
A Multi-Omics Study Revealing the Metabolic Effects of Estrogen in Liver Cancer Cells HepG2

 ,
, _Zhu.png) and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Treatment
2.3. Cell Number, Cytotoxicity, Viability, and Apoptosis
2.4. ERα and ERβ Protein Detection by Western Blot Analysis
2.5. Transcriptome Functional Analysis
2.6. Reverse Transcription Quantitative PCR (qPCR)
2.7. HPLC-MS/MS Targeted Metabolite Analysis
2.8. Metabolic Pathways Analysis
2.9. Multi-Omics Integration Analysis
2.10. Statistical Analysis
3. Results
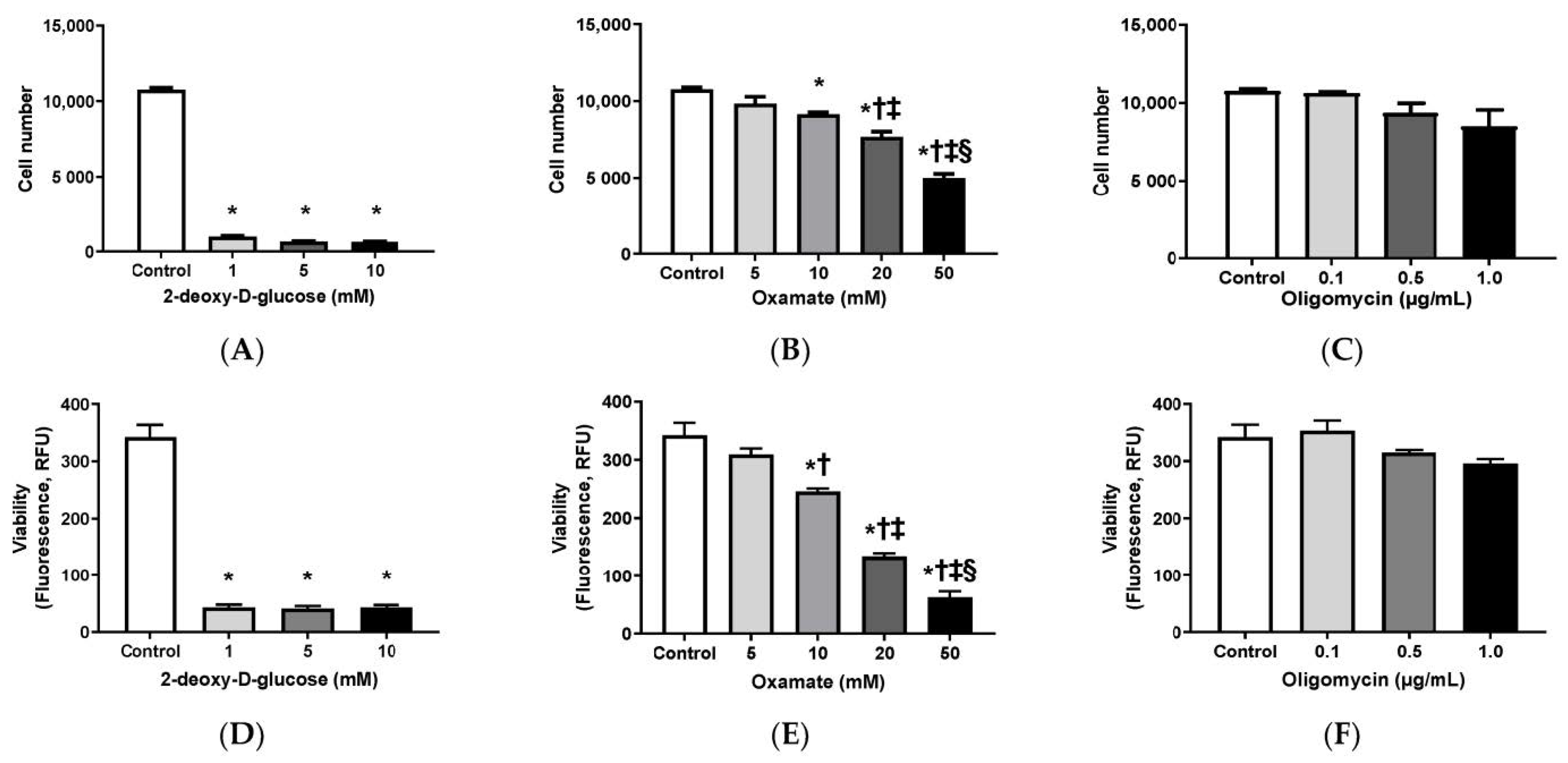
