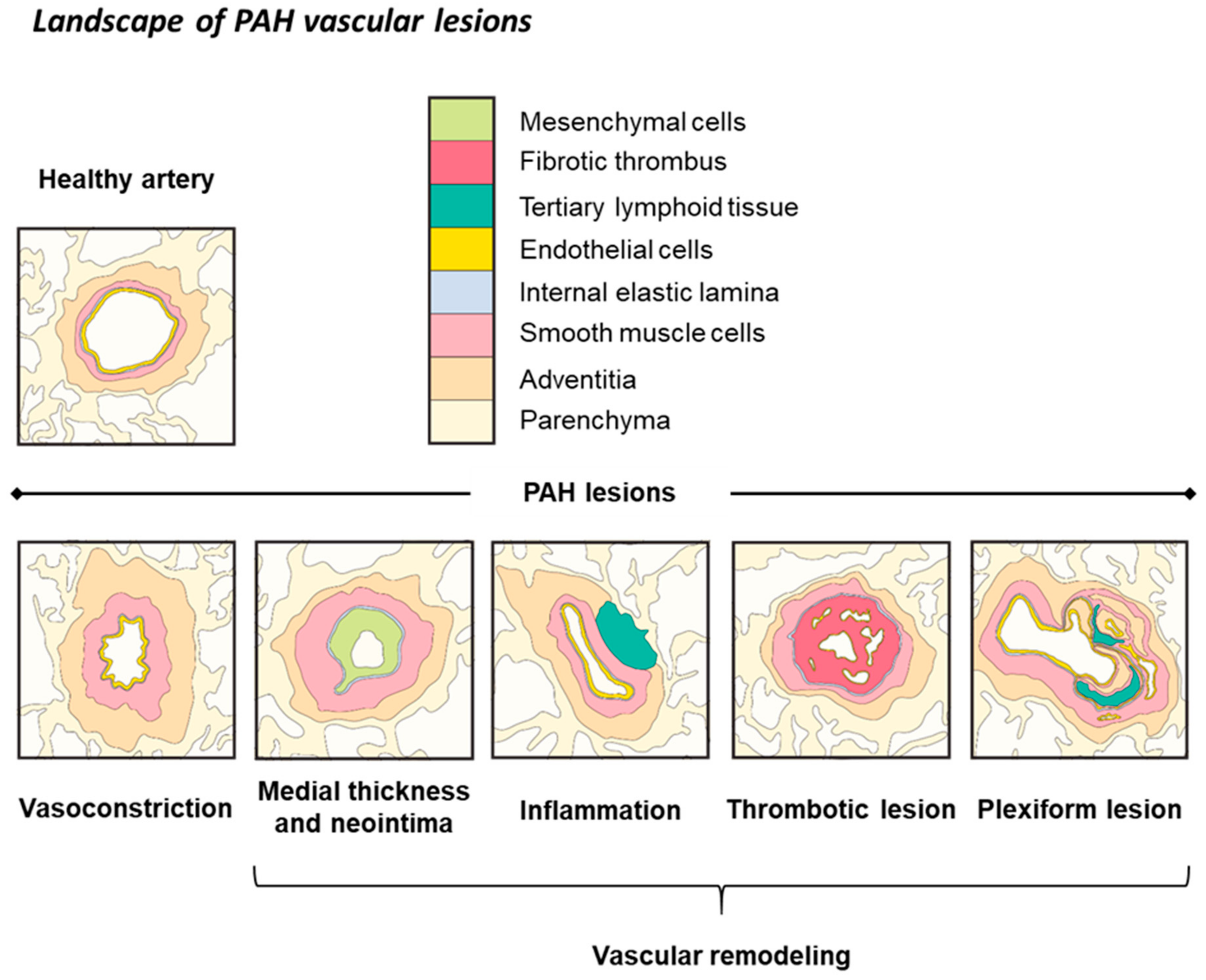
4.1. Inflammation
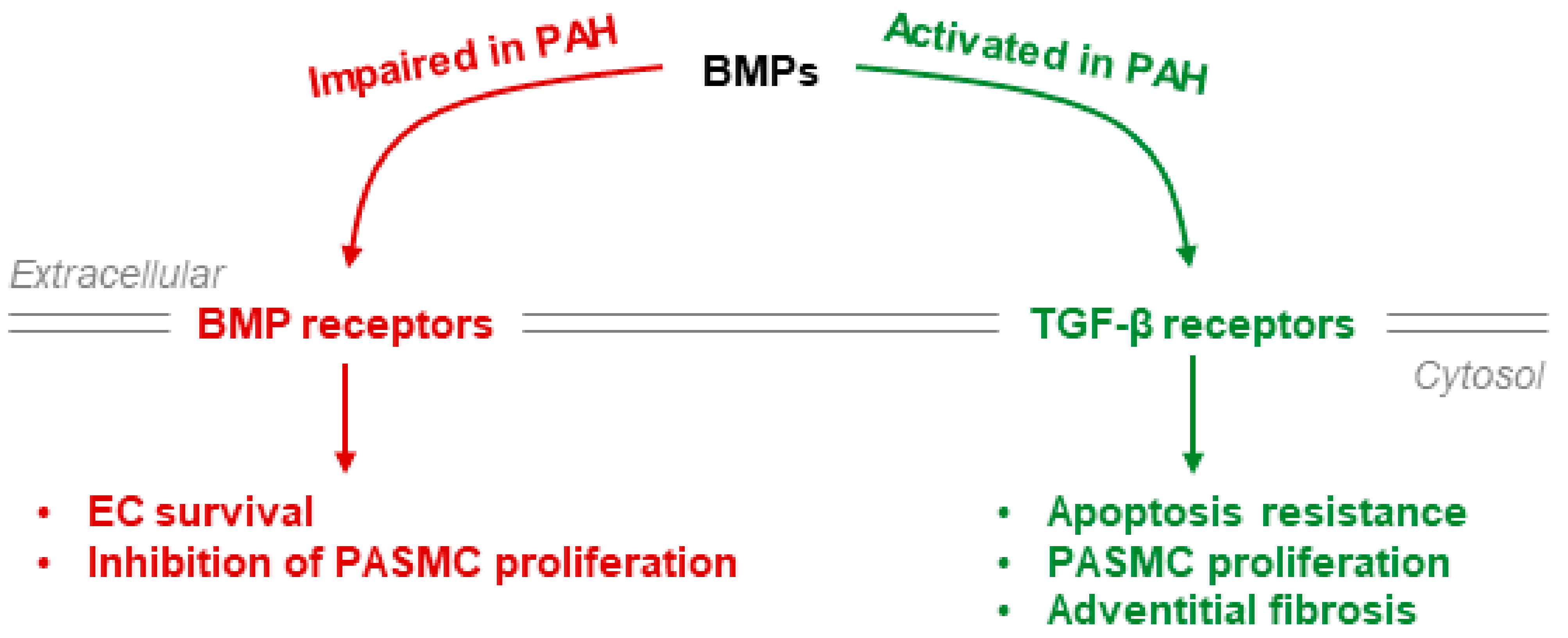
4.2. BMPR-II Signaling
4.3. Hypoxia-Inducible Factors
5. Clinical Implications
6. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.L.; Lopez, A.D. Measuring the Global Burden of Disease. N. Engl. J. Med. 2013, 369, 448–457. [Google Scholar] [CrossRef]
- Camaschella, C. Iron Deficiency: New Insights into Diagnosis and Treatment. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 8–13. [Google Scholar] [CrossRef]
- Ramakrishnan, U.; Yip, R. Experiences and Challenges in Industrialized Countries: Control of Iron Deficiency in Industrialized Countries. J. Nutr. 2002, 132. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.B.; Hurrell, R.F. Nutritional Iron Deficiency. Lancet 2007, 370, 511–520. [Google Scholar] [CrossRef]
- Maio, N.; Rouault, T.A. Iron–Sulfur Cluster Biogenesis in Mammalian Cells: New Insights into the Molecular Mechanisms of Cluster Delivery. Biochim. Biophysica Acta (BBA) Mol. Cell Res. 2015, 1853, 1493–1512. [Google Scholar] [CrossRef]
- Meneghini, R. Iron Homeostasis, Oxidative Stress, and DNA Damage. Free Radic. Biol. Med. 1997, 23, 783–792. [Google Scholar] [CrossRef]
- Wardman, P.; Candeias, L.P. Fenton Chemistry: An Introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef]
- Zhang, C. Essential Functions of Iron-Requiring Proteins in DNA Replication, Repair and Cell Cycle Control. Protein Cell 2014, 5, 750–760. [Google Scholar] [CrossRef]
- von Haehling, S.; Jankowska, E.A.; van Veldhuisen, D.J.; Ponikowski, P.; Anker, S.D. Iron Deficiency and Cardiovascular Disease. Nat. Rev. Cardiol. 2015, 12, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Howard, L.S.; Busbridge, M.; Ashby, D.; Kondili, E.; Gibbs, J.S.R.; Wharton, J.; Wilkins, M.R. Iron Deficiency and Raised Hepcidin in Idiopathic Pulmonary Arterial Hypertension: Clinical Prevalence, Outcomes, and Mechanistic Insights. J. Am. Coll. Cardiol. 2011, 58, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, G.; Lankhorst, S.; Boonstra, A.; Postmus, P.E.; Zweegman, S.; Westerhof, N.; van der Laarse, W.J.; Vonk-Noordegraaf, A. Iron Deficiency Is Common in Idiopathic Pulmonary Arterial Hypertension. Eur. Respir. J. 2011, 37, 1386–1391. [Google Scholar] [CrossRef]
- Soon, E.; Treacy, C.M.; Toshner, M.R.; MacKenzie-Ross, R.; Manglam, V.; Busbridge, M.; Sinclair-McGarvie, M.; Arnold, J.; Sheares, K.K.; Morrell, N.W.; et al. Unexplained Iron Deficiency in Idiopathic and Heritable Pulmonary Arterial Hypertension. Thorax 2011, 66, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Charlton, R.; Seftel, H.; Bothwell, T.; Mayet, F.; Adams, B.; Finch, C.; Layrisse, M. Body Iron Excretion in Man: A Collaborative Study. Am. J. Med. 1968, 45, 336–353. [Google Scholar] [CrossRef]
- Bacon, B.R.; Powell, L.W.; Adams, P.C.; Kresina, T.F.; Hoofnagle, J.H. Molecular Medicine and Hemochromatosis: At the Crossroads. Gastroenterology 1999, 116, 193–207. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Laftah, A.H.; Latunde-Dada, G.O.; Fakih, S.; Hider, R.C.; Simpson, R.J.; McKie, A.T. Haem and Folate Transport by Proton-Coupled Folate Transporter/Haem Carrier Protein 1 (SLC46A1). Br. J. Nutr. 2009, 101, 1150–1156. [Google Scholar] [CrossRef]
- Theil, E.C. Ferritin Protein Nanocages—The Story. Nanotechnol. Percept. 2012, 8, 7–16. [Google Scholar] [CrossRef]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A Novel Duodenal Iron-Regulated Transporter, IREG1, Implicated in the Basolateral Transfer of Iron to the Circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional Cloning of Zebrafish Ferroportin1 Identifies a Conserved Vertebrate Iron Exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- Roy, C.N.; Enns, C.A. Iron Homeostasis: New Tales from the Crypt. Blood 2000, 96, 4020–4027. [Google Scholar] [CrossRef]
- Zhang, A.-S.; Enns, C.A. Iron Homeostasis: Recently Identified Proteins Provide Insight into Novel Control Mechanisms. J. Biol. Chem. 2009, 284, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Koorts, A.M.; Viljoen, M. Ferritin and Ferritin Isoforms I: Structure-Function Relationships, Synthesis, Degradation and Secretion. Arch. Physiol. Biochem. 2007, 113, 30–54. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Poli, M.; Luscieti, S.; Gandini, V.; Maccarinelli, F.; Finazzi, D.; Silvestri, L.; Roetto, A.; Arosio, P. Transferrin Receptor 2 and HFE Regulate Furin Expression via Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase (MAPK/Erk) Signaling. Implications for Transferrin-Dependent Hepcidin Regulation. Haematologica 2010, 95, 1832–1840. [Google Scholar] [CrossRef]
