
The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease

 , , ,
, , ,
Abstract
1. Introduction
2. DJ-1 in Parkinson’s Disease
2.1. DJ-1 in Other Diseases
2.2. DJ-1 and Its Chaperone Function
2.3. DJ-1 and Its Enzymatic Function
2.4. DJ-1 and Mitochondrial Function
2.5. DJ-1 and ROS Signaling
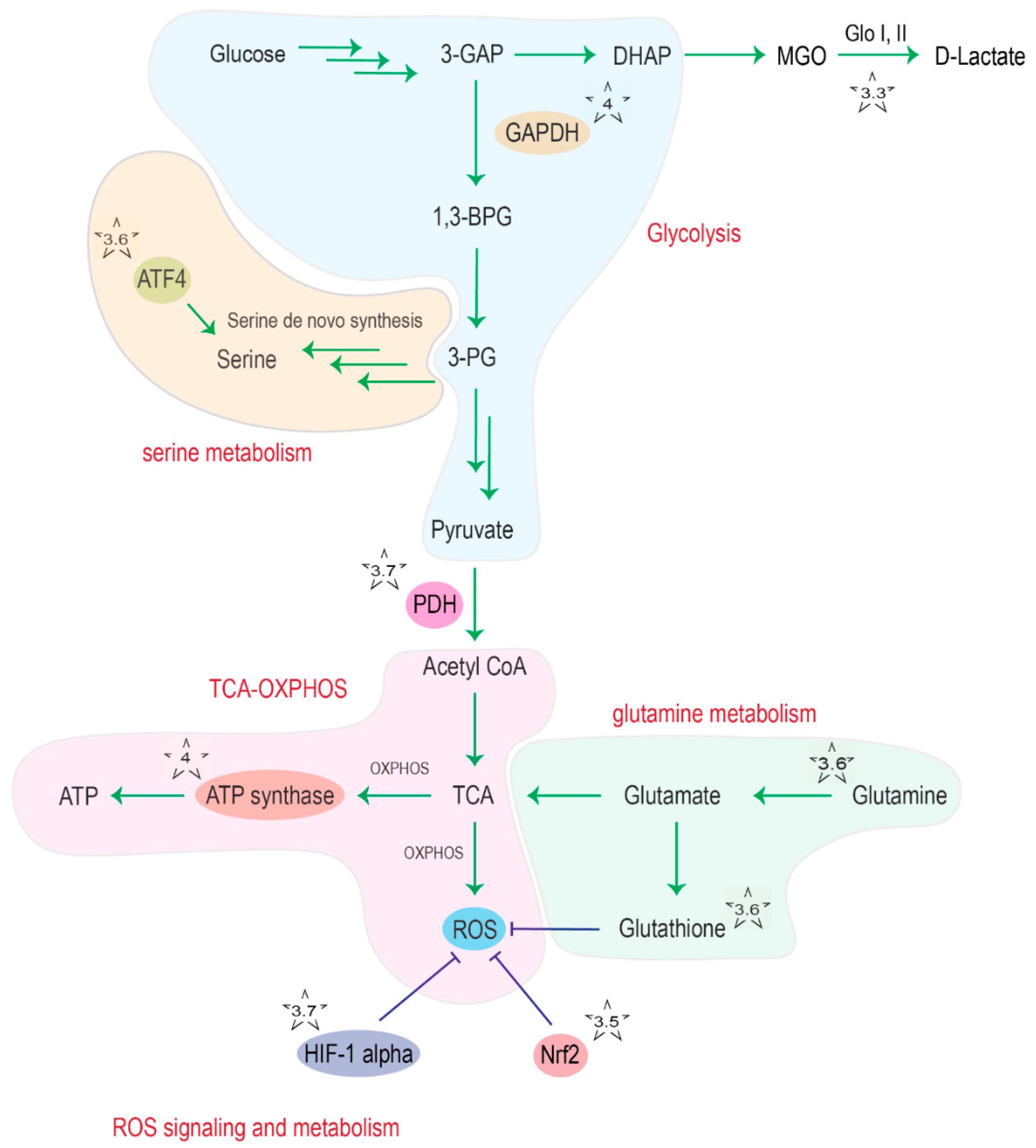
2.6. DJ-1 and Serine/Glutathione/Glutamine Metabolism
2.7. DJ-1 and the Regulation of Glycolysis and the TCA Cycle
3. DJ-1 and Pathophysiological Implications of Altered Metabolism in PD
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ariga, H. Common mechanisms of onset of cancer and neurodegenerative diseases. Biol. Pharm. Bull. 2015, 38, 795–808. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; van Swieten, J.C.; Brice, A.; van Duijn, C.M.; Oostra, B.; Meco, G.; et al. DJ-1 (PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef]
- Rizzu, P.; Hinkle, D.A.; Zhukareva, V.; Bonifati, V.; Severijnen, L.A.; Martinez, D.; Ravid, R.; Kamphorst, W.; Eberwine, J.H.; Lee, V.M.Y.; et al. DJ-1 Colocalizes with Tau Inclusions: A Link between Parkinsonism and Dementia. Ann. Neurol. 2004, 55, 113–118. [Google Scholar] [CrossRef]
- Bader, V.; Zhu, X.R.; Lübbert, H.; Stichel, C.C. Expression of DJ-1 in the adult mouse CNS. Brain Res. 2005, 1041, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.; Wilson, M.A. Structural biology of the DJ-1 superfamily. Adv. Exp. Med. Biol. 2017, 1037, 5–24. [Google Scholar]
- Wilson, M.A.; Collins, J.L.; Hod, Y.; Ringe, D.; Petsko, G.A. The 1.1-Å Resolution Crystal Structure of DJ-1, the Protein Mutated in Autosomal Recessive Early Onset Parkinson’s Disease. 2003. Available online: www.rcsb.org (accessed on 24 December 2020).
- Wilson, M.A. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxidants Redox Signal. 2011, 15, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Oh, S.E.; Mouradian, M.M. Regulation of Signal Transduction by DJ-1. In DJ-1/PARK7 Protein; Advances in Experimental Medicine and Biology; Springer: Singapore, 2017; Volume 1037, pp. 97–131. ISBN 9789811065835. [Google Scholar]
- Waak, J.; Weber, S.S.; Waldenmaier, A.; Görner, K.; Alunni-Fabbroni, M.; Schell, H.; Vogt-Weisenhorn, D.; Pham, T.T.; Reumers, V.; Baekelandt, V.; et al. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 2009, 23, 2478–2489. [Google Scholar] [CrossRef]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 Is a redox-dependent molecular chaperone that inhibits α-synuclein aggregate formation. PLoS Biol. 2004, 2, e362. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Jun, Y.W.; Kool, E.T. Small Substrate or Large? Debate Over the Mechanism of Glycation Adduct Repair by DJ-1. Cell Chem. Biol. 2020, 27, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Jacome, M.S.; Lei, S.; Hernandez-Franco, P.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Metabolic Dysfunction in Parkinson’s Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res. Bull. 2017, 133, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Powers, R.; Lei, S.; Anandhan, A.; Marshall, D.D.; Worley, B.; Cerny, R.L.; Dodds, E.D.; Huang, Y.; Panayiotidis, M.I.; Pappa, A.; et al. Metabolic investigations of the molecular mechanisms associated with Parkinson’s disease. Metabolites 2017, 7, 22. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Mencke, P.; Hanss, Z.; Boussaad, I.; Sugier, P.E.; Elbaz, A.; Krüger, R. Bidirectional Relation Between Parkinson’s Disease and Glioblastoma Multiforme. Front. Neurol. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Jain, D.; Jain, R.; Eberhard, D.; Eglinger, J.; Bugliani, M.; Piemonti, L.; Marchetti, P.; Lammert, E. Age-and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J. Mol. Cell Biol. 2012, 4, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, D.; Lammert, E. The role of the antioxidant protein DJ-1 in type 2 diabetes mellitus. Adv. Exp. Med. Biol. 2017, 1037, 173–186. [Google Scholar] [PubMed]
- Zhang, L.; Wang, J.; Wang, J.; Yang, B.; He, Q.; Weng, Q. Role of DJ-1 in Immune and Inflammatory Diseases. Front. Immunol. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Lev, N.; Roncevich, D.; Ickowicz, D.; Melamed, E.; Offen, D. Role of DJ-1 in Parkinson ’ s Disease. Mol. Neurosci. 2006, 29, 215–225. [Google Scholar] [CrossRef]
- Mhyre, T.R.; Nw, R.; Boyd, J.T.; Hall, G.; Room, C. Parkinson’s Disease. In Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease; Springer: Dordrecht, The Netherlands, 2012; Volume 65, ISBN 978-94-007-5415-7. [Google Scholar]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Bereczki, D. The description of all four cardinal signs of Parkinson’s disease in a Hungarian medical text published in 1690. Park. Relat. Disord. 2010, 16, 290–293. [Google Scholar] [CrossRef]
- Inamdar, N.; Arulmozhi, D.; Tandon, A.; Bodhankar, S. Parkinsons Disease: Genetics and Beyond. Curr. Neuropharmacol. 2007, 5, 99–113. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Brown, K.; Wilkinson, K.D.; Rees, H.D.; Huai, Q.; Ke, H.; Levey, A.I.; Li, L.; Chin, L.S. Familial Parkinson’s Disease-associated L166P Mutation Disrupts DJ-1 Protein Folding and Function. J. Biol. Chem. 2004, 279, 8506–8515. [Google Scholar] [CrossRef]
- Honbou, K.; Suzuki, N.N.; Horiuchi, M.; Niki, T.; Taira, T.; Ariga, H.; Inagaki, F. The crystal structure of DJ-1, a protein related to male fertility and Parkinson’s disease. J. Biol. Chem. 2003, 278, 31380–31384. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Tong, L. Crystal structure of human DJ-1, a protein associated with early onset Parkinson’s disease. J. Biol. Chem. 2003, 278, 31372–31379. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; Zhang, L.; Dawson, T.M.; Dawson, V.L. A missense mutation (L166P) in DJ-1, linked to familial Parkinson’s disease, confers reduced protein stability and impairs homo-oligomerization. J. Neurochem. 2003, 87, 1558–1567. [Google Scholar] [CrossRef]
- Repici, M.; Straatman, K.R.; Balduccio, N.; Enguita, F.J.; Outeiro, T.F.; Giorgini, F. Parkinson’s disease-associated mutations in DJ-1 modulate its dimerization in living cells. J. Mol. Med. 2013, 91, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, N.M.; Catazaro, J.; Lin, J.; Halouska, S.; Kizziah, J.L.; Basiaga, S.; Cerny, R.L.; Powers, R.; Wilson, M.A. Transient sampling of aggregation-prone conformations causes pathogenic instability of a parkinsonian mutant of DJ-1 at physiological temperature. Protein Sci. 2015, 24, 1671–1685. [Google Scholar] [CrossRef] [PubMed]