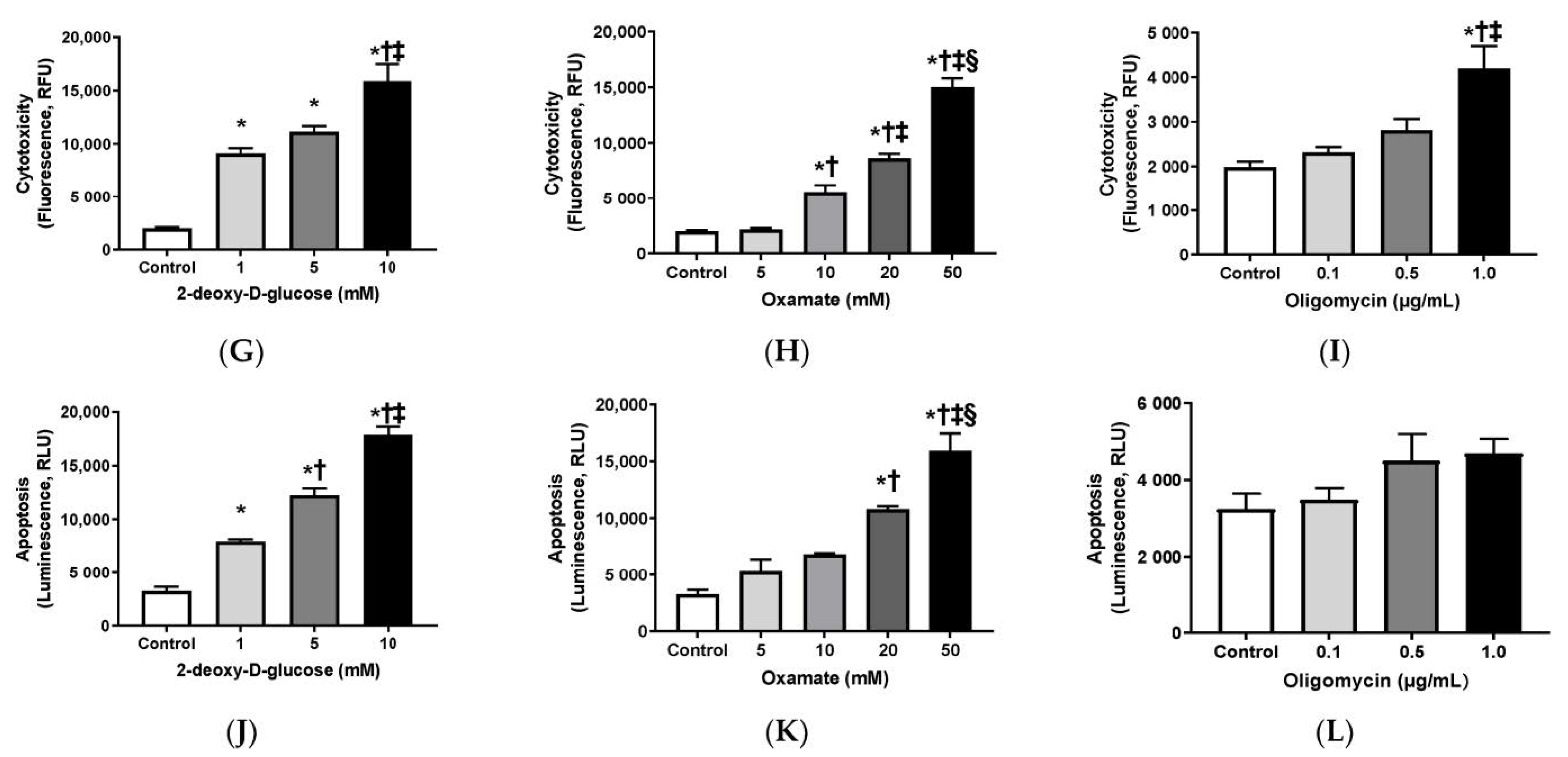
3.1. Effects of 2-DG, Oxamate, and Oligomycin on Cell Growth
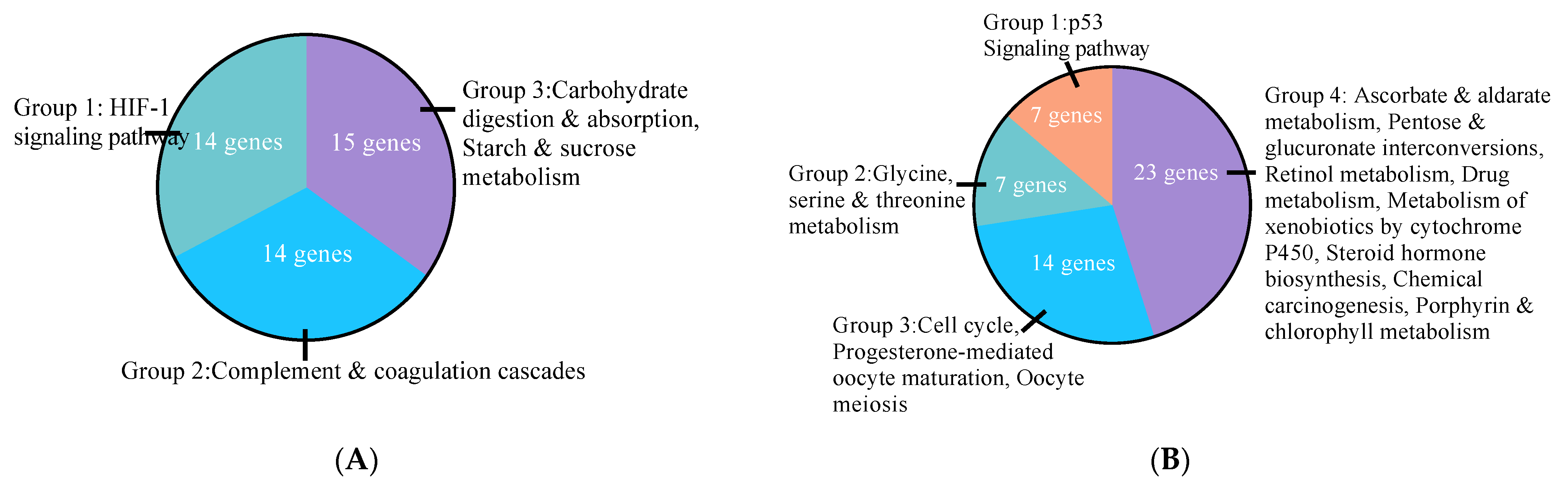
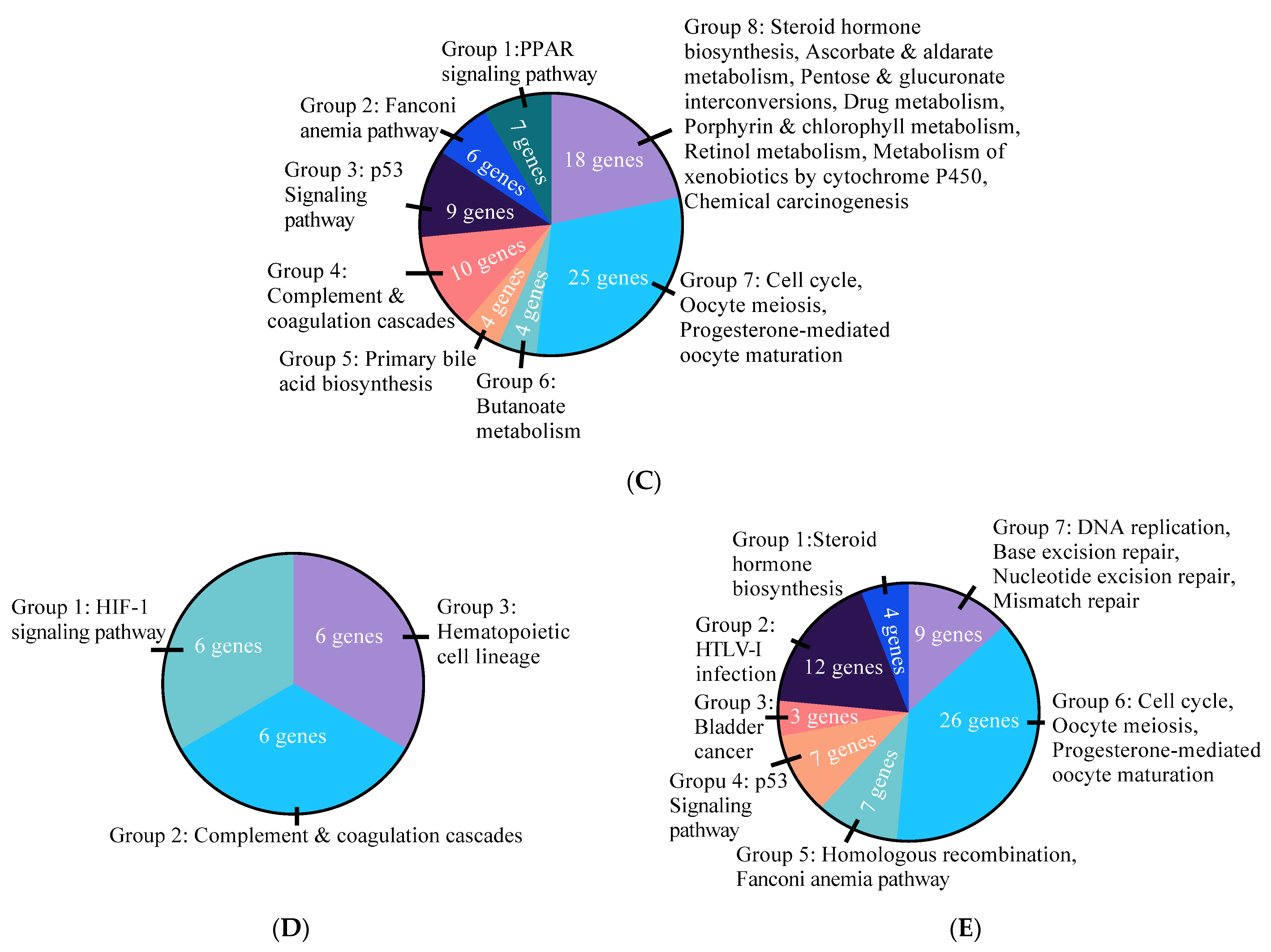
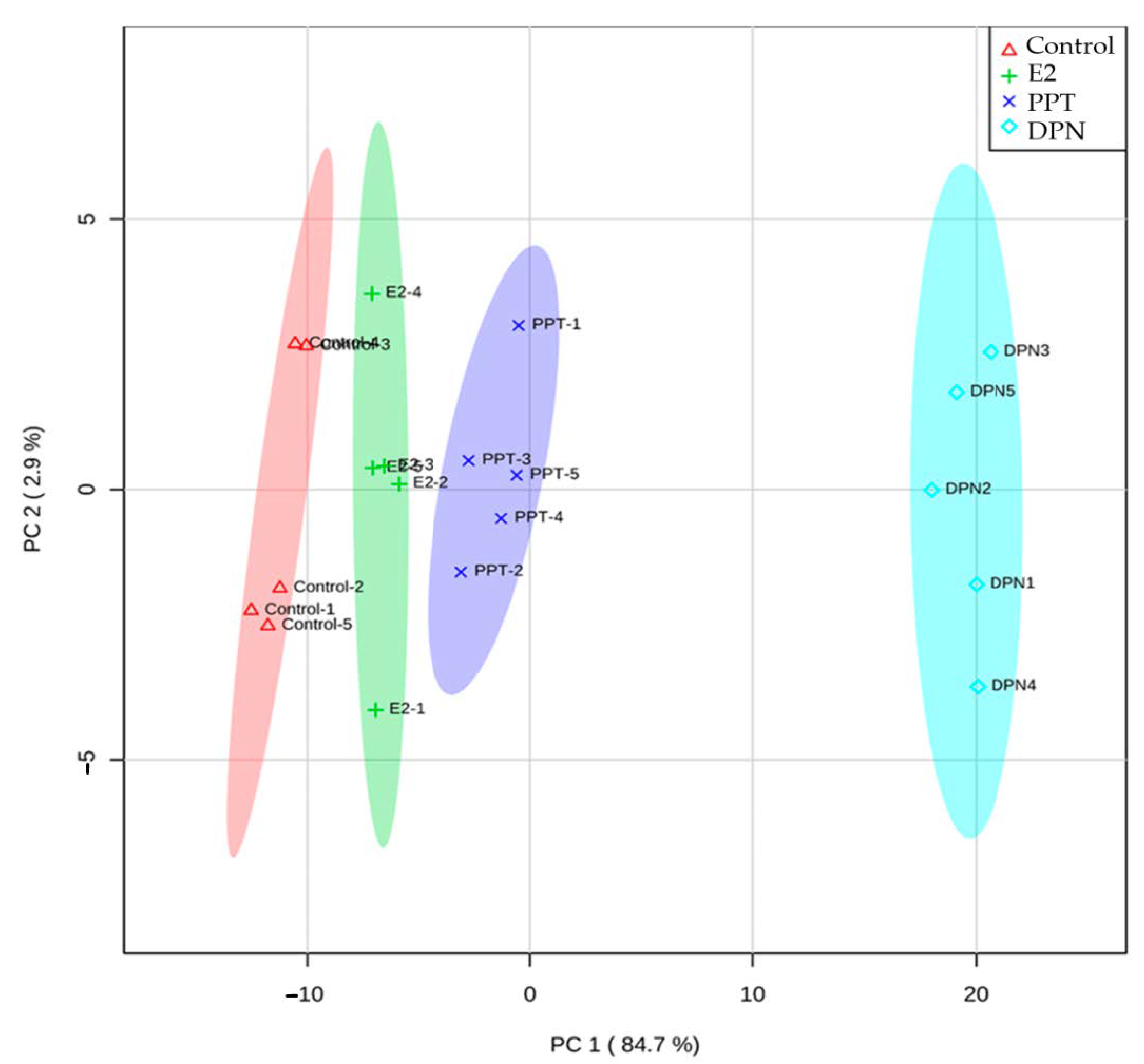
3.2. Effects of E2 and ER Agonists on Transcriptome Functional Pathways
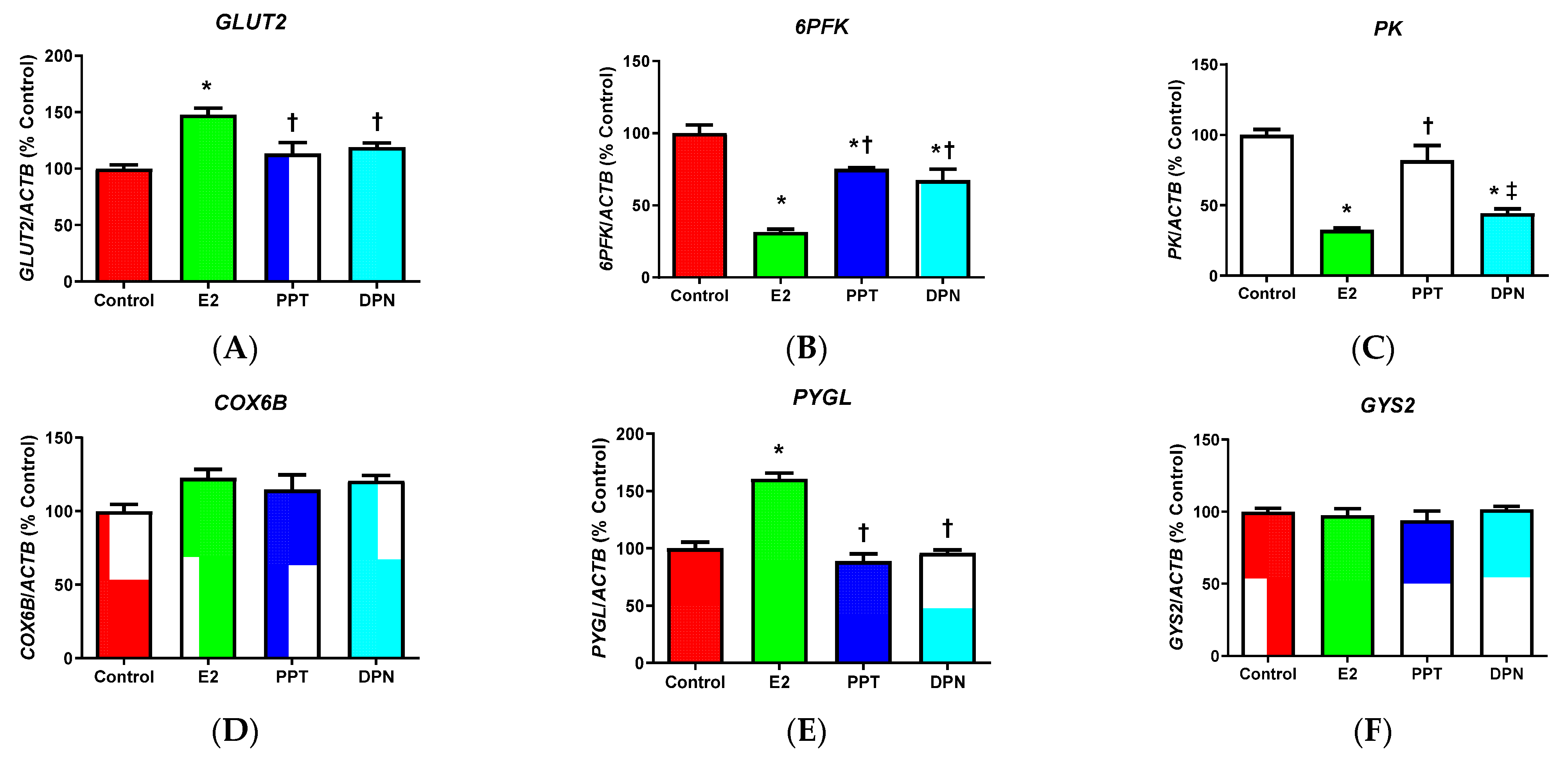
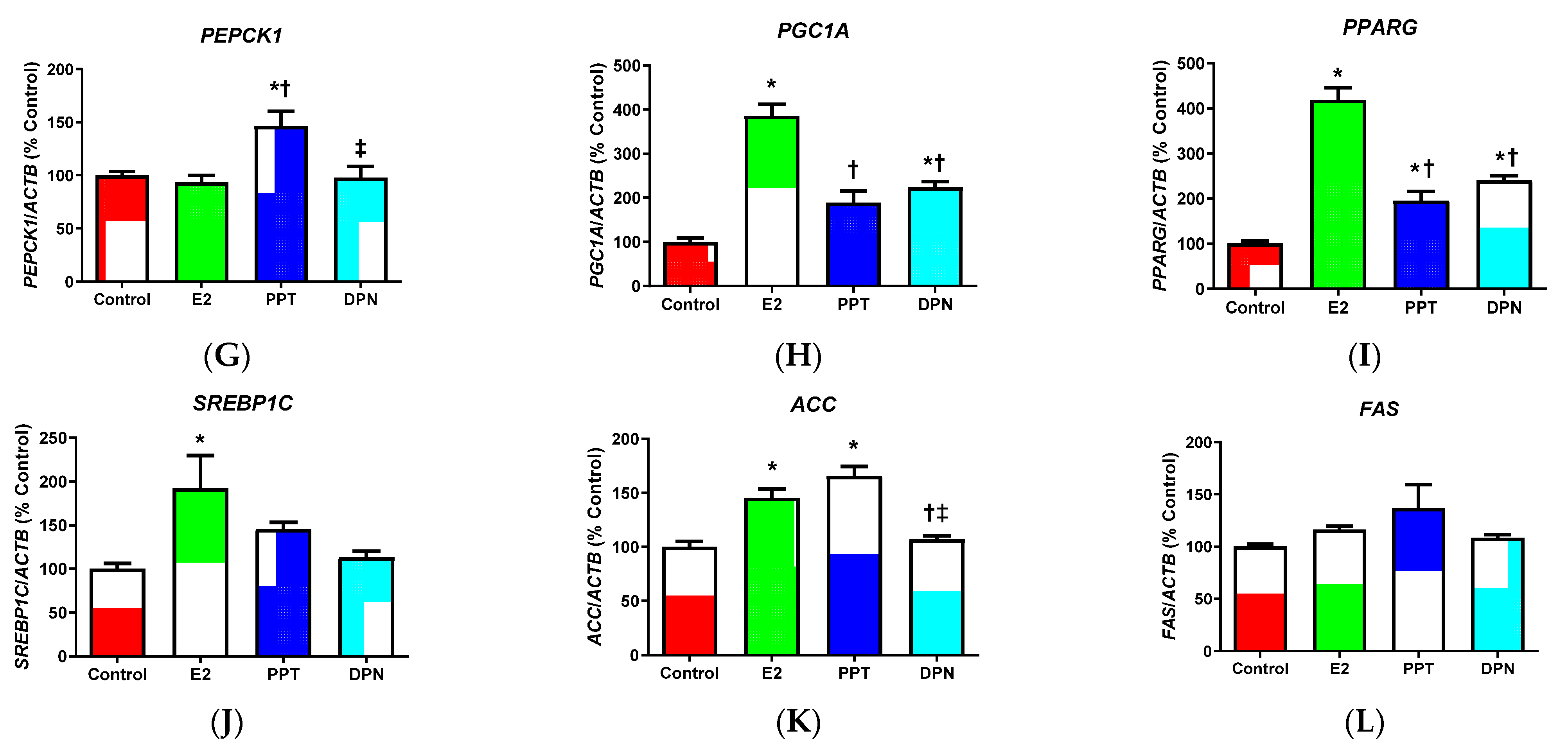
3.3. Effects of E2 and ER Agonists on Metabolic Genes
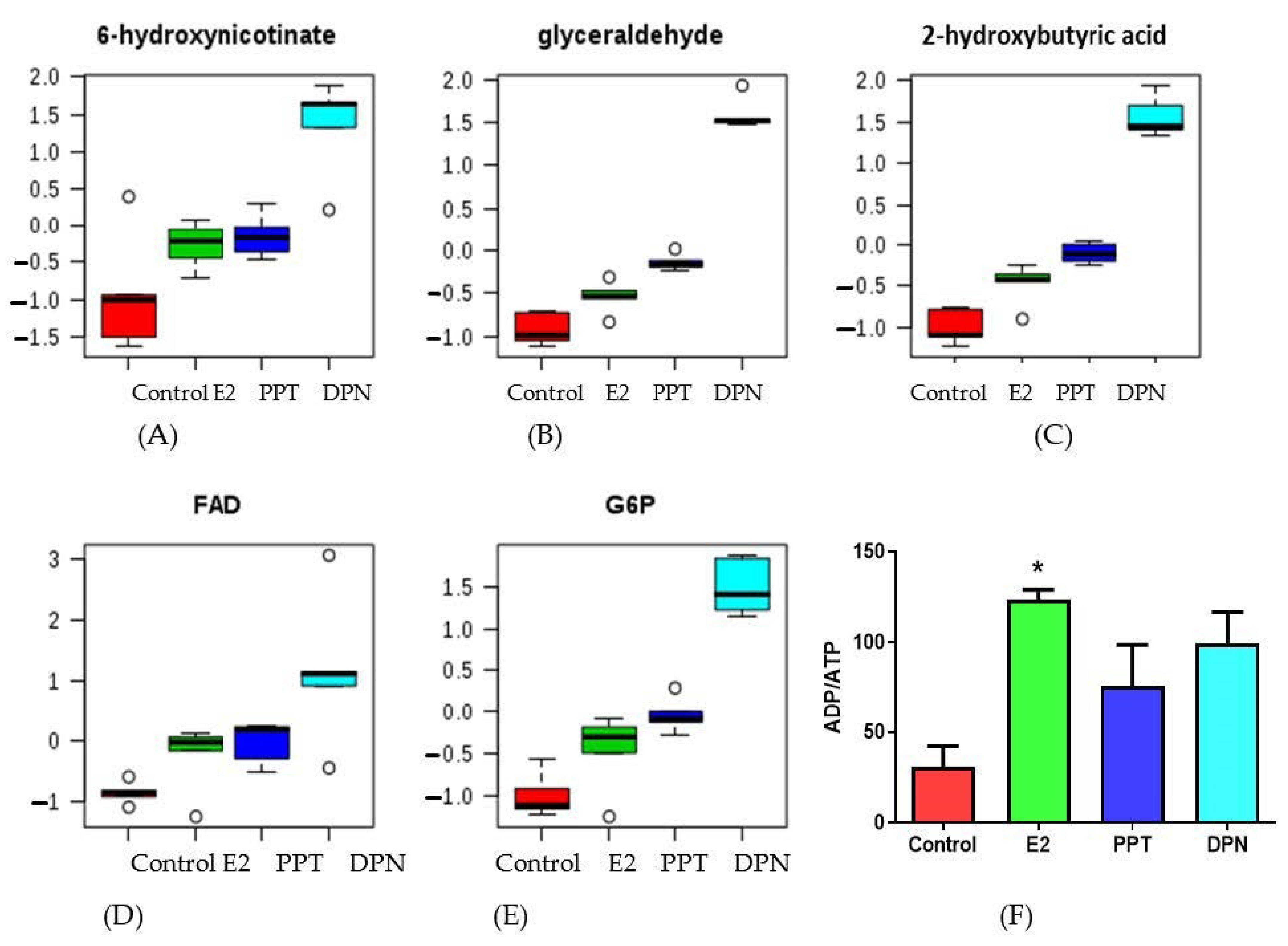
3.4. Effects of E2 and ER Agonists on Metabolic Profiles