- Wallace, D.F.; Summerville, L.; Crampton, E.M.; Frazer, D.M.; Anderson, G.J.; Subramaniam, V.N. Combined Deletion of Hfe and Transferrin Receptor 2 in Mice Leads to Marked Dysregulation of Hepcidin and Iron Overload. Hepatology 2009, 50, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Truksa, J.; Peng, H.; Lee, P.; Beutler, E. Bone Morphogenetic Proteins 2, 4, and 9 Stimulate Murine Hepcidin 1 Expression Independently of Hfe, Transferrin Receptor 2 (Tfr2), and IL-6. Proc. Natl. Acad. Sci. USA 2006, 103, 10289–10293. [Google Scholar] [CrossRef]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone Morphogenetic Protein Signaling by Hemojuvelin Regulates Hepcidin Expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef]
- Kautz, L.; Besson-Fournier, C.; Meynard, D.; Latour, C.; Roth, M.-P.; Coppin, H. Iron Overload Induces BMP6 Expression in the Liver but Not in the Duodenum. Haematologica 2011, 96, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.-S.; Gao, J.; Koeberl, D.D.; Enns, C.A. The Role of Hepatocyte Hemojuvelin in the Regulation of Bone Morphogenic Protein-6 and Hepcidin Expression in Vivo. J. Biol. Chem. 2010, 285, 16416–16423. [Google Scholar] [CrossRef]
- Wang, R.-H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A Role of SMAD4 in Iron Metabolism through the Positive Regulation of Hepcidin Expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.-P. Lack of the Bone Morphogenetic Protein BMP6 Induces Massive Iron Overload. Nat. Genet. 2009, 41, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Maurer, E.; Gütschow, M.; Stirnberg, M. Matriptase-2 (TMPRSS6) Is Directly up-Regulated by Hypoxia Inducible Factor-1: Identification of a Hypoxia-Responsive Element in the TMPRSS6 Promoter Region. Biol. Chem. 2012, 393, 535–540. [Google Scholar] [CrossRef]
- Zhang, A.-S.; Anderson, S.A.; Wang, J.; Yang, F.; DeMaster, K.; Ahmed, R.; Nizzi, C.P.; Eisenstein, R.S.; Tsukamoto, H.; Enns, C.A. Suppression of Hepatic Hepcidin Expression in Response to Acute Iron Deprivation Is Associated with an Increase of Matriptase-2 Protein. Blood 2011, 117, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Huang, F.W.; Xia, Y.; Sidis, Y.; Andrews, N.C.; Lin, H.Y. Modulation of Bone Morphogenetic Protein Signaling in Vivo Regulates Systemic Iron Balance. J. Clin. Investig. 2007, 117, 1933–1939. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The Serine Protease Matriptase-2 (TMPRSS6) Inhibits Hepcidin Activation by Cleaving Membrane Hemojuvelin. Cell Metabolism. 2008, 8, 502–511. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Camaschella, C. Furin-Mediated Release of Soluble Hemojuvelin: A New Link between Hypoxia and Iron Homeostasis. Blood 2008, 111, 924–931. [Google Scholar] [CrossRef]
- Kautz, L.; Meynard, D.; Besson-Fournier, C.; Darnaud, V.; Al Saati, T.; Coppin, H.; Roth, M.-P. BMP/Smad Signaling Is Not Enhanced in Hfe-Deficient Mice despite Increased Bmp6 Expression. Blood 2009, 114, 2515–2520. [Google Scholar] [CrossRef]
- Corradini, E.; Garuti, C.; Montosi, G.; Ventura, P.; Andriopoulos, B.; Lin, H.Y.; Pietrangelo, A.; Babitt, J.L. Bone Morphogenetic Protein Signaling Is Impaired in an HFE Knockout Mouse Model of Hemochromatosis. Gastroenterology 2009, 137, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Schmidt, P.J.; Meynard, D.; Garuti, C.; Montosi, G.; Chen, S.; Vukicevic, S.; Pietrangelo, A.; Lin, H.Y.; Babitt, J.L. BMP6 Treatment Compensates for the Molecular Defect and Ameliorates Hemochromatosis in Hfe Knockout Mice. Gastroenterology 2010, 139, 1721–1729. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 Mediates Hypoferremia of Inflammation by Inducing the Synthesis of the Iron Regulatory Hormone Hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, M.J.; Chow, D.; Brevnova, E.E.; Garcia, K.C. Hexameric Structure and Assembly of the Interleukin-6/IL-6 Alpha-Receptor/Gp130 Complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef]
- Mayeur, C.; Leyton, P.A.; Kolodziej, S.A.; Yu, B.; Bloch, K.D. BMP Type II Receptors Have Redundant Roles in the Regulation of Hepatic Hepcidin Gene Expression and Iron Metabolism. Blood 2014, 124, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Mayeur, C.; Lohmeyer, L.K.; Leyton, P.; Kao, S.M.; Pappas, A.E.; Kolodziej, S.A.; Spagnolli, E.; Yu, B.; Galdos, R.L.; Yu, P.B.; et al. The Type I BMP Receptor Alk3 Is Required for the Induction of Hepatic Hepcidin Gene Expression by Interleukin-6. Blood 2014, 123, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Scindia, Y.; Leeds, J.; Swaminathan, S. Sundararaman Iron Homeostasis in Healthy Kidney and Its Role in Acute Kidney Injury. Seminars Nephrol. 2019, 39, 76–84. [Google Scholar] [CrossRef]
