Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma


 , ,
, ,  ,
, 
Abstract
Simple Summary
Abstract
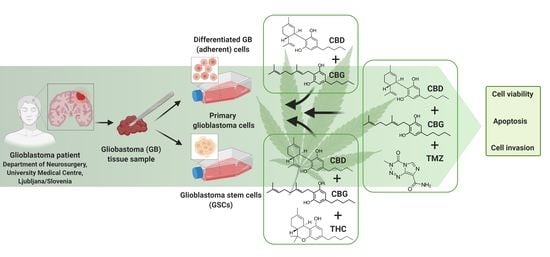
1. Introduction
2. Materials and Methods
2.1. Cannabinoids
2.2. Cell Cultures
2.3. Establishment of Primary GB Cells
2.4. Establishment of GSC Lines
2.5. Cell Viability Assay
2.6. Three-Dimensional (3D) Tumour Spheroid Invasion Assay
2.7. Immunofluorescence of GSC Spheroids
2.8. Immunocytochemistry
2.9. Cell Cycle Analyses
2.10. Apoptosis Analyses
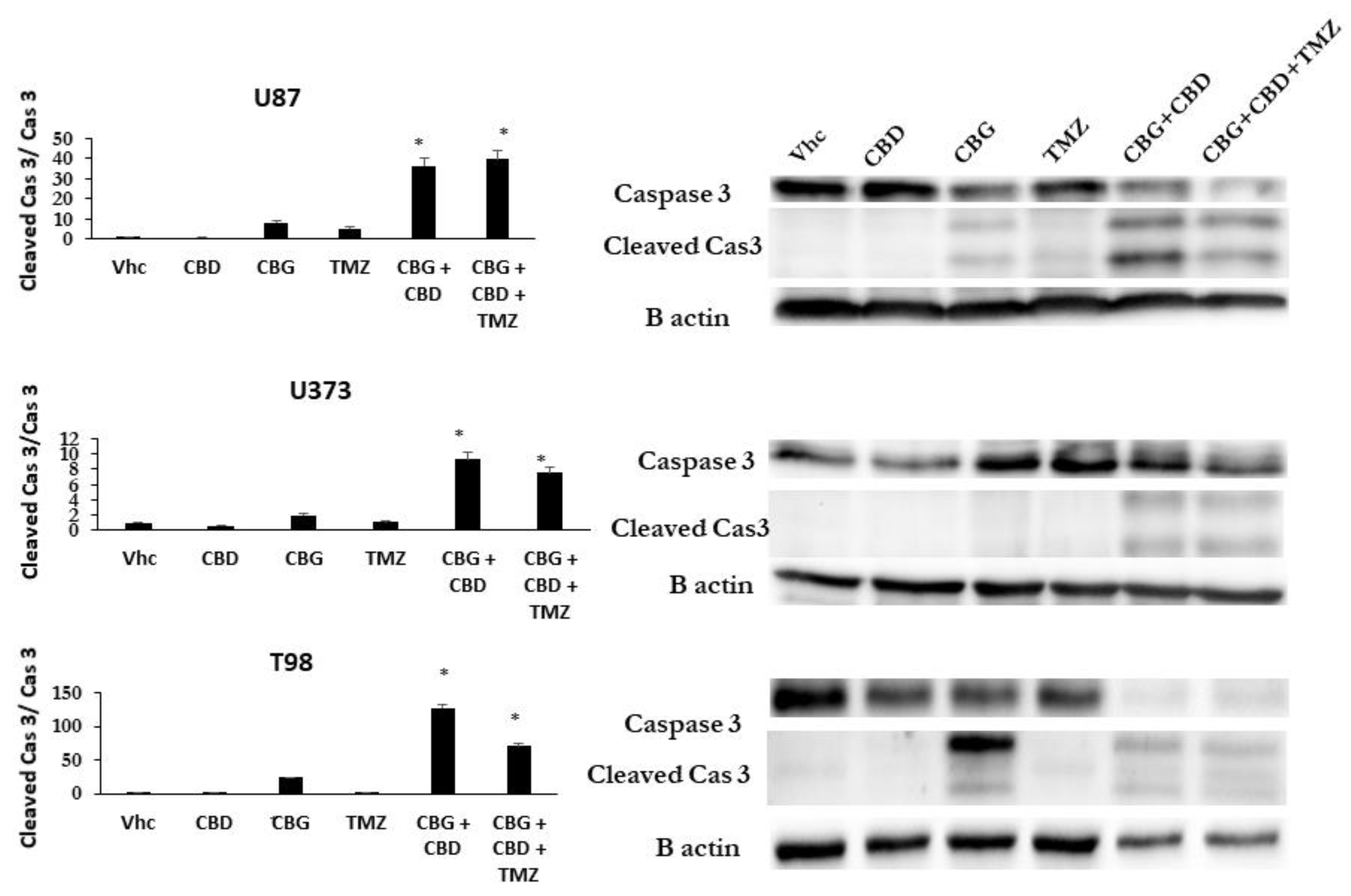
2.11. Caspase-3-Dependent Apoptosis
3. Results
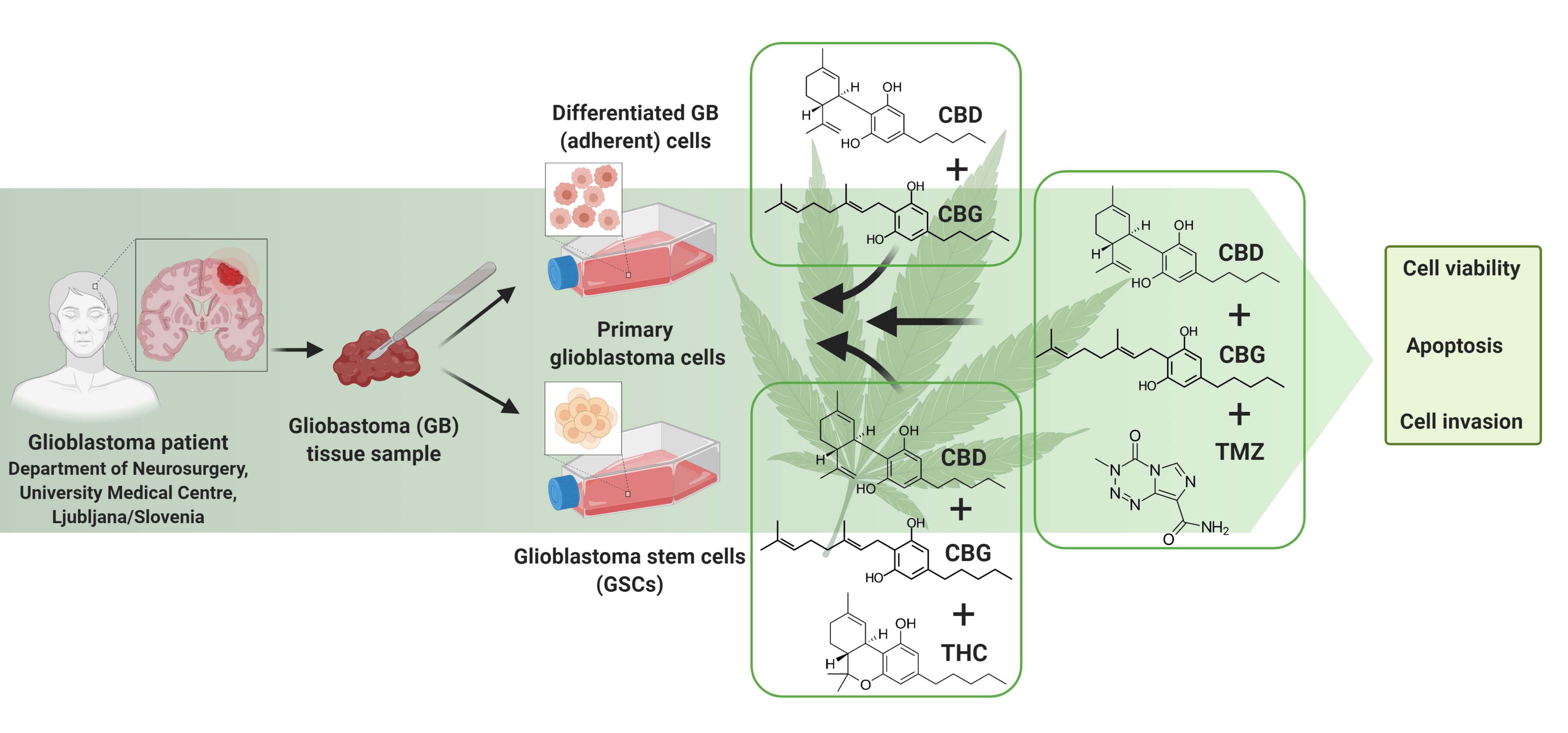
3.1. The Cannabinoids CBG, CBD, and THC Affect the Viability of Primary GB Cells and GSCs
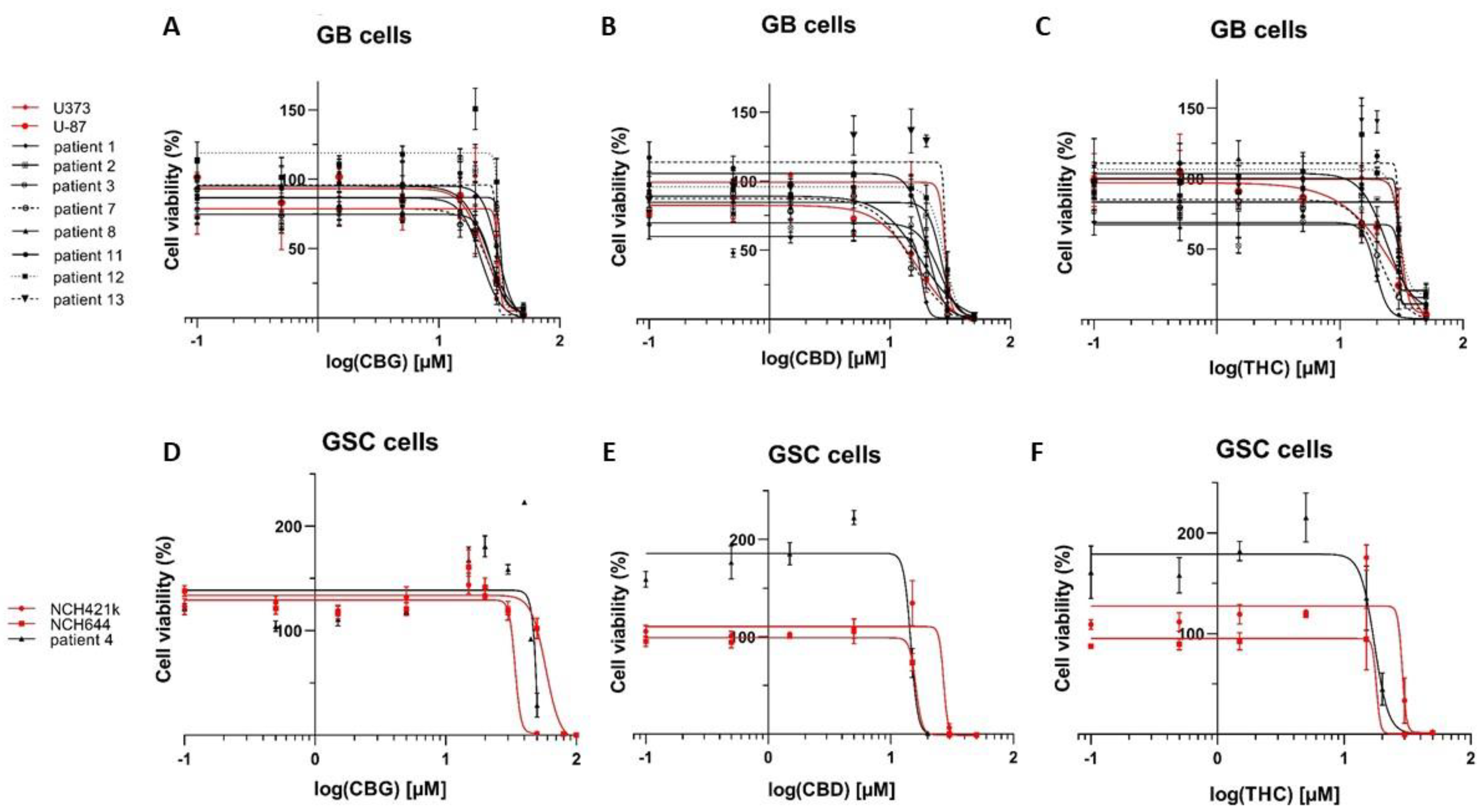
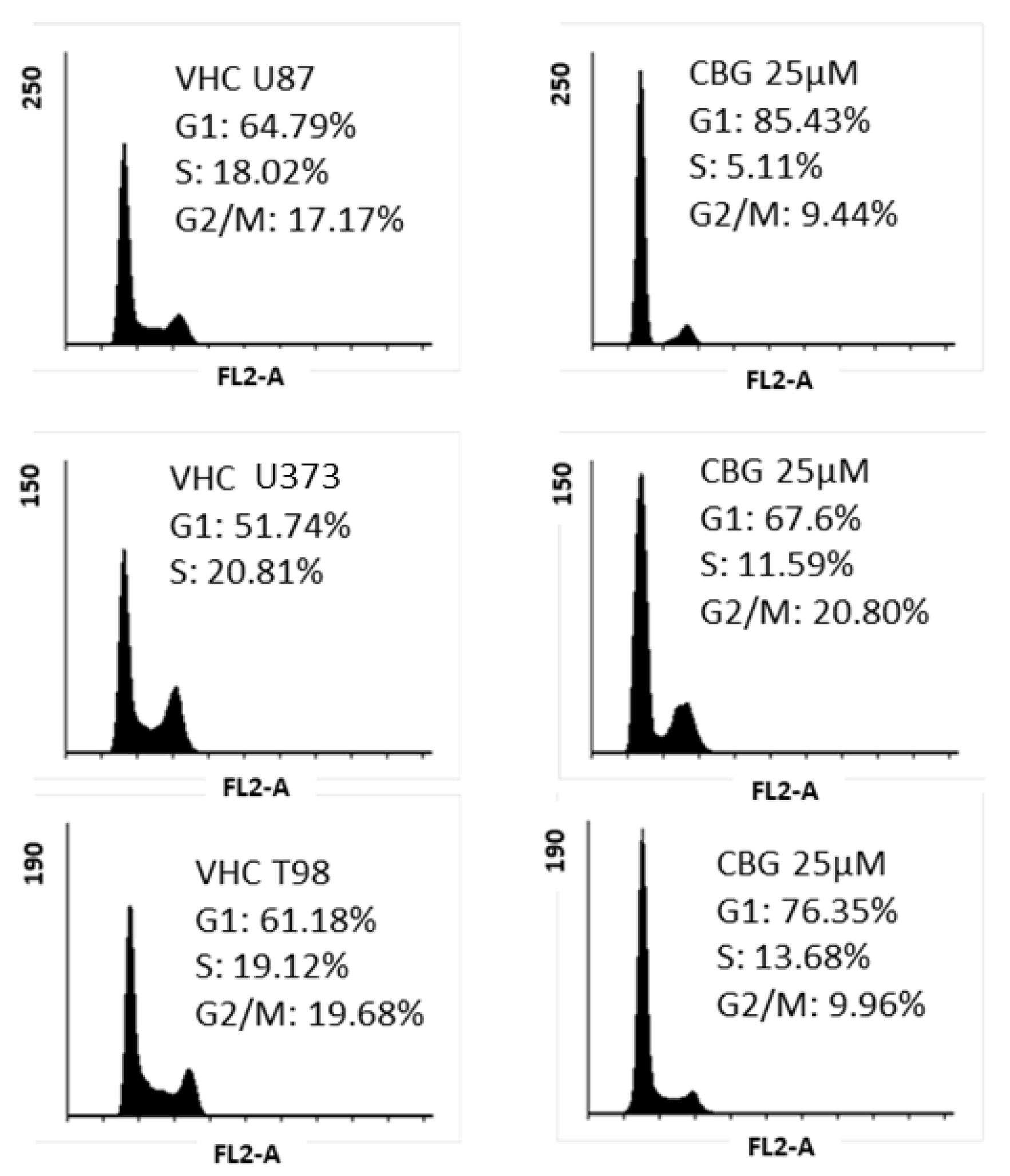
3.2. CBG Exerts a Cytostatic Effect
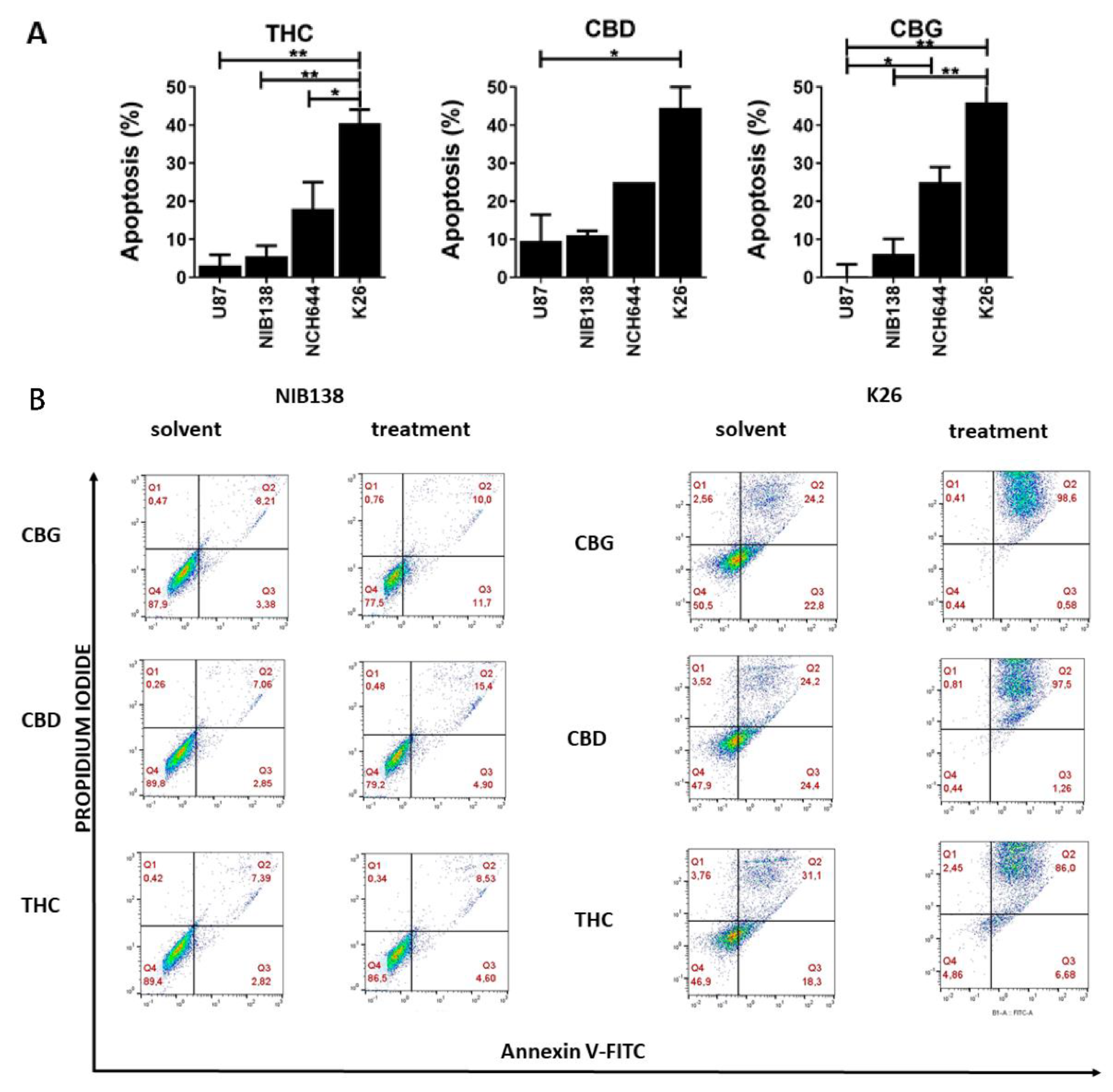
3.3. The Cannabinoids CBG, CBD, and THC Affect Apoptosis of Primary GB Cells and GSCs
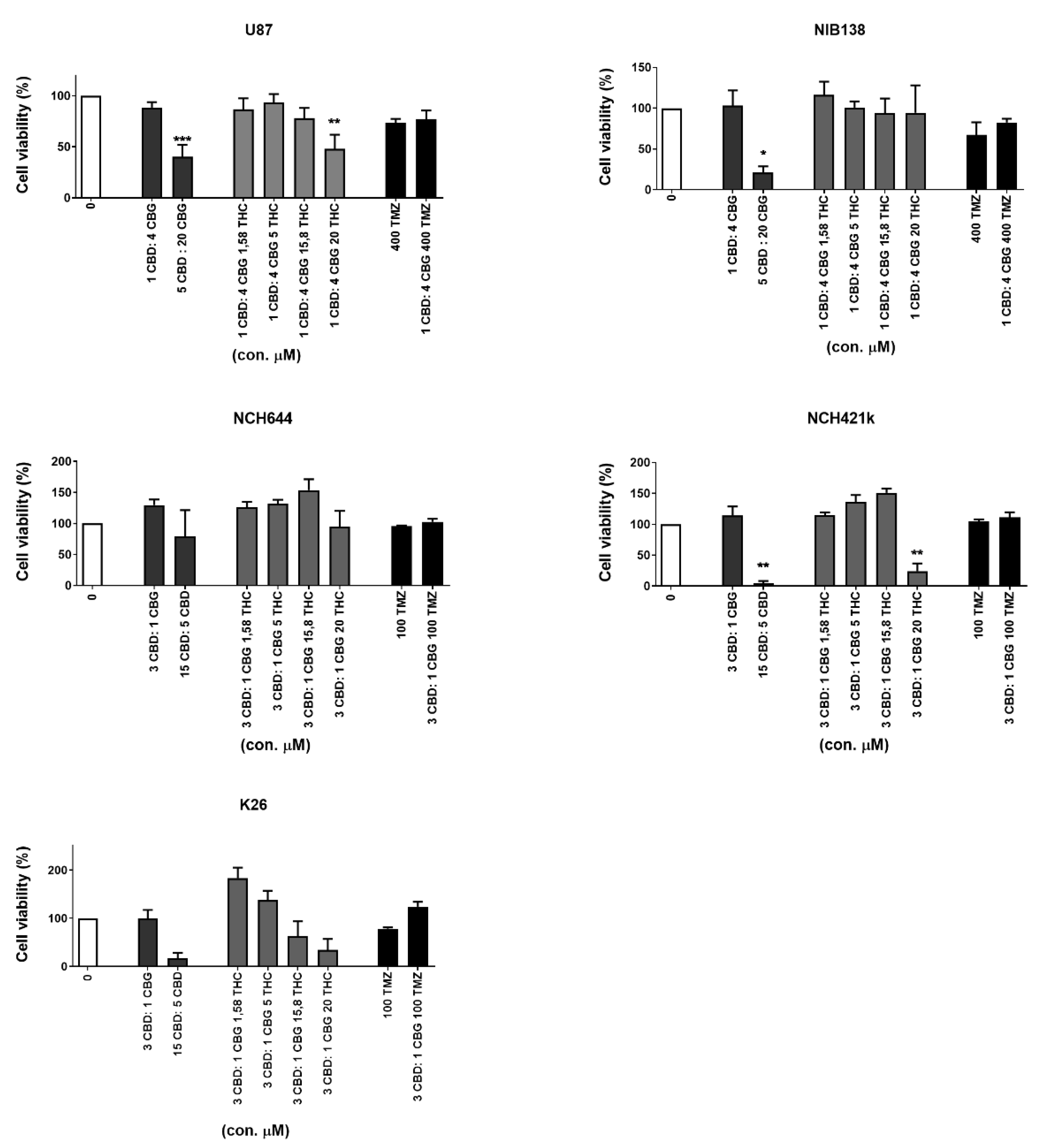
3.4. Combinations of CBG and CBD Reduced the Viability of GB Cells and GSCs in an Additive Manner
3.5. Inhibition of GB Cells and GSCs Viability by Combinations of CBG and CBD with THC and TMZ
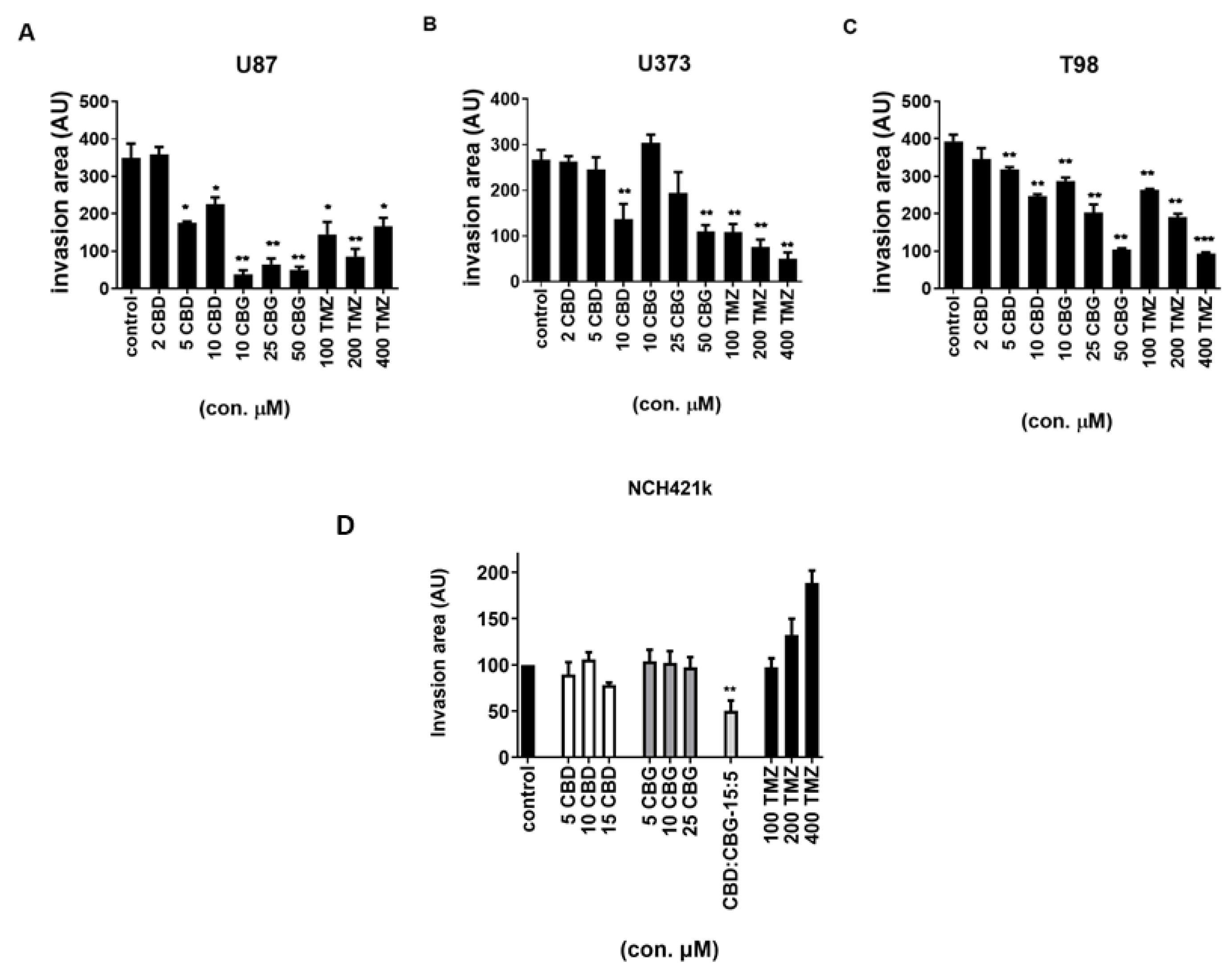
3.6. The Effect of Cannabinoids CBG, CBD and TMZ on Invasion of GB and GSC Cells
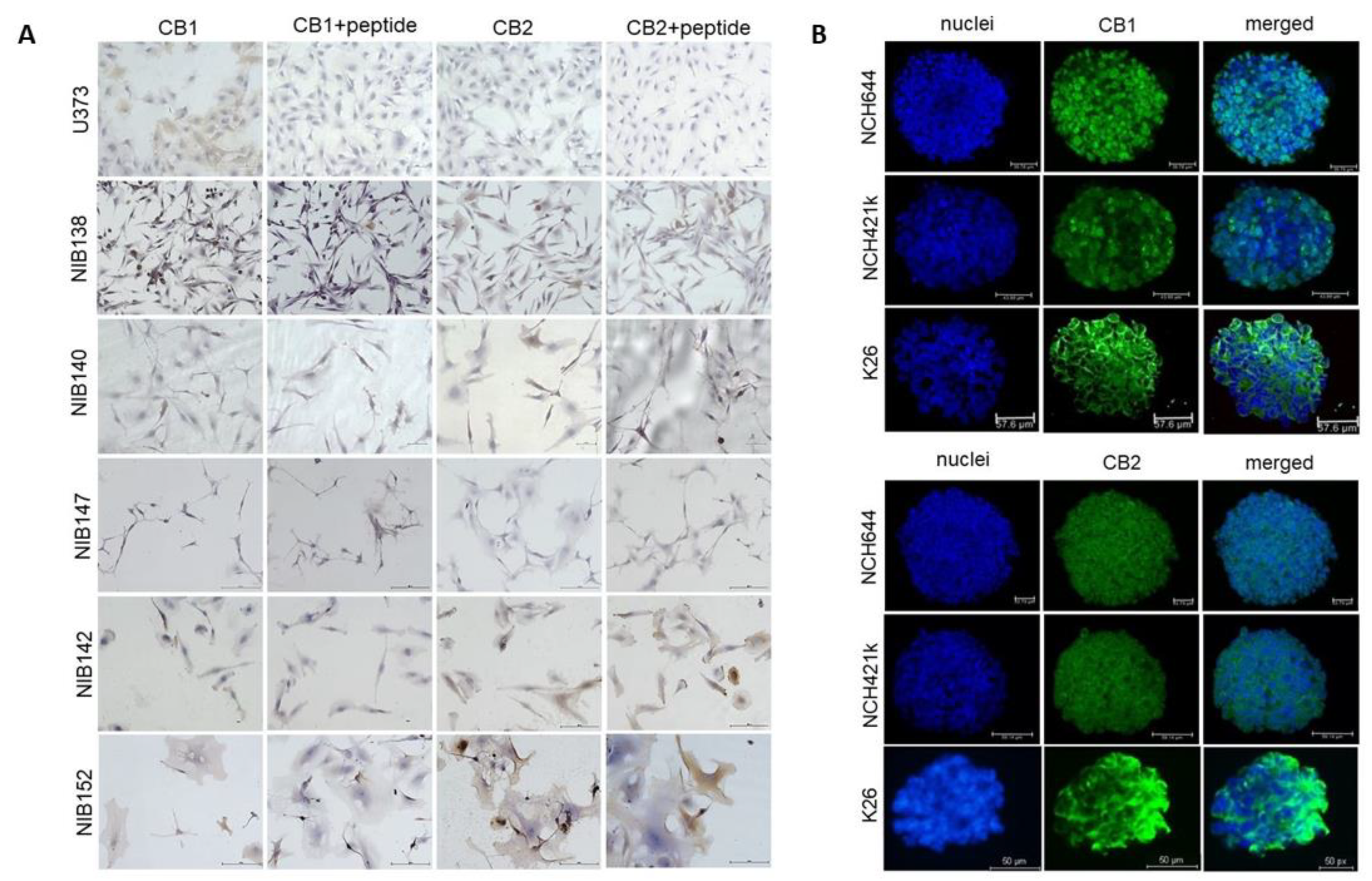
3.7. The Cannabinoid Receptors CB1 and CB2 are Highly but Differentially Expressed in Patient GB Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor cell invasion in glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef]
- Yool, A.J.; Ramesh, S. Molecular targets for combined therapeutic strategies to limit glioblastoma cell migration and invasion. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.-K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Molina, E.S.; Pillat, M.M.; Moura-Neto, V.; Lah, T.T.; Ulrich, H. Glioblastoma stem-like cells: Approaches for isolation and characterization. J. Cancer Stem Cell Res. 2017, 1, 1. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Herbener, V.J.; Burster, T.; Goreth, A.; Pruss, M.; von Bandemer, H.; Baisch, T.; Fitzel, R.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; et al. Considering the experimental use of temozolomide in glioblastoma research. Biomedicines 2020, 8, 151. [Google Scholar] [CrossRef]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. [Google Scholar] [CrossRef]
- Russo, E.B. Cannabis therapeutics and the future of neurology. Front. Integr. Neurosci. 2018, 12, 51. [Google Scholar] [CrossRef]
- Pacher, P.; Kogan, N.M.; Mechoulam, R. Beyond THC and endocannabinoids. Annu. Rev. Pharmacol. Toxicol. 2019, 60, 637–659. [Google Scholar] [CrossRef] [PubMed]
- Afrin, F.; Chi, M.; Eamens, A.L.; Duchatel, R.J.; Douglas, A.M.; Schneider, J.; Gedye, C.; Woldu, A.S.; Dun, M.D. Can hemp help? Low-THC cannabis and non-THC cannabinoids for the treatment of cancer. Cancers 2020, 12, 1033. [Google Scholar] [CrossRef]
- Abrams, D.I. Should oncologists recommend cannabis? Curr. Treat. Options Oncol. 2019, 20, 59. [Google Scholar] [CrossRef]
- Abrams, D.I.; Guzman, M. Cannabis in cancer care. Clin. Pharmacol. Ther. 2015, 97, 575–586. [Google Scholar] [CrossRef]
- Luís, Â.; Marcelino, H.; Rosa, C.; Domingues, F.; Pereira, L.; Cascalheira, J.F. The effects of cannabinoids on glioblastoma growth: A systematic review with meta-analysis of animal model studies. Eur. J. Pharmacol. 2020, 876. [Google Scholar] [CrossRef]
- De Meijer, E.P.M.; Hammond, K.M. The inheritance of chemical phenotype in Cannabis sativa L. (II): Cannabigerol predominant plants. Euphytica 2005, 145, 189–198. [Google Scholar] [CrossRef]
- Likar, R.; Nahler, G. The use of cannabis in supportive care and treatment of brain tumor. Neuro Oncol. Pract. 2017, 4, 151–160. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Bianchessi, S.; Costa, B.; Macchi, P.; Parolaro, D. The non-psychoacti cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell. Mol. Life Sci. 2006, 63, 2057–2066. [Google Scholar] [CrossRef]