- Boussaad, I.; Obermaier, C.D.; Hanss, Z.; Bobbili, D.R.; Bolognin, S.; Glaab, E.; Wolynska, K.; Weisschuh, N.; De Conti, L.; May, C.; et al. A patient-based model of RNA mis-splicing uncovers treatment targets in Parkinson’s disease. Sci. Transl. Med. 2020, 1–12. [Google Scholar]
- Hauser, D.N.; Primiani, C.T.; Cookson, M.R. The Effects of Variants in the Parkin, PINK1, and DJ-1 Genes along with Evidence for their Pathogenicity. Curr. Protein Pept. Sci. 2016, 18, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Annesi, G.; Savettieri, G.; Pugliese, P.; D’Amelio, M.; Tarantino, P.; Ragonese, P.; La Bella, V.; Piccoli, T.; Civitelli, D.; Annesi, F.; et al. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann. Neurol. 2005, 58, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, T.; Joly, N.; Liebart, J.C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P.; et al. Guanine glycation repair by DJ-1/ Park7 and its bacterial homologs. Science 2017, 357, 208–211. [Google Scholar] [CrossRef]
- Richarme, G.; Mihoub, M.; Dairou, J.; Chi Bui, L.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [Google Scholar] [CrossRef]
- Matsuda, N.; Kimura, M.; Queliconi, B.B.; Kojima, W.; Mishima, M.; Takagi, K.; Koyano, F.; Yamano, K.; Mizushima, T.; Ito, Y.; et al. Parkinson’s disease-related DJ-1 functions in thiol quality control against aldehyde attack in vitro. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.C.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [PubMed]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Cantley, L.C.; DeNicola, G.M. NRF2 Rewires Cellular Metabolism to Support the Antioxidant Response. Master Regul. Oxidative Stress Transcr. Factor Nrf2 2016, 1–28. [Google Scholar]
- Giaime, E.; Yamaguchi, H.; Gautier, C.A.; Kitada, T.; Shen, J. Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLoS ONE 2012, 7, e40501. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Meulener, M.; Whitworth, A.J.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.D.; Pallanck, L.J.; Bonini, N.M. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr. Biol. 2005, 15, 1572–1577. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Gan, L.; Johnson, D.A.; Johnson, J.A. Keap1-Nrf2 activation in the presence and absence of DJ-1. Eur. J. Neurosci. 2010, 31, 967–977. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, H.; Zhang, L.; Liu, X.; Zhang, C.; Wang, Y.; He, Q.; Zhang, Y.; Li, Y.; Chen, Q.; et al. DJ-1 promotes colorectal cancer progression through activating PLAGL2/Wnt/BMP4 axis. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- De Lazzari, F.; Bisaglia, M. DJ-1 as a deglycating enzyme: A unique function to explain a multifaceted protein? Neural Regen. Res. 2017, 12, 1797–1798. [Google Scholar]
- Santiago, J.A.; Potashkin, J.A. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol. Med. 2013, 19, 176–186. [Google Scholar] [CrossRef]