- Kulaksiz, H.; Theilig, F.; Bachmann, S.; Gehrke, S.G.; Rost, D.; Janetzko, A.; Cetin, Y.; Stremmel, W. The Iron-Regulatory Peptide Hormone Hepcidin: Expression and Cellular Localization in the Mammalian Kidney. J. Endocrinol. 2005, 184, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of Iron Homeostasis by the Hypoxia-Inducible Transcription Factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef]
- Gammella, E.; Diaz, V.; Recalcati, S.; Buratti, P.; Samaja, M.; Dey, S.; Noguchi, C.T.; Gassmann, M.; Cairo, G. Erythropoietin’s Inhibiting Impact on Hepcidin Expression Occurs Indirectly. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R330–R335. [Google Scholar] [CrossRef]
- Krijt, J.; Jonásová, A.; Neuwirtová, R.; Necas, E. Effect of Erythropoietin on Hepcidin Expression in Hemojuvelin-Mutant Mice. Blood Cells Mol. Dis. 2010, 44, 257–261. [Google Scholar] [CrossRef]
- Mühlenhoff, U.; Hoffmann, B.; Richter, N.; Rietzschel, N.; Spantgar, F.; Stehling, O.; Uzarska, M.A.; Lill, R. Compartmentalization of Iron between Mitochondria and the Cytosol and Its Regulation. Eur. J. Cell Biol. 2015, 94, 292–308. [Google Scholar] [CrossRef]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. Macrophages: Central Regulators of Iron Balance. Metallomics 2014, 6, 1336–1345. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a Ferrireductase Required for Efficient Transferrin-Dependent Iron Uptake in Erythroid Cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef]
- Paul, V.D.; Lill, R. Biogenesis of Cytosolic and Nuclear Iron-Sulfur Proteins and Their Role in Genome Stability. Biochim. Biophys. Acta 2015, 1853, 1528–1539. [Google Scholar] [CrossRef]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Mühlenhoff, U. The Role of Mitochondria in Cellular Iron–Sulfur Protein Biogenesis and Iron Metabolism. Biochim. Biophysica Acta (BBA) Mol. Cell Res. 2012, 1823, 1491–1508. [Google Scholar] [CrossRef]
- De Domenico, I.; McVey Ward, D.; Kaplan, J. Regulation of Iron Acquisition and Storage: Consequences for Iron-Linked Disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of Iron Homeostasis by an Iron-Regulated Ubiquitin Ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef]
- Rouault, T.A. The Role of Iron Regulatory Proteins in Mammalian Iron Homeostasis and Disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Mechanisms and Regulation of Iron Trafficking across the Capillary Endothelial Cells of the Blood-Brain Barrier. Front. Mol. Neurosci. 2015, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and Pathobiology of Pulmonary Hypertension: State of the Art and Research Perspectives. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef] [PubMed]
- Frise, M.C.; Robbins, P.A. Iron, Oxygen, and the Pulmonary Circulation. J. Appl. Physiol. 2015, 119, 1421–1431. [Google Scholar] [CrossRef]
- Smith, T.G.; Balanos, G.M.; Croft, Q.P.P.; Talbot, N.P.; Dorrington, K.L.; Ratcliffe, P.J.; Robbins, P.A. The Increase in Pulmonary Arterial Pressure Caused by Hypoxia Depends on Iron Status. J. Physiol. 2008, 586, 5999–6005. [Google Scholar] [CrossRef]
- Balanos, G.M.; Dorrington, K.L.; Robbins, P.A. Desferrioxamine Elevates Pulmonary Vascular Resistance in Humans: Potential for Involvement of HIF-1. J. Appl. Physiol. 2002, 92, 2501–2507. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Dorrington, K.L.; Maxwell, P.H.; Robbins, P.A. Effects of Desferrioxamine on Serum Erythropoietin and Ventilatory Sensitivity to Hypoxia in Humans. J. Appl. Physiol. 2000, 89, 680–686. [Google Scholar] [CrossRef]
- Bart, N.K.; Curtis, M.K.; Cheng, H.-Y.; Hungerford, S.L.; McLaren, R.; Petousi, N.; Dorrington, K.L.; Robbins, P.A. Elevation of Iron Storage in Humans Attenuates the Pulmonary Vascular Response to Hypoxia. J. Appl. Physiol. 2016, 121, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, G.; Manders, E.; Happé, C.M.; Schalij, I.; Groepenhoff, H.; Howard, L.S.; Wilkins, M.R.; Bogaard, H.J.; Westerhof, N.; van der Laarse, W.J.; et al. Intravenous Iron Therapy in Patients with Idiopathic Pulmonary Arterial Hypertension and Iron Deficiency. Pulm. Circ. 2015, 5, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Viethen, T.; Gerhardt, F.; Dumitrescu, D.; Knoop-Busch, S.; ten Freyhaus, H.; Rudolph, T.K.; Baldus, S.; Rosenkranz, S. Ferric Carboxymaltose Improves Exercise Capacity and Quality of Life in Patients with Pulmonary Arterial Hypertension and Iron Deficiency: A Pilot Study. Int. J. Cardiol. 2014, 175, 233–239. [Google Scholar] [CrossRef]