- Velasco, G.; Carracedo, A.; Blazquez, C.; Lorente, M.; Aguado, T.; Haro, A.; Sanchez, C.; Galve-Roperh, I.; Guzman, M. Cannabinoids and gliomas. Mol. Neurobiol. 2007, 36, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Lorente, M.; Rodríguez-Fornés, F.; Hernández-Tiedra, S.; Salazar, M.; García-Taboada, E.; Barcia, J.; Guzman, M.; Velasco, G. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Iappelli, M.; Verde, R.; Stott, C.G.; Cristino, L.; Orlando, P.; Di Marzo, V. Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: Pro-Apoptotic effects and underlying mechanisms. Br. J. Pharmacol. 2012, 168, 79–102. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Nabissi, M.; Santoni, G.; Ligresti, A. Actions and regulation of ionotropic cannabinoid receptors. Adv. Pharmacol. 2017, 80, 249–289. [Google Scholar] [PubMed]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2012, 4, 48–57. [Google Scholar] [CrossRef]
- Baek, S.-H.; Han, S.D.; Yook, C.N.; Kim, Y.C.; Kwak, J.S. Synthesis and antitumour activity of cannabigerol. Arch. Pharmacol. Res. 1996, 19, 228–230. [Google Scholar] [CrossRef]
- Deiana, S. Potential medicinal uses of cannabigerol: A brief overview. In Handbook of Cannabis and Related Pathologies: Biology, Pharmacology, Diagnosis, and Treatment, 1st ed.; Preedy, V., Ed.; Academic Press: London, UK, 2017; pp. 958–967. [Google Scholar]
- Pena Almidon, A.M. Evaluation of Cannabigerol Activity in Human Glioblastoma Cell Lines. Master’s Thesis, Scuola di Scienze del Farmaco e dei Prodotti Della Salute, Universita’ Degli Studi di Camerino, Camerino, Italy, 2019. [Google Scholar]
- Ligresti, A.; Moriello, A.S.; Starowicz, K.; Matias, I.; Pisanti, S.; De Petrocellis, L.; Laezza, C.; Portella, G.; Bifulco, M.; Di Marzo, V. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.-Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer Ther. 2007, 6, 2921–2927. [Google Scholar] [CrossRef]