- Cao, J.; Lou, S.; Ying, M.; Yang, B. DJ-1 as a human oncogene and potential therapeutic target. Biochem. Pharmacol. 2015, 93, 241–250. [Google Scholar] [CrossRef]
- Hinkle, D.A.; Mullett, S.J.; Gabris, B.E.; Hamilton, R.L. DJ-1 expression in glioblastomas shows positive correlation with p53 expression and negative correlation with epidermal growth factor receptor amplification. Neuropathology 2011, 31, 29–37. [Google Scholar] [CrossRef]
- Skoneczna, A.; Miciałkiewicz, A.; Skoneczny, M. Saccharomyces cerevisiae Hsp31p, a stress response protein conferring protection against reactive oxygen species. Free Radic. Biol. Med. 2007, 42, 1409–1420. [Google Scholar] [CrossRef]
- Tsai, C.J.; Aslam, K.; Drendel, H.M.; Asiago, J.M.; Goode, K.M.; Paul, L.N.; Rochet, J.C.; Hazbun, T.R. Hsp31 is a stress response chaperone that intervenes in the protein misfolding process. J. Biol. Chem. 2015, 290, 24816–24834. [Google Scholar] [CrossRef]
- Guerrero, E.; Vasudevaraju, P.; Hegde, M.L.; Britton, G.B.; Rao, K.S. Recent advances in α-synuclein functions, advanced glycation, and toxicity: Implications for Parkinson’s disease. Mol. Neurobiol. 2013, 47, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Gonc¸alves, S.; Tenreiro, S.; Rosado-Ramos, R.; Betzer, C.; Straatman, K.R.; Jensen, P.H.; Giorgini, F.; et al. DJ-1 interactions with α-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cit. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Kumar, S.; Hanpude, P.; Singh, A.K.; Johari, T.; Majumder, S.; Maiti, T.K. Partially oxidized DJ-1 inhibits α-synuclein nucleation and remodels mature α-synuclein fibrils in vitro. Commun. Biol. 2019, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; Pereira, C.; Reis, I.; Alonso, I.; Bastos-Lima, A.; Melo-Pires, M.; Magalhães, M. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016, 139, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Solti, K.; Kuan, W.L.; Fórizs, B.; Kustos, G.; Mihály, J.; Varga, Z.; Herberth, B.; Moravcsik, É.; Kiss, R.; Kárpáti, M.; et al. DJ-1 can form β-sheet structured aggregates that co-localize with pathological amyloid deposits. Neurobiol. Dis. 2020, 134, 104629. [Google Scholar] [CrossRef]
- Maillard, L.C.; Maillard, L.C.; Maillard, L.; Maillard, L. Action des acides aminés sur les sucres: Formation des mélanoïdines par voie méthodique. C. R. Acad. Sci. (Paris) 1912, 154, 66–68. [Google Scholar]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems—Role in ageing and disease. Drug Metab. Drug Interact. 2009, 23, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990, 269, 1–11. [Google Scholar] [CrossRef]
- Misra, K.; Banerjee, A.B.; Ray, S.; Raytt, M. Glyoxalase III from Escherichia coli: A single novel enzyme for the conversion of methylglyoxal into D-lactate without reduced glutathione. Biochem. J. 2000, 1003, 999–1003. [Google Scholar] [CrossRef]
- Subedi, K.P.; Choi, D.; Kim, I.; Min, B.; Park, C. Hsp31 of Escherichia coli K-12 is glyoxalase III. Mol. Microbiol. 2011, 81, 926–936. [Google Scholar] [CrossRef]