- Sonnweber, T.; Nairz, M.; Theurl, I.; Petzer, V.; Tymoszuk, P.; Haschka, D.; Rieger, E.; Kaessmann, B.; Deri, M.; Watzinger, K.; et al. The Crucial Impact of Iron Deficiency Definition for the Course of Precapillary Pulmonary Hypertension. PLoS ONE 2018, 13, e0203396. [Google Scholar] [CrossRef]
- White, K.; Lu, Y.; Annis, S.; Hale, A.E.; Chau, B.N.; Dahlman, J.E.; Hemann, C.; Opotowsky, A.R.; Vargas, S.O.; Rosas, I.; et al. Genetic and Hypoxic Alterations of the MicroRNA-210-ISCU1/2 Axis Promote Iron-Sulfur Deficiency and Pulmonary Hypertension. EMBO Mol. Med. 2015, 7, 695–713. [Google Scholar] [CrossRef]
- Yu, Q.; Tai, Y.-Y.; Tang, Y.; Zhao, J.; Negi, V.; Culley, M.K.; Pilli, J.; Sun, W.; Brugger, K.; Mayr, J.; et al. BOLA (BolA Family Member 3) Deficiency Controls Endothelial Metabolism and Glycine Homeostasis in Pulmonary Hypertension. Circulation 2019, 139, 2238–2255. [Google Scholar] [CrossRef]
- Ahting, U.; Mayr, J.A.; Vanlander, A.V.; Hardy, S.A.; Santra, S.; Makowski, C.; Alston, C.L.; Zimmermann, F.A.; Abela, L.; Plecko, B.; et al. Clinical, Biochemical, and Genetic Spectrum of Seven Patients with NFU1 Deficiency. Front. Genet. 2015, 6. [Google Scholar] [CrossRef]
- Hickey, M.M.; Richardson, T.; Wang, T.; Mosqueira, M.; Arguiri, E.; Yu, H.; Yu, Q.-C.; Solomides, C.C.; Morrisey, E.E.; Khurana, T.S.; et al. The von Hippel-Lindau Chuvash Mutation Promotes Pulmonary Hypertension and Fibrosis in Mice. J. Clin. Investig. 2010, 120, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Caravita, S.; Deboeck, G.; Vachiery, J.-L.; Naeije, R. Pulmonary Arterial Hypertension Associated with a von Hippel-Lindau Gene Mutation. J. Heart Lung Transplant. 2016, 35, 1138–1139. [Google Scholar] [CrossRef]
- Sarangi, S.; Lanikova, L.; Kapralova, K.; Acharya, S.; Swierczek, S.; Lipton, J.M.; Wolfe, L.; Prchal, J.T. The Homozygous VHL(D126N) Missense Mutation Is Associated with Dramatically Elevated Erythropoietin Levels, Consequent Polycythemia, and Early Onset Severe Pulmonary Hypertension. Pediatr. Blood Cancer 2014, 61, 2104–2106. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Gale, D.P.; Connor, T.; Adams, S.; de Boer, J.; Gascoyne, D.M.; Williams, O.; Maxwell, P.H.; Ancliff, P.J. Dysregulation of the HIF Pathway Due to VHL Mutation Causing Severe Erythrocytosis and Pulmonary Arterial Hypertension. Blood 2011, 117, 3699–3701. [Google Scholar] [CrossRef] [PubMed]
- Molteni, A.; Ward, W.F.; Ts’ao, C.H.; Fitzsimons, E.J. Serum Copper Concentration as an Index of Cardiopulmonary Injury in Monocrotaline-Treated Rats. Ann. Clin. Lab. Sci. 1988, 18, 476–483. [Google Scholar]
- Xiao, R.; Su, Y.; Feng, T.; Sun, M.; Liu, B.; Zhang, J.; Lu, Y.; Li, J.; Wang, T.; Zhu, L.; et al. Monocrotaline Induces Endothelial Injury and Pulmonary Hypertension by Targeting the Extracellular Calcium-Sensing Receptor. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Naito, Y.; Hosokawa, M.; Hao, H.; Sawada, H.; Hirotani, S.; Iwasaku, T.; Okuhara, Y.; Eguchi, A.; Hirota, S.; Ohyanagi, M.; et al. Impact of Dietary Iron Restriction on the Development of Monocrotaline-Induced Pulmonary Vascular Remodeling and Right Ventricular Failure in Rats. Biochem. Biophys. Res. Commun. 2013, 436, 145–151. [Google Scholar] [CrossRef]
- Naito, Y.; Hosokawa, M.; Sawada, H.; Oboshi, M.; Iwasaku, T.; Okuhara, Y.; Eguchi, A.; Nishimura, K.; Soyama, Y.; Hirotani, S.; et al. Iron Is Associated with the Development of Hypoxia-Induced Pulmonary Vascular Remodeling in Mice. Heart Vessels 2016, 31, 2074–2079. [Google Scholar] [CrossRef] [PubMed]
- Shannahan, J.H.; Schladweiler, M.C.J.; Richards, J.H.; Ledbetter, A.D.; Ghio, A.J.; Kodavanti, U.P. Pulmonary Oxidative Stress, Inflammation, and Dysregulated Iron Homeostasis in Rat Models of Cardiovascular Disease. J. Toxicol. Environ. Health Part A 2010, 73, 641–656. [Google Scholar] [CrossRef]
- Kong, W.-N.; Niu, Q.-M.; Ge, L.; Zhang, N.; Yan, S.-F.; Chen, W.-B.; Chang, Y.-Z.; Zhao, S.-E. Sex Differences in Iron Status and Hepcidin Expression in Rats. Biol. Trace Elem. Res. 2014, 160, 258–267. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Wharton, J.; Howard, L.; Gibbs, J.S.R.; Vonk-Noordegraaf, A.; Wilkins, M.R. Iron Deficiency in Pulmonary Arterial Hypertension: A Potential Therapeutic Target. Eur. Respir. J. 2011, 38, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-M.; Preston, I.R.; Hill, N.S.; Suzuki, Y.J. Iron Chelation Inhibits the Development of Pulmonary Vascular Remodeling. Free Radic. Biol. Med. 2012, 53, 1738–1747. [Google Scholar] [CrossRef]