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From phytocannabinoids to cannabinoid receptors and endocannabinoids: Pleiotropic physiological and pathological roles through complex pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [PubMed]
- Brierley, D.I.; Harman, J.R.; Giallourou, N.; Leishman, E.; Roashan, A.E.; Mellows, B.A.D.; Bradshaw, H.B.; Swann, J.R.; Patel, K.; Whalley, B.J.; et al. Chemotherapy-Induced cachexia dysregulates hypothalamic and systemic lipoamines and is attenuated by cannabigerol. J. Cachexia Sarcopenia Muscle 2019, 10, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Pagano, E.; Romano, B.; Panzera, S.; Maiello, F.; Coppola, D.; De Petrocellis, L.; Buono, L.; Orlando, P.; Izzo, A.A. Colon carcinogenesis is inhibited by the TRPM8 antagonist cannabigerol, a Cannabis-Derived non-psychotic cannabinoid. Carcinogenesis 2014, 35, 2787–2797. [Google Scholar] [CrossRef] [PubMed]
- Deiana, S.; Watanabe, A.; Yamasaki, Y.; Amada, N.; Arthur, M.; Fleming, S.; Woodcock, H.; Dorward, P.; Pigliacampo, B.; Close, S.; et al. Plasma and brain pharmacokinetic profile of cannabidiol (CBD), cannabidivarine (CBDV), Δ9-tetrahydrocannabivarin (THCV) and cannabigerol (CBG) in rats and mice following oral and intraperitoneal administration and CBD action on obsessive-compulsive behaviour. Psychopharmacology 2011, 219, 859–873. [Google Scholar]
- Kološa, K.; Motaln, H.; Herold-Mende, C.; Koršič, M.; Lah, T.T. Paracrine effects of mesenchymal stem cells induce senescence and differentiation of glioblastoma stem-like cells. Cell Transplant. 2015, 24, 631–644. [Google Scholar] [CrossRef]
- Breznik, B.; Motaln, H.; Vittori, M.; Rotter, A.; Turnšek, L.T. Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget 2017, 8, 25482–25499. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2020, 8. [Google Scholar] [CrossRef]