- Bankapalli, K.; Saladi, S.D.; Awadia, S.S.; Goswami, A.V.; Samaddar, M.; D’Silva, P. Robust Glyoxalase activity of Hsp31, a ThiJ/DJ-1/PfpI Family Member Protein, Is Critical for Oxidative Stress Resistance in Saccharomyces cerevisiae. J. Biol. Chem. 2015, 290, 26491–26507. [Google Scholar] [CrossRef]
- Zhao, Q.; Su, Y.; Wang, Z.; Chen, C.; Wu, T.; Huang, Y. Identification of glutathione (GSH)-independent glyoxalase III from Schizosaccharomyces pombe. BMC Evol. Biol. 2014, 14, 1–18. [Google Scholar] [CrossRef]
- Hasim, S.; Hussin, N.A.; Alomar, F.; Bidasee, K.R.; Nickerson, K.W.; Wilson, M.A. A glutathione-independent glyoxalase of the DJ-1 superfamily plays an important role in managing metabolically generated methylglyoxal in candida albicans. J. Biol. Chem. 2014, 289, 1662–1674. [Google Scholar] [CrossRef]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Yang, Y.; Gehrke, S.; Imai, Y.; Huang, Z.; Ouyang, Y.; Wang, J.W.; Yang, L.; Beal, M.F.; Vogel, H.; Lu, B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA 2006, 103, 10793–10798. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, D.; Calì, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Cookson, M.R. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy 2011, 7, 531–532. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Paik, J.Y.; Jung, K.H.; Lee, J.H.; Park, J.W.; Lee, K.H. Reactive oxygen species-driven HIF1α triggers accelerated glycolysis in endothelial cells exposed to low oxygen tension. Nucl. Med. Biol. 2017, 45, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Parsanejad, M.; Zhang, Y.; Qu, D.; Irrcher, I.; Rousseaux, M.W.C.; Aleyasin, H.; Kamkar, F.; Callaghan, S.; Slack, R.S.; Mak, T.W.; et al. Regulation of the VHL/HIF-1 pathway by DJ-1. J. Neurosci. 2014, 34, 8043–8050. [Google Scholar] [CrossRef] [PubMed]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M.M. Neuroprotective function of dj-1 in Parkinson’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 683920. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y. Oxidized DJ1 as a possible biomarker of Parkinson’s disease. J. Clin. Biochem. Nutr. 2013, 54, 31–38. [Google Scholar]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef]
- Piston, D.; Alvarez-Erviti, L.; Bansal, V.; Gargano, D.; Yao, Z.; Szabadkai, G.; Odell, M.; Rhyan Puno, M.; Björkblom, B.; Maple-Grødem, J.; et al. Corrigendum: DJ-1 is a redox sensitive adapter protein for high molecular weight complexes involved in regulation of catecholamine homeostasis. Hum. Mol. Genet. 2017, 26, 4028–4041. [Google Scholar] [CrossRef]
- Piston, D.; Gegg, M.E. The role of DJ-1 complexes and catecholamine metabolism: Relevance for familial and idiopathic Parkinson’s disease. Neural Regen. Res. 2018, 13, 815–816. [Google Scholar]