- Cotroneo, E.; Ashek, A.; Wang, L.; Wharton, J.; Dubois, O.; Bozorgi, S.; Busbridge, M.; Alavian, K.N.; Wilkins, M.R.; Zhao, L. Iron Homeostasis and Pulmonary Hypertension: Iron Deficiency Leads to Pulmonary Vascular Remodeling in the Rat. Circ. Res. 2015, 116, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Lakhal-Littleton, S.; Crosby, A.; Frise, M.C.; Mohammad, G.; Carr, C.A.; Loick, P.A.M.; Robbins, P.A. Intracellular Iron Deficiency in Pulmonary Arterial Smooth Muscle Cells Induces Pulmonary Arterial Hypertension in Mice. Proc. Natl. Acad. Sci. USA 2019, 116, 13122–13130. [Google Scholar] [CrossRef]
- Paulin, R.; Michelakis, E.D. The Metabolic Theory of Pulmonary Arterial Hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef] [PubMed]
- Culley, M.K.; Perk, D.; Chan, S.Y. NFU1, Iron-Sulfur Biogenesis, and Pulmonary Arterial Hypertension: A (Metabolic) Shift in Our Thinking. Am. J. Respir. Cell Mol. Biol. 2020, 62, 136–138. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.-Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 Controls Mitochondrial Metabolism during Hypoxia by Repressing the Iron-Sulfur Cluster Assembly Proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef]
- Park, A.-M.; Wong, C.-M.; Jelinkova, L.; Liu, L.; Nagase, H.; Suzuki, Y.J. Pulmonary Hypertension-Induced GATA4 Activation in the Right Ventricle. Hypertension 2010, 56, 1145–1151. [Google Scholar] [CrossRef]
- Bär, H.; Kreuzer, J.; Cojoc, A.; Jahn, L. Upregulation of Embryonic Transcription Factors in Right Ventricular Hypertrophy. Basic Res. Cardiol. 2003, 98, 285–294. [Google Scholar] [CrossRef]
- Kobak, K.; Kasztura, M.; Dziegala, M.; Bania, J.; Kapuśniak, V.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Iron Limitation Promotes the Atrophy of Skeletal Myocytes, Whereas Iron Supplementation Prevents This Process in the Hypoxic Conditions. Int. J. Mol. Med. 2018, 41, 2678–2686. [Google Scholar] [CrossRef]
- Peters, E.L.; Offringa, C.; Kos, D.; Van der Laarse, W.J.; Jaspers, R.T. Regulation of Myoglobin in Hypertrophied Rat Cardiomyocytes in Experimental Pulmonary Hypertension. Pflugers Arch. 2016, 468, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, G.; Ying Wong, Y.; de Man, F.S.; Louis Handoko, M.; Jaspers, R.T.; Postmus, P.E.; Westerhof, N.; Niessen, H.W.M.; van der Laarse, W.J.; Vonk-Noordegraaf, A. Right Ventricular Oxygen Supply Parameters Are Decreased in Human and Experimental Pulmonary Hypertension. J. Heart Lung Transplant. 2013, 32, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T. Iron Deficiency beyond Erythropoiesis: Should We Be Concerned? Curr. Med. Res. Opin. 2018, 34, 81–93. [Google Scholar] [CrossRef]
- Finch, C.A.; Miller, L.R.; Inamdar, A.R.; Person, R.; Seiler, K.; Mackler, B. Iron Deficiency in the Rat. Physiological and Biochemical Studies of Muscle Dysfunction. J. Clin. Investig. 1976, 58, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron Deficiency Impairs Contractility of Human Cardiomyocytes through Decreased Mitochondrial Function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef]
- Gu, M.; Shao, N.-Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific IPSC-Derived Endothelial Cells Uncover Pathways That Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490–504.e5. [Google Scholar] [CrossRef] [PubMed]
- Hautefort, A.; Mendes-Ferreira, P.; Sabourin, J.; Manaud, G.; Bertero, T.; Rucker-Martin, C.; Riou, M.; Adão, R.; Manoury, B.; Lambert, M.; et al. Bmpr2 Mutant Rats Develop Pulmonary and Cardiac Characteristics of Pulmonary Arterial Hypertension. Circulation 2019, 139, 932–948. [Google Scholar] [CrossRef]
- Bertoli, S.R.; Marques, V.B.; Rossi, E.M.; Krause, M.; Carneiro, M.T.W.D.; Simões, M.R.; Dos Santos, L. Chronic Iron Overload Induces Vascular Dysfunction in Resistance Pulmonary Arteries Associated with Right Ventricular Remodeling in Rats. Toxicol. Lett. 2018, 295, 296–306. [Google Scholar] [CrossRef]
- Mehmood, M.; Agarwal, R.; Raina, A.; Correa-Jaque, P.; Benza, R.L. Hemodynamic Response to Treatment of Iron Deficiency Anemia in Pulmonary Arterial Hypertension: Longitudinal Insights from an Implantable Hemodynamic Monitor. Pulm. Circ. 2016, 6, 616–618. [Google Scholar] [CrossRef][Green Version]
- Xu, C.; Dong, C.; Xu, C.; Han, T.; Bao, S.; Gao, X. Effect of Iron Supplementation on the Expression of Hypoxia-Inducible Factor and Antioxidant Status in Rats Exposed to High-Altitude Hypoxia Environment. Biol. Trace Elem. Res. 2014, 162, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Kobak, K.A.; Radwańska, M.; Dzięgała, M.; Kasztura, M.; Josiak, K.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Structural and Functional Abnormalities in Iron-Depleted Heart. Heart Fail. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D.; Walter, P.B.; Ames, B.N.; Viteri, F.E. Both Iron Deficiency and Daily Iron Supplements Increase Lipid Peroxidation in Rats. J. Nutr. 2000, 130, 621–628. [Google Scholar] [CrossRef]