- Podergajs, N.; Motaln, H.; Rajčević, U.; Verbovšek, U.; Koršič, M.; Obad, N.; Espedal, H.; Vittori, M.; Herold-Mende, C.; Miletic, H.; et al. Transmembrane protein CD9 is glioblastoma biomarker, relevant for maintenance of glioblastoma stem cells. Oncotarget 2016, 7, 593–609. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ng, L.; Ozawa, T.; Stella, N. Quantitative analyses of synergistic responses between cannabidiol and DNA-damaging agents on the proliferation and viability of glioblastoma and neural progenitor cells in culture. J. Pharmacol. Exp. Ther. 2017, 360, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Orhan, G.; Bayram, A.; Zer, Y.; Balci, I. Synergy tests by E test and checkerboard methods of antimicrobial combinations against Brucella melitensis. J. Clin. Microbiol. 2005, 43, 140–143. [Google Scholar] [CrossRef]
- Hira, V.V.; Breznik, B.; Van Noorden, C.J.F.; Turnšek, L.T.; Molenaar, R.J. 2D and 3D in vitro assays to quantify the invasive behavior of glioblastoma stem cells in response to SDF-1α. BioTechniques 2020, 69, 5. [Google Scholar] [CrossRef]
- Kenig, S.; Frangež, R.; Pucer, A.; Lah, T. Inhibition of cathepsin L lowers the apoptotic threshold of glioblastoma cells by up-regulating p53 and transcription of caspases 3 and 7. Apoptosis Int. J. Program. Cell Death 2011, 16, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Carracedo, A.; Julien, B.; Velasco, G.; Milman, G.; Mechoulamluis, R.; Alvarez, L.; Guzman, M.; Galve-Roperth, I. Cannabinoids induce glioma stem-like cell differentiation and inhibit gliomagenesis. J. Biol. Chem. 2007, 282, 6854–6862. [Google Scholar] [CrossRef]
- Zhu, L.X.; Sharma, S.; Stolina, M.; Gardner, B.; Roth, M.D.; Tashkin, D.P.; Dubinett, S.M. Δ-9-Tetrahydrocannabinol inhibits antitumor immunity by a CB2 receptor-mediated, cytokine-dependent pathway. J. Immunol. 2000, 165, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Kaul, K.; Mishra, S.; Charan, M.; Ganju, R.K. Cannabinoid signaling in cancer. Adv. Exp. Med. Biol. 2019, 1162, 51–61. [Google Scholar] [PubMed]
