
Intestinal Stem Cell Development in the Neonatal Gut: Pathways Regulating Development and Relevance to Necrotizing Enterocolitis


{kind=link}
Abstract
1. Introduction
2. Intestinal Stem Cells
3. Neonatal Intestinal Ontogeny
4. Paracrine Signaling Regulating Stem Cell Development
4.1. Hedgehog(Hh) Signaling
4.2. BMP Signaling
4.3. Wnt Signaling
4.4. Notch Signaling
5. Endocrine Signaling
6. Transcription Factors
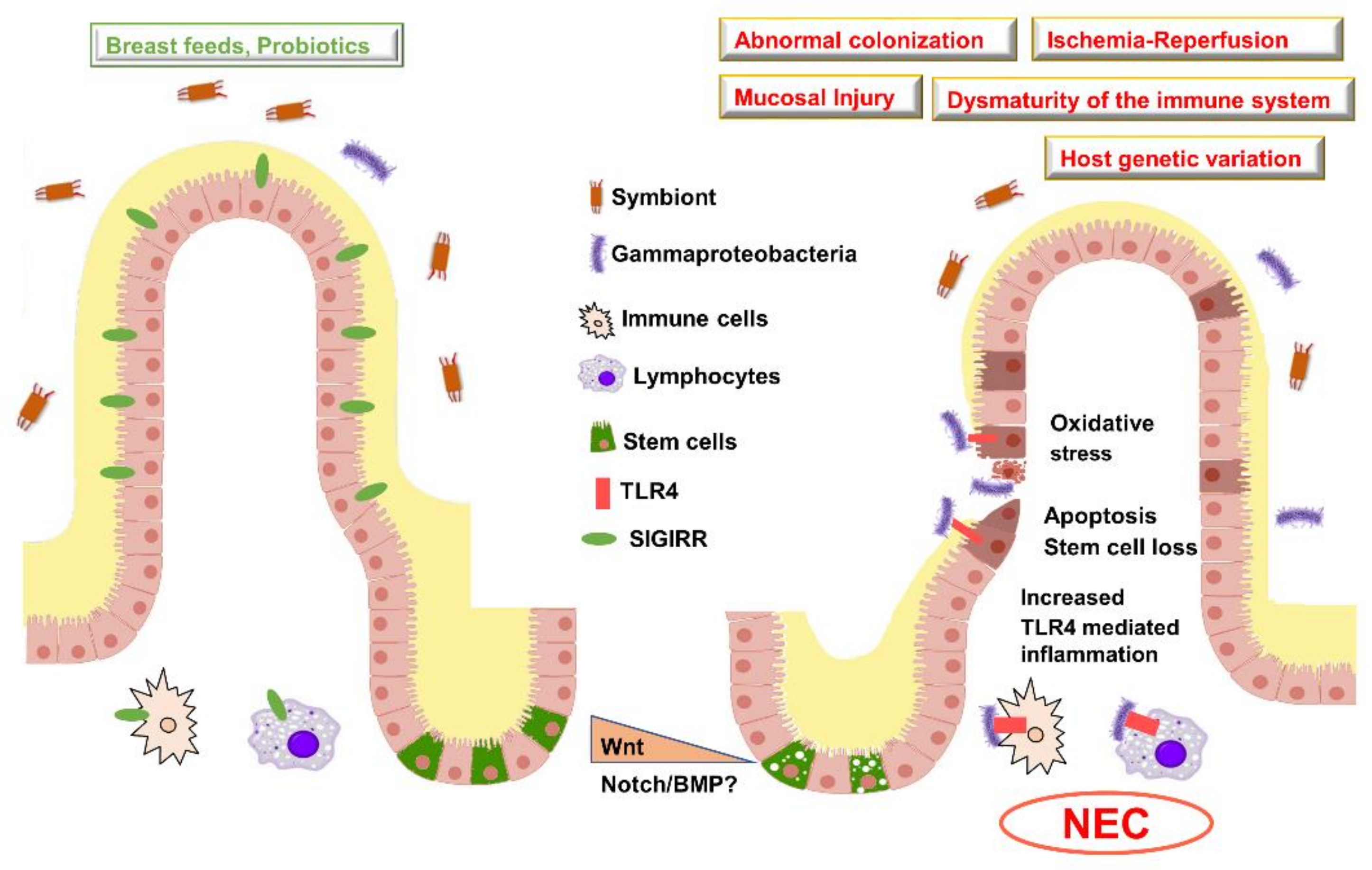
7. Necrotizing Enterocolitis Pathophysiology
7.1. Dysmaturity of the Intestinal Immune System in Preterm Neonates
7.2. TLR4 Signaling and NEC Pathogenesis
7.3. Alternate Pathways Involved in NEC
7.4. Role of the Gut Microbiome in Programming NEC Vulnerability
7.5. Genetic Susceptibility to NEC in Preterm Neonates
8. Morphogenetic Pathways and NEC
9. Stem Cell Therapy for NEC—Sources and Mechanism of Action
10. Stem Cell Therapy in Preclinical Models
11. Conclusions
Funding
Conflicts of Interest
References
- Qi, D.; Shi, W.; Black, A.R.; Kuss, M.A.; Pang, X.; He, Y.; Liu, B.; Duan, B. Repair and regeneration of small intestine: A review of current engineering approaches. Biomaterials 2020, 240, 119832. [Google Scholar] [CrossRef] [PubMed]
- Halpern, M.D.; Denning, P.W. The role of intestinal epithelial barrier function in the development of NEC. Tissue Barriers 2015, 3, e1000707. [Google Scholar] [CrossRef] [PubMed]
- Paimela, H.; Goddard, P.J.; Silen, W. Present views on restitution of gastrointestinal epithelium. Dig. Dis. Sci. 1995, 40, 2495–2496. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van de Wetering, M.; Clevers, H. The intestinal stem cell. Genes Dev. 2008, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef]
- Bjerknes, M.; Cheng, H. The stem-cell zone of the small intestinal epithelium. I. Evidence from Paneth cells in the adult mouse. Am. J. Anat. 1981, 160, 51–63. [Google Scholar] [CrossRef]
- Kim, C.K.; Yang, V.W.; Bialkowska, A.B. The Role of Intestinal Stem Cells in Epithelial Regeneration Following Radiation-Induced Gut Injury. Curr. Stem Cell Rep. 2017, 3, 320–332. [Google Scholar] [CrossRef]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef]
- Santos, A.J.M.; Lo, Y.H.; Mah, A.T.; Kuo, C.J. The Intestinal Stem Cell Niche: Homeostasis and Adaptations. Trends Cell Biol. 2018, 28, 1062–1078. [Google Scholar] [CrossRef]
- Takahashi, T.; Shiraishi, A. Stem Cell Signaling Pathways in the Small Intestine. Int. J. Mol. Sci. 2020, 21, 2032. [Google Scholar] [CrossRef] [PubMed]
- Drozdowski, L.A.; Clandinin, T.; Thomson, A.B. Ontogeny, growth and development of the small intestine: Understanding pediatric gastroenterology. World J. Gastroenterol. 2010, 16, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Bry, L.; Falk, P.; Huttner, K.; Ouellette, A.; Midtvedt, T.; Gordon, J.I. Paneth cell differentiation in the developing intestine of normal and transgenic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 10335–10339. [Google Scholar] [CrossRef] [PubMed]