- Ramakrishnan, L.; Pedersen, S.L.; Toe, Q.K.; West, L.E.; Mumby, S.; Casbolt, H.; Issitt, T.; Garfield, B.; Lawrie, A.; Wort, S.J.; et al. The Hepcidin/Ferroportin Axis Modulates Proliferation of Pulmonary Artery Smooth Muscle Cells. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaminsky, S.; Hautefort, A.; Price, L.; Humbert, M.; Perros, F. Inflammation in Pulmonary Hypertension: What We Know and What We Could Logically and Safely Target First. Drug Discov. Today 2014, 19, 1251–1256. [Google Scholar] [CrossRef]
- Huertas, A.; Perros, F.; Tu, L.; Cohen-Kaminsky, S.; Montani, D.; Dorfmüller, P.; Guignabert, C.; Humbert, M. Immune Dysregulation and Endothelial Dysfunction in Pulmonary Arterial Hypertension: A Complex Interplay. Circulation 2014, 129, 1332–1340. [Google Scholar] [CrossRef]
- Dorfmüller, P.; Humbert, M. Progress in Pulmonary Arterial Hypertension Pathology: Relighting a Torch inside the Tunnel. Am. J. Respir. Crit. Care Med. 2012, 186, 210–212. [Google Scholar] [CrossRef]
- Perros, F.; Dorfmüller, P.; Montani, D.; Hammad, H.; Waelput, W.; Girerd, B.; Raymond, N.; Mercier, O.; Mussot, S.; Cohen-Kaminsky, S.; et al. Pulmonary Lymphoid Neogenesis in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Konijn, A.M. Iron Metabolism in Inflammation. Baillieres Clin. Haematol. 1994, 7, 829–849. [Google Scholar] [CrossRef]
- Jasiewicz, M.; Knapp, M.; Waszkiewicz, E.; Ptaszynska-Kopczynska, K.; Szpakowicz, A.; Sobkowicz, B.; Musial, W.J.; Kaminski, K.A. Enhanced IL-6 Trans-Signaling in Pulmonary Arterial Hypertension and Its Potential Role in Disease-Related Systemic Damage. Cytokine 2015, 76, 187–192. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated Levels of Inflammatory Cytokines Predict Survival in Idiopathic and Familial Pulmonary Arterial Hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef]
- Lee, P.; Peng, H.; Gelbart, T.; Wang, L.; Beutler, E. Regulation of Hepcidin Transcription by Interleukin-1 and Interleukin-6. Proc. Natl. Acad. Sci. USA 2005, 102, 1906–1910. [Google Scholar] [CrossRef]
- Montani, D.; Girerd, B.; Jaïs, X.; Laveneziana, P.; Lau, E.M.T.; Bouchachi, A.; Hascoët, S.; Günther, S.; Godinas, L.; Parent, F.; et al. Screening for Pulmonary Arterial Hypertension in Adults Carrying a BMPR2 Mutation. Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary Pulmonary Hypertension Is Associated with Reduced Pulmonary Vascular Expression of Type II Bone Morphogenetic Protein Receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Leyton, P.A.; Beppu, H.; Pappas, A.; Martyn, T.M.; Derwall, M.; Baron, D.M.; Galdos, R.; Bloch, D.B.; Bloch, K.D. Deletion of the Sequence Encoding the Tail Domain of the Bone Morphogenetic Protein Type 2 Receptor Reveals a Bone Morphogenetic Protein 7-Specific Gain of Function. PLoS ONE 2013, 8, e76947. [Google Scholar] [CrossRef]
- Yu, P.B.; Beppu, H.; Kawai, N.; Li, E.; Bloch, K.D. Bone Morphogenetic Protein (BMP) Type II Receptor Deletion Reveals BMP Ligand-Specific Gain of Signaling in Pulmonary Artery Smooth Muscle Cells. J. Biol. Chem. 2005, 280, 24443–24450. [Google Scholar] [CrossRef] [PubMed]
- Pauk, M.; Grgurevic, L.; Brkljacic, J.; Kufner, V.; Bordukalo-Niksic, T.; Grabusic, K.; Razdorov, G.; Rogic, D.; Zuvic, M.; Oppermann, H.; et al. Exogenous BMP7 Corrects Plasma Iron Overload and Bone Loss in Bmp6-/- Mice. Int. Orthop. 2015, 39, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, B.-X.; Sun, N.; Yan, Y.; Yuan, P.; Qu, J.-M.; Jing, Z.-C. Elevated Levels of Circulating Bone Morphogenetic Protein 7 Predict Mortality in Pulmonary Arterial Hypertension. Chest 2016, 150, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Karamanian, V.A.; Harhay, M.; Grant, G.R.; Palevsky, H.I.; Grizzle, W.E.; Zamanian, R.T.; Ihida-Stansbury, K.; Taichman, D.B.; Kawut, S.M.; Jones, P.L. Erythropoietin Upregulation in Pulmonary Arterial Hypertension. Pulm. Circ. 2014, 4, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Jilwan, F.N.; Escourrou, P.; Garcia, G.; Jaïs, X.; Humbert, M.; Roisman, G. High Occurrence of Hypoxemic Sleep Respiratory Disorders in Precapillary Pulmonary Hypertension and Mechanisms. Chest 2013, 143, 47–55. [Google Scholar] [CrossRef]
- Rafanan, A.L.; Golish, J.A.; Dinner, D.S.; Hague, L.K.; Arroliga, A.C. Nocturnal Hypoxemia Is Common in Primary Pulmonary Hypertension. Chest 2001, 120, 894–899. [Google Scholar] [CrossRef]