- Galve-Roperh, I.; Chiurchiù, V.; Díaz-Alonso, J.; Bari, M.; Guzmán, M.; Maccarrone, M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog. Lipid Res. 2013, 52, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol stimulates AML-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-Dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Sandalcioglu, I.E.; Karsak, M. Cannabinoids in glioblastoma therapy: New applications for old drugs. Front. Mol. Neurosci. 2018, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ladin, D.A.; Soliman, E.; Griffin, L.T.; Van Dross, R. Preclinical and clinical assessment of cannabinoids as anti-cancer agents. Front. Pharmacol. 2016, 7, 1–18. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [PubMed]
- Cascio, M.G.; Gauson, L.A.; Stevenson, L.A.; Ross, R.A.; Pertwee, R.G. Evidence that the plant cannabinoid cannabigerol is a highly potent α2-adrenoceptor agonist and moderately potent 5HT1A receptor antagonist. Br. J. Pharmacol. 2010, 159, 129–141. [Google Scholar] [CrossRef]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef]
- Spiteri, I.; Caravagna, G.; Cresswell, G.D.; Vatsiou, A.; Nichol, D.; Acar, A.; Ermini, L.; Chkhaidze, K.; Werner, B.; Mair, R.; et al. Evolutionary dynamics of residual disease in human glioblastoma. Ann. Oncol. 2019, 30, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Murase, R.; Limbad, C.; Singer, E.; Allison, J.; Adrados, I.; Kawamura, R.; Pakdel, A.; Fukuyo, Y.; Nguyen, D.; et al. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res. 2013, 73, 1559–1569. [Google Scholar] [CrossRef]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef]