- Meiser, J.; Delcambre, S.; Wegner, A.; Jäger, C.; Ghelfi, J.; d’Herouel, A.F.; Dong, X.; Weindl, D.; Stautner, C.; Nonnenmacher, Y.; et al. Loss of DJ-impairs antioxidant response by altered glutamine and serine metabolism. Neurobiol. Dis. 2016, 89, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, K.S.; Iyirhiaro, G.O.; Marcogliese, P.C.; Callaghan, S.M.; Qu, D.; Kim, W.J.; Slack, R.S.; Park, D.S. DJ-1 modulates the unfolded protein response and cell death via upregulation of ATF4 following ER stress. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, B.; Azuelos, I.; Platé, M.; Guillotin, D.; Forty, E.J.; Contento, G.; Woodcock, H.V.; Redding, M.; Taylor, A.; Brunori, G.; et al. MTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-1-induced collagen biosynthesis. Sci. Signal. 2019, 12, eaav3048. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T α-synuclein toxicity. J. Biol. Chem. 2005, 280, 43150–43158. [Google Scholar] [CrossRef]
- Saeed, U.; Ray, A.; Valli, R.K.; Kumar, A.M.R.; Ravindranath, V. DJ-1 Loss by glutaredoxin but not glutathione depletion triggers daxx translocation and cell death. Antioxidants Redox Signal. 2010, 13, 127–144. [Google Scholar] [CrossRef]
- Lopert, P.; Patel, M. Brain mitochondria from DJ-1 knockout mice show increased respiration-dependent hydrogen peroxide consumption. Redox Biol. 2014, 2, 667–672. [Google Scholar] [CrossRef]
- Ozawa, K.; Tsumoto, H.; Miura, Y.; Yamaguchi, J.; Iguchi-Ariga, S.M.M.; Sakuma, T.; Yamamoto, T.; Uchiyama, Y. DJ-1 is indispensable for the S-nitrosylation of Parkin, which maintains function of mitochondria. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Danileviciute, E.; Zeng, N.; Capelle, C.; Paczia, N.; Kurniawan, H.; Gillespie, M.A.; Coowar, D.; Maria, D.; Weisenhorn, V.; Grusdat, M.; et al. PARK7/DJ-1 promotes pyruvate dehydrogenase activity and maintains Treg homeostasis. bioRxiv 2019, 4–6. [Google Scholar] [CrossRef]
- Malty, R.H.; Aoki, H.; Kumar, A.; Phanse, S.; Amin, S.; Zhang, Q.; Minic, Z.; Goebels, F.; Musso, G.; Wu, Z.; et al. A Map of Human Mitochondrial Protein Interactions Linked to Neurodegeneration Reveals New Mechanisms of Redox Homeostasis and NF-κB Signaling. Cell Syst. 2017, 5, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; et al. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Hum. Mol. Genet. 2005, 14, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Ottolini, D.; Soriano, M.E.; Brini, M. A new split-GFP-based probe reveals DJ-1 translocation into the mitochondrial matrix to sustain ATP synthesis upon nutrient deprivation. Hum. Mol. Genet. 2015, 24, 1045–1060. [Google Scholar] [CrossRef]
- Chen, R.; Park, H.A.; Mnatsakanyan, N.; Niu, Y.; Licznerski, P.; Wu, J.; Miranda, P.; Graham, M.; Tang, J.; Boon, A.J.W.; et al. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef]
- Weinert, M.; Millet, A.; Jonas, E.A.; Alavian, K.N. The mitochondrial metabolic function of DJ-1 is modulated by 14-3-3b. FASEB J. 2019, 33, 8925–8934. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Berry, E.M.; Growdon, J.H.; Wurtman, J.J.; Caballero, B.; Wurtman, R.J. A balanced carbohydrate: Protein diet in the management of Parkinson’s disease. Neurology 1991, 41, 1295. [Google Scholar] [CrossRef]
- VanItallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tian, B. Metabolic syndrome: An important risk factor for Parkinson’s disease. Oxid. Med. Cell. Longev. 2014, 2014, 729194. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Richarme, G.; Abdallah, J.; Mathas, N.; Gautier, V.; Dairou, J. Further characterization of the Maillard deglycase DJ-1 and its prokaryotic homologs, deglycase 1/Hsp31, deglycase 2/YhbO, and deglycase 3/YajL. Biochem. Biophys. Res. Commun. 2018, 503, 703–709. [Google Scholar] [CrossRef]