- Kaiser, R.; Seiler, S.; Held, M.; Bals, R.; Wilkens, H. Prognostic Impact of Renal Function in Precapillary Pulmonary Hypertension. J. Intern. Med. 2014, 275, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Farha, S.; Asosingh, K.; Xu, W.; Sharp, J.; George, D.; Comhair, S.; Park, M.; Tang, W.H.W.; Loyd, J.E.; Theil, K.; et al. Hypoxia-Inducible Factors in Human Pulmonary Arterial Hypertension: A Link to the Intrinsic Myeloid Abnormalities. Blood 2011, 117, 3485–3493. [Google Scholar] [CrossRef]
- Gale, D.P.; Harten, S.K.; Reid, C.D.L.; Tuddenham, E.G.D.; Maxwell, P.H. Autosomal Dominant Erythrocytosis and Pulmonary Arterial Hypertension Associated with an Activating HIF2 Alpha Mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Kerestes, H.; Percy, M.J.; Pietrofesa, R.; Chen, L.; Khurana, T.S.; Christofidou-Solomidou, M.; Lappin, T.R.J.; Lee, F.S. Erythrocytosis and Pulmonary Hypertension in a Mouse Model of Human HIF2A Gain of Function Mutation. J. Biol. Chem. 2013, 288, 17134–17144. [Google Scholar] [CrossRef]
- Ghosh, M.C.; Zhang, D.-L.; Jeong, S.Y.; Kovtunovych, G.; Ollivierre-Wilson, H.; Noguchi, A.; Tu, T.; Senecal, T.; Robinson, G.; Crooks, D.R.; et al. Deletion of Iron Regulatory Protein 1 Causes Polycythemia and Pulmonary Hypertension in Mice through Translational Derepression of HIF2α. Cell Metab. 2013, 17, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.-Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Wharton, J.; Howard, L.S.; Gibbs, J.S.R.; Wilkins, M.R. Red Cell Distribution Width Outperforms Other Potential Circulating Biomarkers in Predicting Survival in Idiopathic Pulmonary Arterial Hypertension. Heart 2011, 97, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Hampole, C.V.; Mehrotra, A.K.; Thenappan, T.; Gomberg-Maitland, M.; Shah, S.J. Usefulness of Red Cell Distribution Width as a Prognostic Marker in Pulmonary Hypertension. Am. J. Cardiol. 2009, 104, 868–872. [Google Scholar] [CrossRef]
- Lakhal, S.; Talbot, N.P.; Crosby, A.; Stoepker, C.; Townsend, A.R.M.; Robbins, P.A.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. Regulation of Growth Differentiation Factor 15 Expression by Intracellular Iron. Blood 2009, 113, 1555–1563. [Google Scholar] [CrossRef]
- Ulrich, A.; Wharton, J.; Thayer, T.E.; Swietlik, E.M.; Assad, T.R.; Desai, A.A.; Gräf, S.; Harbaum, L.; Humbert, M.; Morrell, N.W.; et al. Mendelian Randomisation Analysis of Red Cell Distribution Width in Pulmonary Arterial Hypertension. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [PubMed]
- Ottolenghi, S.; Zulueta, A.; Caretti, A. Iron and Sphingolipids as Common Players of (Mal)Adaptation to Hypoxia in Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 307. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric Carboxymaltose in Patients with Heart Failure and Iron Deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current Understanding of Iron Homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Wilkinson, N.; Pantopoulos, K. Pharmacological Targeting of the Hepcidin/Ferroportin Axis. Front. Pharmacol. 2016, 7, 160. [Google Scholar] [CrossRef]
- Vadhan-Raj, S.; Abonour, R.; Goldman, J.W.; Smith, D.A.; Slapak, C.A.; Ilaria, R.L.; Tiu, R.V.; Wang, X.; Callies, S.; Cox, J.; et al. A First-in-Human Phase 1 Study of a Hepcidin Monoclonal Antibody, LY2787106, in Cancer-Associated Anemia. J. Hematol. Oncol. 2017, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Sánchez, J.; Harlow, L.; Church, C.; Gaine, S.; Knightbridge, E.; Bunclark, K.; Gor, D.; Bedding, A.; Morrell, N.; Corris, P.; et al. Clinical Trial Protocol for TRANSFORM-UK: A Therapeutic Open-Label Study of Tocilizumab in the Treatment of Pulmonary Arterial Hypertension. Pulm. Circ. 2018, 8, 2045893217735820. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-N.J.; Iwahashi, M.; Tomosugi, N.; Uno, K.; Yamana, J.; Yamana, S.; Isobe, T.; Ito, H.; Kawabata, H.; Yoshizaki, K. Comparative Evaluation of the Effects of Treatment with Tocilizumab and TNF-α Inhibitors on Serum Hepcidin, Anemia Response and Disease Activity in Rheumatoid Arthritis Patients. Arthritis Res. Ther. 2013, 15, R141. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.R.; Liu, J.; Pearsall, R.S.; Li, G.; Kumar, R. ACTRIIA-Fc (Sotatercept) Reverses Pulmonary Vascular Remodeling to Attenuate Pulmonary Arterial Hypertension (PAH) by Rebalancing TGF-b/BMP Signaling in a Preclinical Model. In C26. Let it Bleed: Endothelial Injury and Angiogenesis in Pulmonary Hypertension; American Thoracic Society International Conference Abstracts; American Thoracic Society: New York, NY, USA, 2019; p. A4395. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Trials | Status | ID |
|---|---|---|