- Galanti, G.; Fisher, T.; Kventsel, I.; Shoham, J.; Gallily, R.; Mechoulam, R.; Lavie, G.; Amariglio, N.; Rechavi, G.; Toren, A. Δ9-Tetrahydrocannabinol inhibits cell cycle progression by downregulation of E2F1 in human glioblastoma multiforme cells. Acta Oncol. 2008, 47, 1062–1070. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Offidani, M.; Amantini, C.; Gentili, S.; Soriani, A.; Cardinali, C.; Leoni, P.; Santoni, G. Cannabinoids synergize with carfilzomib, reducing multiple myeloma cells viability and migration. Oncotarget 2016, 7, 77543–77557. [Google Scholar] [CrossRef]
- Wolff, V.; Schlagowski, A.-I.; Rouyer, O.; Charles, A.-L.; Singh, F.; Auger, C.; Schini-Kerth, V.; Marescaux, C.; Raul, J.-S.; Zoll, J.; et al. Tetrahydrocannabinol induces brain mitochondrial respiratory chain dysfunction and increases oxidative stress: A potential mechanism involved in cannabis-related stroke. Biomed. Res. Int. 2015, 323706. [Google Scholar] [CrossRef]
- Rai, Y.; Pathak, R.; Kumari, N.; Sah, D.K.; Pandey, S.; Kalra, N.; Soni, R.; Dwarakanath, B.S.; Bhatt, A.N. Mitochondrial biogenesis and metabolic hyperactivation limits the application of MTT assay in the estimation of radiation induced growth inhibition. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.S. Roles of TRPM8 ion channels in cancer: Proliferation, survival, and invasion. Cancers 2015, 7, 2134–2146. [Google Scholar] [CrossRef] [PubMed]
- Verbovšek, U.; Van Noorden, C.J.F.; Lah, T.T. Complexity of cancer protease biology: Cathepsin K expression and function in cancer progression. Semin. Cancer Biol. 2015, 71–84. [Google Scholar] [CrossRef]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous activation of induced heterodimerization between CXCR4 chemokine receptor and cannabinoid receptor 2 (CB2) reveals a mechanism for regulation of tumor progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef]