- Pfaff, D.H.; Fleming, T.; Nawroth, P.; Teleman, A.A. Evidence Against a Role for the Parkinsonism-associated Protein DJ-1 in Methylglyoxal Detoxification. J. Biol. Chem. 2017, 292, 685–690. [Google Scholar] [CrossRef]
- Andreeva, A.; Bekkhozhin, Z.; Omertassova, N.; Baizhumanov, T.; Yeltay, G.; Akhmetali, M.; Toibazar, D.; Utepbergenov, D. The apparent deglycase activity of DJ-1 results from the conversion of free methylglyoxal present in fast equilibrium with hemithioacetals and hemiaminals. J. Biol. Chem. 2019, 294, 18863–18872. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Weber, D.; Raupbach, J.; Dakal, T.C.; Fließbach, K.; Ramirez, A.; Grune, T.; Wüllner, U. Advanced glycation end products and protein carbonyl levels in plasma reveal sex-specific differences in Parkinson’s and Alzheimer’s disease. Redox Biol. 2020, 34, 101546. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K.; Tilley, B.C.; Elm, J.J.; Babcock, D.; Hauser, R.; Ross, G.W.; Augustine, A.H.; Augustine, E.U.; Aminoff, M.J.; Bodis-Wollner, I.G.; et al. Effect of creatine monohydrate on clinical progression in patients with parkinson disease: A randomized clinical trial. JAMA J. Am. Med. Assoc. 2015, 313, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Nickels, S.L.; Walter, J.; Bolognin, S.; Gérard, D.; Jaeger, C.; Qing, X.; Tisserand, J.; Jarazo, J.; Hemmer, K.; Harms, A.; et al. Impaired serine metabolism complements LRRK2-G2019S pathogenicity in PD patients. Park. Relat. Disord. 2019, 67, 48–55. [Google Scholar] [CrossRef]
- Nuzzo, T.; Punzo, D.; Devoto, P.; Rosini, E.; Paciotti, S.; Sacchi, S.; Li, Q.; Thiolat, M.L.; Véga, C.; Carella, M.; et al. The levels of the NMDA receptor co-agonist D-serine are reduced in the substantia nigra of MPTP-lesioned macaques and in the cerebrospinal fluid of Parkinson’s disease patients. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Gelfin, E.; Kaufman, Y.; Korn-Lubetzki, I.; Bloch, B.; Kremer, I.; Javitt, D.C.; Heresco-Levy, U. D-serine adjuvant treatment alleviates behavioural and motor symptoms in Parkinson’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef]
- Prasad, R.K.; Ismail-Beigi, F. Mechanism of stimulation of glucose transport by H2O2: Role of phospholipase C. Arch. Biochem. Biophys. 1999, 362, 113–122. [Google Scholar] [CrossRef]
- Liemburg-Apers, D.C.; Willems, P.H.G.M.; Koopman, W.J.H.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; LeWitt, P.A.; Xu, K.; Eberly, S.; Watts, A.; Matson, W.R.; Marras, C.; Kieburtz, K.; Rudolph, A.; Bogdanov, M.B.; et al. Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch. Neurol. 2009, 66, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Schwarzschild, M.A.; Schwid, S.R.; Marek, K.; Watts, A.; Lang, A.E.; Oakes, D.; Shoulson, I.; Ascherio, A.; Hyson, C.; Gorbold, E.; et al. Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch. Neurol. 2008, 65, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Shoulsen, I.; Fann, S.; Oakes, D.; Kieburtz, K. Effects-of-Tocopherol-and-Deprenyl-on-the-Progression-of-Disabil-1993. N. Engl. J. Med. 1993, 328, 176–183. [Google Scholar]