3.5. Effects of E2 and ER Agonists on Metabolic Pathways
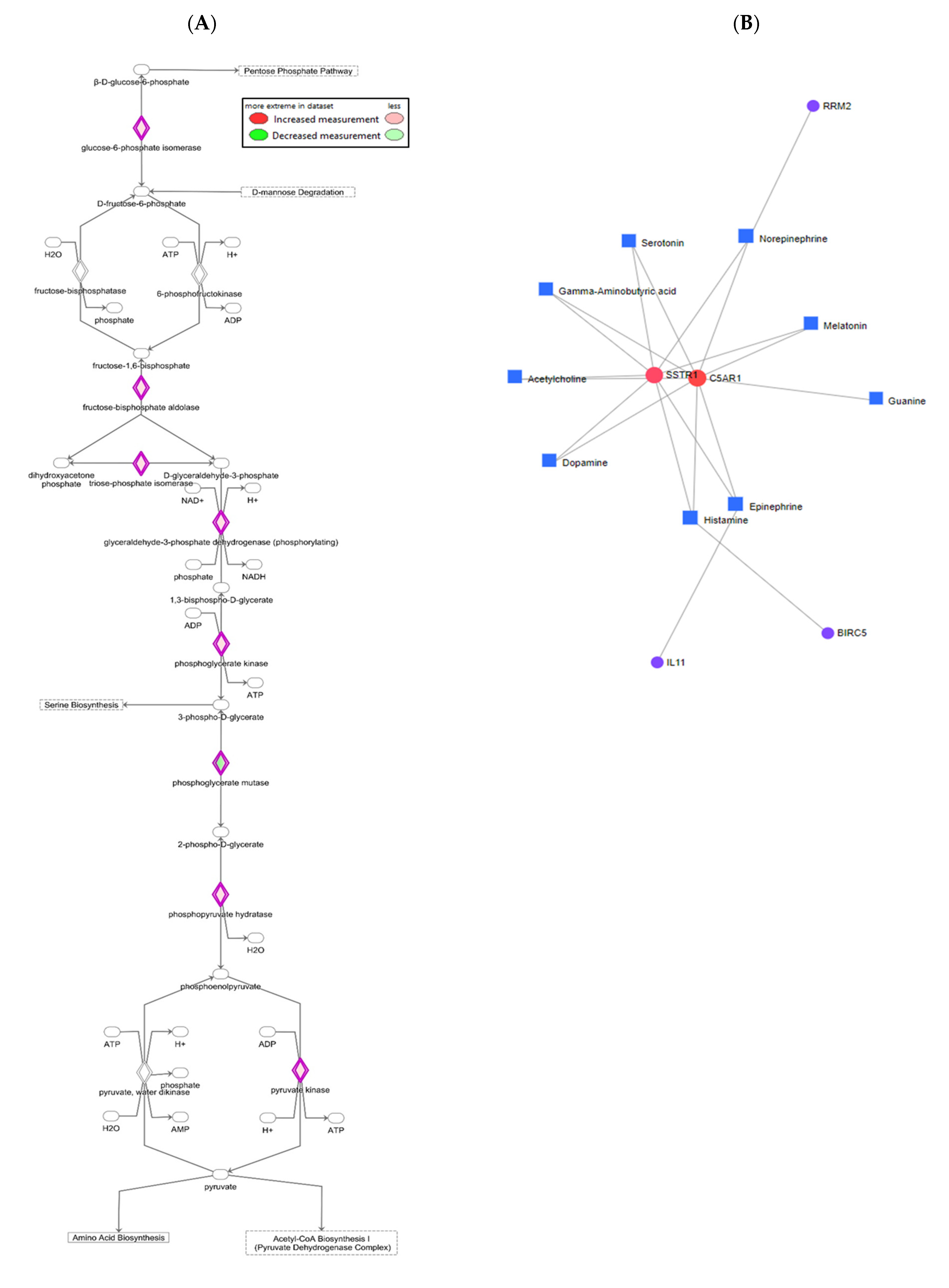
3.6. Effects of E2 and ER Agonists on Gene–Metabolite Interaction
4. Discussion
4.1. Summary of Findings
4.2. Metabolism Reprograming in HepG2 Cells
4.3. Effects of E2 and ER Agonists on Metabolic Gene Regulation in HepG2 Cells
4.4. Effects of E2 and ER Agonists on Metabolite Regulation in HepG2 Cells
4.5. Multi-Omics Integration Analysis Revealed Other Pathway Impacts in Addition to Glycolysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Njei, B.; Rotman, Y.; Ditah, I.; Lim, J.K. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015, 61, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Vanni, E.; Bugianesi, E. Obesity and Liver Cancer. Clin. Liver Dis. 2014, 18, 191–203. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Kanwal, F. Epidemiology of hepatocellular carcinoma in the United States: Where are we? Where do we go? Hepatology 2014, 60, 1767–1775. [Google Scholar] [CrossRef]
- Wands, J. Hepatocellular Carcinoma and Sex. N. Engl. J. Med. 2007, 357, 1974–1976. [Google Scholar] [CrossRef]
- Liu, P.; Xie, S.-H.; Hu, S.; Cheng, X.; Gao, T.; Zhang, C.; Song, Z. Age-specific sex difference in the incidence of hepatocellular carcinoma in the United States. Oncotarget 2017, 8, 68131–68137. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.-H.; Chen, P.-J. Gender Disparity of Hepatocellular Carcinoma: The Roles of Sex Hormones. Oncology 2010, 78, 172–179. [Google Scholar] [CrossRef]
- Cancer Research UK. Liver Cancer Incidence Trends over Time. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/liver-cancer/incidence#heading-Two (accessed on 19 February 2021).