- Lane, S.W.; Scadden, D.T.; Gilliland, D.G. The leukemic stem cell niche: Current concepts and therapeutic opportunities. Blood 2009, 114, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; Westhoff, M.-A. Temozolomide and other alkylating agents in glioblastoma therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines (GB) ** | CBG [µM] | CBD [µM] | THC [µM] |
|---|---|---|---|
| U87 | 24.2 | 17.4 | 25.5 |
| U373 | 31.1 | 29.7 | 34.9 |
| NIB138 | 22.3 | 18.3 | 19.5 |
| NIB140 | 32.0 | 26.7 | 29.9 |
| NIB142 | 27.4 | 25.9 | 34.4 |
| NIB160 | 26.3 | 15.0 | 21.1 |
| NIB167 | 25.6 | 12.6 | 23.7 |
| NIB180 | 30.8 | 20.1 | 29.6 |
| NIB182 | 31.6 | 28.1 | 30.2 |
| NIB185 | 29.8 | 28.8 | 30.0 |
| Mean ± S.E.M. | 28.1 ± 1.1 | 22.2 ± 2.1 | 27.9 ± 1.8 |
| Stem Cell Lines (GSCs) | CBG [µM] | CBD [µM] | THC [µM] |
| NCH644 | 58.3 | 15.9 | 22.3 |
| NCH421k | 34.0 | 27.9 | 28.7 |
| K26 | 84.8 | 14.6 | 17.4 |
| Mean ± S.E.M. | 59.0 ± 14.7 | 19.5 ± 4.2 | 22.8 ± 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lah, T.T.; Novak, M.; Pena Almidon, M.A.; Marinelli, O.; Žvar Baškovič, B.; Majc, B.; Mlinar, M.; Bošnjak, R.; Breznik, B.; Zomer, R.; et al. Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma. Cells 2021, 10, 340. https://doi.org/10.3390/cells10020340
Lah TT, Novak M, Pena Almidon MA, Marinelli O, Žvar Baškovič B, Majc B, Mlinar M, Bošnjak R, Breznik B, Zomer R, et al. Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma. Cells. 2021; 10(2):340. https://doi.org/10.3390/cells10020340
Chicago/Turabian StyleLah, Tamara T., Metka Novak, Milagros A. Pena Almidon, Oliviero Marinelli, Barbara Žvar Baškovič, Bernarda Majc, Mateja Mlinar, Roman Bošnjak, Barbara Breznik, Roby Zomer, and et al. 2021. "Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma" Cells 10, no. 2: 340. https://doi.org/10.3390/cells10020340
APA StyleLah, T. T., Novak, M., Pena Almidon, M. A., Marinelli, O., Žvar Baškovič, B., Majc, B., Mlinar, M., Bošnjak, R., Breznik, B., Zomer, R., & Nabissi, M. (2021). Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma. Cells, 10(2), 340. https://doi.org/10.3390/cells10020340