- Shoulson, I.; Oakes, D.; Fahn, S.; Lang, A.; William Langston, J.; LeWitt, P.; Warren Olanow, C.; Penney, J.B.; Tanner, C.; Kieburtz, K.; et al. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: A randomized placebo-controlled extension of the Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism trial. Ann. Neurol. 2002, 51, 604–612. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.J.; Murphy, M.P.; Taylor, K.M. A double-blind, placebo-controlled study to assess the mitochondria- targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Flint Beal, M.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early parkinson disease no evidence of benefit. JAMA Neurol. 2014, 75, 543–552. [Google Scholar]


{kind=link}
| Metabolic Alteration | DJ-1 Status or Mutation | Model | Reference |
|---|---|---|---|
| Nucleotide/DNA/RNA glycation | siRNA knockdown | HeLa cells | [38] |
| Amino acid/protein glycation | C106S, C53S, and C46S DJ-1 mutants | - | [39] |
| Dicarbonyl-adduct damage | L10P, M26I, A104T, D149A, and L166P | - | [40] |
| Abnormal mitochondrial morphology mitochondrial/neuronal dysfunction mitochondrial/neuronal dysfunction | Loss of protein | M17 human neuroblastoma cells Mouse embryonic fibroblasts (MEFs) PD patient iPSC-derived neurons | [37] [41] [42] [43] [33] |
| Compromised mitochondrial uncoupling | Loss of protein | primary murine neurons | [44] |
| Increased ROS levels | Loss of protein | primary mouse embryonic fibroblasts brains from DJ- KO mice | [45] |
| Decreased PDH protein levels in DJ-1 KO compared to WT mice | Loss of protein | brain tissue from DJ-1 deficient mice | [46] |
| Decreased HIF1α level upon hypoxia | Loss of protein | primary cortical neurons derived from DJ-1 KO mouse embryos | [47] |
| Reduced serine biosynthesis | Loss of protein | LUHMES cells, a dopaminergic neuronal culture model | [48] |
| Decreased ATF4 transcript expression | Loss of protein | mouse embryonic fibroblasts | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mencke, P.; Boussaad, I.; Romano, C.D.; Kitami, T.; Linster, C.L.; Krüger, R. The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease. Cells 2021, 10, 347. https://doi.org/10.3390/cells10020347
Mencke P, Boussaad I, Romano CD, Kitami T, Linster CL, Krüger R. The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease. Cells. 2021; 10(2):347. https://doi.org/10.3390/cells10020347
Chicago/Turabian StyleMencke, Pauline, Ibrahim Boussaad, Chiara D. Romano, Toshimori Kitami, Carole L. Linster, and Rejko Krüger. 2021. "The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease" Cells 10, no. 2: 347. https://doi.org/10.3390/cells10020347
APA StyleMencke, P., Boussaad, I., Romano, C. D., Kitami, T., Linster, C. L., & Krüger, R. (2021). The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease. Cells, 10(2), 347. https://doi.org/10.3390/cells10020347