- Bosch, F.; Ribes, J.; Díaz, M.; Cléries, R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology 2004, 127, S5–S16. [Google Scholar] [CrossRef]
- Shen, M.; Shi, H. Sex Hormones and Their Receptors Regulate Liver Energy Homeostasis. Int. J. Endocrinol. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Lampertico, P.; Seletti, C.; Francesca Donato, M.; Ronchi, G.; Del Ninno, E.; Colombo, M. The clinical and pathogenetic significance of estrogen receptor-β expression in chronic liver diseases and liver carcinoma. Cancer 2003, 98, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Mazzarella, L. Why does obesity promote cancer? Epidemiology, biology, and open questions. Ecancermedicalscience 2015, 9, 554. [Google Scholar] [CrossRef]
- Shen, M.; Shi, H. Estradiol and Estrogen Receptor Agonists Oppose Oncogenic Actions of Leptin in HepG2 Cells. PLoS ONE 2016, 11, e0151455. [Google Scholar] [CrossRef]
- Shen, M.; Cao, J.; Shi, H. Effects of Estrogen and Estrogen Receptors on Transcriptomes of HepG2 Cells: A Preliminary Study Using RNA Sequencing. Int. J. Endocrinol. 2018, 2018, 5789127. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Yasuda, M.; Mizobuchi, Y.; Ma, Y.R.; Liu, F.; Shiba, M.; Horie, T.; Ito, S. Suppressive effect of oestradiol on chemical hepatocarcinogenesis in rats. Gut 1998, 42, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Shimizu, I.; Shiba, M.; Ito, S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999, 29, 719–727. [Google Scholar] [CrossRef]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors (Review). Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Weinhouse, S. On respiratory impairment in cancer cells. Science 1956, 124, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.; Testa, I.; Festa, A.; Bagnarelli, P.; Chang, C.; Carloni, G. Differential response of the human hepatoma-derived cell line HA22T/VGH to polypeptide mitogens. FEBS Lett. 1987, 221, 11–17. [Google Scholar] [CrossRef]
- Jammart, B.; Michelet, M.; Pécheur, E.-I.; Parent, R.; Bartosch, B.; Zoulim, F.; Durantel, D. Very-Low-Density Lipoprotein (VLDL)-Producing and Hepatitis C Virus-Replicating HepG2 Cells Secrete No More Lipoviroparticles than VLDL-Deficient Huh7.5 Cells. J. Virol. 2013, 87, 5065–5080. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Shu, P.-Y.; Chang, C.; Ng, A.-K.; Hu, C.-P. IL-4 Suppresses the Expression and the Replication of Hepatitis B Virus in the Hepatocellular Carcinoma Cell Line Hep3B. J. Immunol. 2003, 171, 4708–4716. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.P.; Archer, T.K.; Deeley, R.G. Effects of estrogen on apolipoprotein secretion by the human hepatocarcinoma cell line, HepG2. J. Biol. Chem 1985, 260, 1670–1675. [Google Scholar] [CrossRef]
- Teng, Y.; Litchfield, L.M.; Ivanova, M.M.; Prough, R.A.; Clark, B.J.; Klinge, C.M. Dehydroepiandrosterone-induces miR-21 transcription in HepG2 cells through estrogen receptor β and androgen receptor. Mol. Cell. Endocrinol. 2014, 392, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Guo, D.-K.; You, T.-G.; Shen, D.-W.; Wang, C.; Tang, L.; Wang, J.; Xu, R.-H.; Zhang, H. Inhibition of hepatocellular carcinoma by fulvestrant involves the estrogen receptor α and Wnt pathways in vitro and in patients. Mol. Med. Rep. 2014, 10, 3125–3131. [Google Scholar] [CrossRef] [PubMed]
- Hishida, M.; Nomoto, S.; Inokawa, Y.; Hayashi, M.; Kanda, M.; Okamura, Y.; Nishikawa, Y.; Tanaka, C.; Kobayashi, D.; Yamada, S.; et al. Estrogen receptor 1 gene as a tumor suppressor gene in hepatocellular carcinoma detected by triple-combination array analysis. Int. J. Oncol. 2013, 43, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Göttel, M.; Le Corre, L.; Dumont, C.; Schrenk, D.; Chagnon, M.-C. Estrogen receptor α and aryl hydrocarbon receptor cross-talk in a transfected hepatoma cell line (HepG2) exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Rep. 2014, 1, 1029–1036. [Google Scholar] [CrossRef]
- Hayashida, K.; Shoji, I.; Deng, L.; Jiang, D.-P.; Ide, Y.-H.; Hotta, H. 17β-estradiol inhibits the production of infectious particles of hepatitis C virus. Microbiol. Immunol. 2010, 54, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Winer, L.S.P.; Wu, M. Rapid Analysis of Glycolytic and Oxidative Substrate Flux of Cancer Cells in a Microplate. PLoS ONE 2014, 9, e109916. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271. [Google Scholar] [CrossRef]
- Zhu, J.; Djukovic, D.; Deng, L.; Gu, H.; Himmati, F.; Chiorean, E.G.; Raftery, D. Colorectal Cancer Detection Using Targeted Serum Metabolic Profiling. J. Proteome Res. 2014, 13, 4120–4130. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Djukovic, D.; Deng, L.; Gu, H.; Himmati, F.; Abu Zaid, M.; Chiorean, E.G.; Raftery, D. Targeted serum metabolite profiling and sequential metabolite ratio analysis for colorectal cancer progression monitoring. Anal. Bioanal. Chem. 2015, 407, 7857–7863. [Google Scholar] [CrossRef]
- Xu, M.; Zhong, F.; Zhu, J. Evaluating metabolic response to light exposure in Lactobacillus species via targeted metabolic profiling. J. Microbiol. Methods 2017, 133, 14–19. [Google Scholar] [CrossRef]
- Zhong, F.; Xu, M.; Bruno, R.S.; Ballard, K.D.; Zhu, J. Targeted High Performance Liquid Chromatography Tandem Mass Spectrometry-based Metabolomics differentiates metabolic syndrome from obesity. Exp. Biol. Med. 2017, 242, 773–780. [Google Scholar] [CrossRef]
- Schelli, K.; Rutowski, J.; Roubidoux, J.; Zhu, J.; Rutowski, J. Staphylococcus aureus methicillin resistance detected by HPLC-MS/MS targeted metabolic profiling. J. Chromatogr. B 2017, 1047, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Bajad, S.U.; Lu, W.; Kimball, E.H.; Yuan, J.; Peterson, C.; Rabinowitz, J.D. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J. Chromatogr. A 2006, 1125, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion–switching, targeted mass spectrometry–based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Xu, Y.; Yang, H.; Shang, D.; Zhang, C.; Zhang, Y.; Sun, Z.; Shi, X.; Feng, L.; Han, J.; et al. Global pri-oritization of disease candidate metabolites based on a multi-omics composite network. Sci. Rep. 2015, 5, 17201. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Res. 2007, 36, D684–D688. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.E.H.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nat. Cell Biol. 2001, 413, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer. Endocr. Relat. Cancer 2015, 22, R1–R31. [Google Scholar] [CrossRef]
- Omoto, Y.; Iwase, H. Clinical significance of estrogen receptor β in breast and prostate cancer from biological aspects. Cancer Sci. 2015, 106, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.P.; Herrington, C.S.; Hollis, R.L.; Gourley, C. Estrogen Signaling and Its Potential as a Target for Therapy in Ovarian Cancer. Cancers 2020, 12, 1647. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Gly-colysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef]