| BMPR2 Mutations and Iron Metabolism in Pulmonary Arterial Hypertension | Recruiting | NCT04086537 |
| IV Iron Replacement for Iron Deficiency in Idiopathic Pulmonary Arterial Hypertension (IPAH) Patients | Completed | NCT01447628 |
| Iron Deficiency in Pulmonary Hypertension | Unknown status | NCT01288651 |
| Oral Iron Supplementation in Pulmonary Hypertension | Completed | NCT01446848 |
| Iron Status and Hypoxic Pulmonary Vascular Responses | Completed | NCT01847352 |
| Eisenmenger Quality Enhancement Research Initiative | Completed | NCT01623492 |
| Study of the Effects of Iron on Lung Blood Pressure at High Altitude | Withdrawn | NCT00960921 |
| ORal IrON Supplementation with Ferric Maltol in Patients With Pulmonary Hypertension (ORION-PH-1) | Terminated | NCT03371173 |
| Study of the Effects of Iron Levels on the Lungs at High Altitude | Completed | NCT00952302 |
| Genetic and Environmental Determinants That Control Metabolism in Pulmonary Hypertension | Recruiting | NCT02594917 |
| Blood Markers Predict Effect of Normobaric Hypoxia at Rest and during Exercise in Patients with Pulmonary Hypertension | Not yet recruiting | NCT04715113 |
| A Study of Sotatercept for the Treatment of Pulmonary Arterial Hypertension (PAH) | Active, not recruiting | NCT03496207 |
| Bardoxolone Methyl in Patients with Connective Tissue Disease-Associated Pulmonary Arterial Hypertension-CATALYST | Terminated | NCT02657356 |
| Outcome Study Assessing a 75 Milligrams (mg) Dose of Macitentan in Patients with Pulmonary Arterial Hypertension | Recruiting | NCT04273945 |
| Clinical Study of Pulsed, Inhaled Nitric Oxide versus Placebo in Symptomatic Subjects with PAH | Terminated | NCT02725372 |
| Use of Inhaled Nitric Oxide to Prevent Pulmonary Hypertension Associated to Stored Blood Transfusion | Withdrawn | NCT02217683 |
| Erythrocyte Glutamine Level Relation to Pulmonary Hypertension Risk in Beta Thalassemia Major Children | Completed | NCT03133169 |
| Extended Access Program to Assess Long-Term Safety of Bardoxolone Methyl in Patients with Pulmonary Hypertension RANGER | Terminated | NCT03068130 |
| Hydroxyurea and Erythropoietin to Treat Sickle Cell Anemia | Completed | NCT00270478 |
| Beta3 Agonist Treatment in Chronic Pulmonary Hypertension Secondary to Heart Failure | Unknown status | NCT02775539 |
| Effects of Inspiratory Muscle Training in Patients with Pulmonary Hypertension | Recruiting | NCT04152187 |
| Sildenafil for Secondary Pulmonary Hypertension Due to Valvular Disease | Completed | NCT00862043 |
| Efficacy, Safety, and Tolerability Study of Pirfenidone in Combination with Sildenafil in Participants with Advanced Idiopathic Pulmonary Fibrosis (IPF) and Intermediate or High Probability of Group 3 Pulmonary Hypertension | Completed | NCT02951429 |
| AZ, MZ, and the Pulmonary System Response to Hypoxia | Completed | NCT02760121 |
| Treatment of Atrial Fibrillation in Patients by Pulmonary Vein Isolation in Combination with Renal Denervation or Pulmonary Vein Isolation Only | Recruiting | NCT02115100 |
| Hemodynamic Effects of Acute Normobaric Hypoxia during Exercise in Patients with Pulmonary Hypertension: Single-Center Randomized Controlled Trial | Not yet recruiting | NCT04715113 |
| A Therapeutic Open Label Study of Tocilizumab in the Treatment of Pulmonary Arterial Hypertension (TRANSFORM-UK) | Completed | NCT02676947 |
| A Phase 1 Study of LY2787106 in Cancer and Anemia | Terminated | NCT01340976 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quatredeniers, M.; Mendes-Ferreira, P.; Santos-Ribeiro, D.; Nakhleh, M.K.; Ghigna, M.-R.; Cohen-Kaminsky, S.; Perros, F. Iron Deficiency in Pulmonary Arterial Hypertension: A Deep Dive into the Mechanisms. Cells 2021, 10, 477. https://doi.org/10.3390/cells10020477
Quatredeniers M, Mendes-Ferreira P, Santos-Ribeiro D, Nakhleh MK, Ghigna M-R, Cohen-Kaminsky S, Perros F. Iron Deficiency in Pulmonary Arterial Hypertension: A Deep Dive into the Mechanisms. Cells. 2021; 10(2):477. https://doi.org/10.3390/cells10020477
Chicago/Turabian StyleQuatredeniers, Marceau, Pedro Mendes-Ferreira, Diana Santos-Ribeiro, Morad K. Nakhleh, Maria-Rosa Ghigna, Sylvia Cohen-Kaminsky, and Frédéric Perros. 2021. "Iron Deficiency in Pulmonary Arterial Hypertension: A Deep Dive into the Mechanisms" Cells 10, no. 2: 477. https://doi.org/10.3390/cells10020477
APA StyleQuatredeniers, M., Mendes-Ferreira, P., Santos-Ribeiro, D., Nakhleh, M. K., Ghigna, M.-R., Cohen-Kaminsky, S., & Perros, F. (2021). Iron Deficiency in Pulmonary Arterial Hypertension: A Deep Dive into the Mechanisms. Cells, 10(2), 477. https://doi.org/10.3390/cells10020477