- Fujita, M.; Imadome, K.; Somasundaram, V.; Kawanishi, M.; Karasawa, K.; Wink, D.A. Metabolic characterization of ag-gressive breast cancer cells exhibiting invasive phenotype: Impact of non-cytotoxic doses of 2-DG on diminishing invasiveness. BMC Cancer 2020, 20, 929. [Google Scholar] [CrossRef] [PubMed]
- Lucantoni, F.; Dussmann, H.; Prehn, J.H.M. Metabolic Targeting of Breast Cancer Cells With the 2-Deoxy-D-Glucose and the Mitochondrial Bioenergetics Inhibitor MDIVI-1. Front. Cell Dev. Biol. 2018, 6, 113. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free. Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Shonk, C.E.; Morris, H.P.; Boxer, G.E. Patterns of glycolytic enzymes in rat liver and hepatoma. Cancer Res. 1965, 25, 671–676. [Google Scholar] [PubMed]
- Weber, G.; Allard, C.; De Lamirande, G.; Cantero, A. Liver glucose-6-phosphatase activity and intracellular distribution after cortisone administration. Endocrinology 1956, 58, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.W.; Pyeritz, E.A.; Morris, H.P.; White, H.B. Proportional activities of glycerol kinase and glycerol 3-phosphate dehydrogenase in rat hepatomas. Biochem. J. 1975, 148, 545–550. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.; Frankel, W.; Ghoshal, K.; Jacob, S.T. Stat3-mediated activation of miR-23a suppresses gluconeogenesis in hepatocellular carcinoma by downregulating G6PC and PGC-1α. Hepatology 2012, 56, 186–197. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Jackowski, S. Coordination of membrane phospholipid synthesis with the cell cycle. J. Biol. Chem. 1994, 269, 3858–3867. [Google Scholar] [CrossRef]
- Koga, H.; Sakisaka, S.; Harada, M.; Takagi, T.; Hanada, S.; Taniguchi, E.; Kawaguchi, T.; Sasatomi, K.; Kimura, R.; Hashimoto, O.; et al. Involvement of p21WAF1/Cip1, p27Kip1, and p18INK4c in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology 2001, 33, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Rumi, M.A.K.; Sato, H.; Ishihara, S.; Kawashima, K.; Hamamoto, S.; Kazumori, H.; Okuyama, T.; Fukuda, R.; Nagasue, N.; Kinoshita, Y. Peroxisome proliferator-activated receptor gamma ligand-induced growth inhibition of human hepatocellular carcinoma. Br. J. Cancer 2001, 84, 1640–1647. [Google Scholar] [CrossRef]
- Toyoda, M.; Takagi, H.; Horiguchi, N.; Kakizaki, S.; Sato, K.; Takayama, H.; Mori, M. A ligand for peroxisome proliferator activated receptor γ inhibits cell growth and induces apoptosis in human liver cancer cells. Gut 2002, 50, 563–567. [Google Scholar] [CrossRef]
- Borbath, I.; Horsmans, Y. The role of PPARγ in hepatocellular carcinoma. PPAR Res. 2008, 2008, 209520. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Ivan, M.; Kaelin, W.G. The EGLN-HIF O 2 -Sensing System: Multiple Inputs and Feedbacks. Mol. Cell 2017, 66, 772–779. [Google Scholar] [CrossRef]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by func-tionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Kung, A.L.; Wang, S.; Klco, J.M.; Kaelin, W.G.; Livingston, D.M. Suppression of tumor growth through disruption of hy-poxia-inducible transcription. Nat. Med. 2000, 6, 1335. [Google Scholar] [CrossRef] [PubMed]
- Maranchie, J.K.; Vasselli, J.R.; Riss, J.; Bonifacino, J.S.; Linehan, W.; Klausner, R.D. The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 2002, 1, 247–255. [Google Scholar] [CrossRef]
- Raval, R.R.; Lau, K.W.; Tran, M.G.B.; Sowter, H.M.; Mandriota, S.J.; Li, J.-L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting Properties of Hypoxia-Inducible Factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-Associated Renal Cell Carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef]
- Brown, E.B.; Campbell, R.B.; Tsuzuki, Y.; Xu, L.; Carmeliet, P.; Fukumura, D.; Jain, R.K. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat. Med. 2001, 7, 864–868. [Google Scholar] [CrossRef]
- Yu, J.L.; Rak, J.W.; Carmeliet, P.; Nagy, A.; Kerbel, R.S.; Coomber, B.L. Heterogeneous Vascular Dependence of Tumor Cell Populations. Am. J. Pathol. 2001, 158, 1325–1334. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.-M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.H.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nat. Cell Biol. 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Acker, T.; Díez-Juan, A.; Aragonés, J.; Tjwa, M.; Brusselmans, K.; Moons, L.; Fukumura, D.; Moreno-Murciano, M.P.; Herbert, J.-M.; Burger, A.; et al. Genetic evidence for a tumor suppressor role of HIF-2α. Cancer Cell 2005, 8, 131–141. [Google Scholar] [CrossRef]
- Kuiper, G.G.J.M.; Carlsson, B.; Grandien, K.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustafsson, J.-Å. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef]
- Kuiper, G.G.J.M.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; Van Der Saag, P.T.; Van Der Burg, B.; Gustafsson, J.-Å. Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor β. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Hagopian, K.; Ramsey, J.J.; Weindruch, R. Fructose metabolizing enzymes from mouse liver: Influence of age and caloric restriction. Biochim. Biophys. Acta Gen. Subj. 2005, 1721, 37–43. [Google Scholar] [CrossRef]
- Nishiumi, S.; Suzuki, M.; Kobayashi, T.; Matsubara, A.; Azuma, T.; Yoshida, M. Metabolomics for Biomarker Discovery in Gastroenterological Cancer. Metabolites 2014, 4, 547–571. [Google Scholar] [CrossRef]
- Gall, W.E.; Beebe, K.; Lawton, K.A.; Adam, K.P.; Mitchell, M.W.; Nakhle, P.J.; Ryals, J.A.; Milburn, M.V.; Nannipieri, M.; Camastra, S.; et al. Alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS ONE 2010, 5, e10883. [Google Scholar] [CrossRef]
- Kim, H.J.; Winge, D.R. Emerging concepts in the flavinylation of succinate dehydrogenase. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 627–636. [Google Scholar] [CrossRef]
- Hu, H.; Zhu, W.; Qin, J.; Chen, M.; Gong, L.; Li, L.; Liu, X.; Tao, Y.; Yin, H.; Zhou, H.; et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 2017, 65, 515–528. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, H.; Zhang, X.; Ding, X.; Huang, M.; Geng, M.; Li, H.; Xie, Z. Phosphoglycerate mutase 1 predicts the poor prognosis of oral squamous cell carcinoma and is associated with cell migration. J. Cancer 2017, 8, 1943–1951. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Glatzle, J.; Bajaeifer, K.; Bühler, S.; Lehmann, T.; Königsrainer, I.; Vollmer, J.-P.; Sipos, B.; Ahmad, S.S.; Northoff, H.; et al. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. Int. J. Oncol. 2013, 43, 586–590. [Google Scholar] [CrossRef]
- Shashni, B.; Sakharkar, K.R.; Nagasaki, Y.; Sakharkar, M.K. Glycolytic enzymes PGK1 and PKM2 as novel transcriptional targets of PPARγ in breast cancer pathophysiology. J. Drug Target. 2012, 21, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Lay, A.J.; Jiang, X.-M.; Kisker, O.; Flynn, E.; Underwood, A.; Condron, R.; Hogg, P.J. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nat. Cell Biol. 2000, 408, 869–873. [Google Scholar] [CrossRef]
- Tang, S.J.; Ho, M.Y.; Cho, H.C.; Lin, Y.C.; Sun, G.H.; Chi, K.H.; Wang, Y.S.; Jhou, R.S.; Yang, W.; Sun, K.H. Phospho-glycerate kinase 1-overexpressing lung cancer cells reduce cyclooxygenase 2 expression and promote anti-tumor immunity in vivo. Int. J. Cancer 2008, 123, 2840–2848. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, C.; Schovanek, J.; Wang, H.; Bullova, P.; Caisova, V.; Gupta, G.; Wolf, K.I.; Semenza, G.L.; Zhuang, Z.; et al. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017, 8, 22313–22324. [Google Scholar] [CrossRef]
- Jiang, Y.; He, R.; Jiang, Y.; Liu, D.; Tao, L.; Yang, M.; Lin, C.; Shen, Y.; Fu, X.; Yang, J.; et al. Transcription factor NFAT5 contributes to the glycolytic phenotype rewiring and pancreatic cancer progression via transcription of PGK1. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, H.; Rombouts, K.K.; Vandermonde, A.A.; Urbain, D.; Kumar, U.U.; Bioulac-Sage, P.P.; Pinzani, M.M.; Rosenbaum, J.J.; Geerts, A.A. Expression of somatostatin receptors in normal and cirrhotic human liver and in hepatocellular carcinoma. Gut 2004, 53, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Koc, E.U.; Ozgur, T.; Yerci, O.; Gurel, S. Somatostatin receptor 1 (SSTR1) and somatostatin receptor 5 (SSTR5) expression in hepatocellular carcinoma. Hepatogastroenterology 2013, 60, 1693–1697. [Google Scholar]
- Hu, W.H.; Hu, Z.; Shen, X.; Dong, L.Y.; Zhou, W.Z.; Yu, X.X. C5a receptor enhances hepatocellular carcinoma cell inva-siveness via activating ERK1/2-mediated epithelial-mesenchymal transition. Exp. Mol. Pathol. 2016, 100, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Towner, L.; Donev, R. Complement in Cancer and Cancer Immunotherapy. Arch. Immunol. Ther. Exp. 2011, 59, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, M.J.; Sughrue, M.E.; Kane, A.J.; Mills, S.A.; Parsa, A.T. Cancer and the Complement Cascade. Mol. Cancer Res. 2010, 8, 1453–1465. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Garber, K. Energy Deregulation: Licensing Tumors to Grow. Science 2006, 312, 1158–1159. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Coding Protein | Forward Primers (5′–3′) | Reverse Primers (5′–3′) |
|---|---|---|---|
| ACC | acetyl-CoA carboxylase | GCTGCTCGGATCACTAGTGAA | TTCTGCTATCAGTCTGTCCAG |
| ACTB | β actin | AGAGCTACGAGCTGCCTGAC | AGCACTGTGTTGGCGTACAG |
| COX6B | cytochrome c oxidase subunit 6B | CTCAACGTGTTCCTCAAGTC | ATGGAGGACAGAGGAAAGG |
| FAS | fatty acid synthase | GAAACTGCAGGAGCTGTC | CACGGAGTTGAGGCGGAT |
| GLUT2 | glucose transporter 2 | AGTTAGATGAGGAAGTCAAAGCAA | TAGGCTGTCGGTAGCTGG |
| GYS2 | glycogen synthase 2 | GCCAGACACCTGACATTAAG | CTCCACTTCATCTTCCACATC |
| PEPCK1 | phosphoenolpyruvate carboxykinase | CCAGGCAGTGAGGGAGTTTCT | ACTGTGTCTCTTTGCTCTTGG |
| 6PFK | 6-phosphofructokinase | CTCACAGGTGCCAACATC | GCCGCAGAAGTCGTTATC |
| PGC1A | PPAR gamma coactivator 1 alpha | GACGACGAAGCAGACAAG | CCAAGGGTAGCTCAGTTTATC |
| PK | pyruvate kinase | TCGTCTTTGCCTCCTTTG | CTCACCTCCAGGATTTCATC |
| PPARG | PPAR gamma | GAAATGACCATGGTTGAC | CCGCTAGTACAAGTCCTTGTA |
| PYGL | glycogen phosphorylase liver form | CCTGTGATGAGGCCATTTAC | GTATCCATAGGCTGCAAGTC |
| SREBP1C | sterol regulatory element-binding transcription factor 1 | CTTTGCCCACCCTGGTGAGT | GGTTCTCCTGCTTGAGTTTCTGG |
| Upregulated Pathways | Treatment Group |
|---|---|
| HIF-1 signaling pathway | E2 DPN |
| Complement & coagulation cascades | E2 DPN |
| Carbohydrate digestion & absorption | E2 |
| Starch & sucrose metabolism | E2 |
| Hematopoietic cell lineage | DPN |
| Downregulated Pathways | Treatment Group |
| Steroid hormone biosynthesis | E2 PPT DPN |
| Cell cycle | E2 PPT DPN |
| Progesterone-mediated oocyte maturation | E2 PPT DPN |
| Oocyte meiosis | E2 PPT DPN |
| p53 Signaling pathway | E2 PPT DPN |
| Ascorbate and aldarate metabolism | E2 PPT |
| Pentose and glucuronate interconversions | E2 PPT |
| Retinol metabolism | E2 PPT |
| Metabolism of xenobiotics by cytochrome P450 | E2 PPT |
| Drug metabolism | E2 PPT |
| Chemical carcinogenesis | E2 PPT |
| Porphyrin and chlorophyll metabolism | E2 PPT |
| Fanconi anemia pathway | PPT DPN |
| Glycine, serine & threonine metabolism | E2 |
| Butanoate metabolism | PPT |
| Primary bile acid biosynthesis | PPT |
| Complement and coagulation cascades | PPT |
| PPAR signaling pathway | PPT |
| DNA replication | DPN |
| Base excision repair | DPN |
| Nucleotide excision repair | DPN |
| Mismatch repair | DPN |
| Homologous recombination | DPN |
| Bladder cancer | DPN |
| HTLV-I infection | DPN |
| Metabolism | Pathways | Treatment Group |
|---|---|---|
| Amino acid | Tyrosine metabolism | E2 PPT DPN |
| Tryptophan metabolism | E2 PPT DPN | |
| Histidine metabolism | E2 PPT DPN | |
| Glycine, serine and threonine metabolism | E2 PPT DPN | |
| Arginine and proline metabolism | E2 PPT DPN | |
| Taurine and hypotaurine metabolism | E2 PPT DPN | |
| Cysteine and methionine metabolism | PPT DPN | |
| beta-Alanine metabolism | PPT DPN | |
| Arginine biosynthesis | PPT DPN | |
| Alanine, aspartate and glutamate metabolism | PPT DPN | |
| Glutathione metabolism | PPT DPN | |
| Carbohydrate | Amino sugar and nucleotide sugar metabolism | E2 PPT DPN |
| Pyruvate metabolism | E2 PPT DPN | |
| Citrate cycle (TCA cycle) | E2 PPT DPN | |
| Glycolysis and gluconeogenesis | E2 PPT DPN | |
| Glyoxylate and dicarboxylate metabolism | PPT DPN | |
| Lipid | Glycerophospholipid metabolism | PPT DPN |
| Cofactors and vitamins | One carbon pool by folate | E2 DPN |
| Vitamin B6 metabolism | PPT DPN | |
| Nicotinate and nicotinamide metabolism | PPT DPN | |
| Biotin metabolism | DPN | |
| Nucleotide | Pyrimidine metabolism | PPT DPN |
| Biosynthesis of other metabolites | Caffeine metabolism | PPT DPN |
| Genetic information translation processing | Aminoacyl-tRNA biosynthesis | PPT DPN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, M.; Xu, M.; Zhong, F.; Crist, M.C.; Prior, A.B.; Yang, K.; Allaire, D.M.; Choueiry, F.; Zhu, J.; Shi, H. A Multi-Omics Study Revealing the Metabolic Effects of Estrogen in Liver Cancer Cells HepG2. Cells 2021, 10, 455. https://doi.org/10.3390/cells10020455
Shen M, Xu M, Zhong F, Crist MC, Prior AB, Yang K, Allaire DM, Choueiry F, Zhu J, Shi H. A Multi-Omics Study Revealing the Metabolic Effects of Estrogen in Liver Cancer Cells HepG2. Cells. 2021; 10(2):455. https://doi.org/10.3390/cells10020455
Chicago/Turabian StyleShen, Minqian, Mengyang Xu, Fanyi Zhong, McKenzie C. Crist, Anjali B. Prior, Kundi Yang, Danielle M. Allaire, Fouad Choueiry, Jiangjiang Zhu, and Haifei Shi. 2021. "A Multi-Omics Study Revealing the Metabolic Effects of Estrogen in Liver Cancer Cells HepG2" Cells 10, no. 2: 455. https://doi.org/10.3390/cells10020455
APA StyleShen, M., Xu, M., Zhong, F., Crist, M. C., Prior, A. B., Yang, K., Allaire, D. M., Choueiry, F., Zhu, J., & Shi, H. (2021). A Multi-Omics Study Revealing the Metabolic Effects of Estrogen in Liver Cancer Cells HepG2. Cells, 10(2), 455. https://doi.org/10.3390/cells10020455