- De Santa Barbara, P.; van den Brink, G.R.; Roberts, D.J. Development and differentiation of the intestinal epithelium. Cell Mol. Life Sci. 2003, 60, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review). Int. J. Mol. Med. 2006, 18, 1019–1023. [Google Scholar] [CrossRef]
- Madison, B.B.; Braunstein, K.; Kuizon, E.; Portman, K.; Qiao, X.T.; Gumucio, D.L. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development 2005, 132, 279–289. [Google Scholar] [CrossRef]
- Karlsson, L.; Lindahl, P.; Heath, J.K.; Betsholtz, C. Abnormal gastrointestinal development in PDGF-A and PDGFR-(alpha) deficient mice implicates a novel mesenchymal structure with putative instructive properties in villus morphogenesis. Development 2000, 127, 3457–3466. [Google Scholar]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone morphogenetic proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef]
- Haramis, A.P.; Begthel, H.; van den Born, M.; van Es, J.; Jonkheer, S.; Offerhaus, G.J.; Clevers, H. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 2004, 303, 1684–1686. [Google Scholar] [CrossRef]
- Woodford-Richens, K.; Williamson, J.; Bevan, S.; Young, J.; Leggett, B.; Frayling, I.; Thway, Y.; Hodgson, S.; Kim, J.C.; Iwama, T.; et al. Allelic loss at SMAD4 in polyps from juvenile polyposis patients and use of fluorescence in situ hybridization to demonstrate clonal origin of the epithelium. Cancer Res. 2000, 60, 2477–2482. [Google Scholar]
- Howe, J.R.; Roth, S.; Ringold, J.C.; Summers, R.W.; Jarvinen, H.J.; Sistonen, P.; Tomlinson, I.P.; Houlston, R.S.; Bevan, S.; Mitros, F.A.; et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998, 280, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Batts, L.E.; Polk, D.B.; Dubois, R.N.; Kulessa, H. Bmp signaling is required for intestinal growth and morphogenesis. Dev. Dyn. 2006, 235, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- He, X.C.; Zhang, J.; Tong, W.G.; Tawfik, O.; Ross, J.; Scoville, D.H.; Tian, Q.; Zeng, X.; He, X.; Wiedemann, L.M.; et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat. Genet. 2004, 36, 1117–1121. [Google Scholar] [CrossRef]
- Mah, A.T.; Yan, K.S.; Kuo, C.J. Wnt pathway regulation of intestinal stem cells. J. Physiol. 2016, 594, 4837–4847. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Crosnier, C.; Stamataki, D.; Lewis, J. Organizing cell renewal in the intestine: Stem cells, signals and combinatorial control. Nat. Rev. Genet. 2006, 7, 349–359. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998, 19, 379–383. [Google Scholar] [CrossRef]
- Van Es, J.H.; Haegebarth, A.; Kujala, P.; Itzkovitz, S.; Koo, B.K.; Boj, S.F.; Korving, J.; van den Born, M.; van Oudenaarden, A.; Robine, S.; et al. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell Biol. 2012, 32, 1918–1927. [Google Scholar] [CrossRef]
- Kuhnert, F.; Davis, C.R.; Wang, H.T.; Chu, P.; Lee, M.; Yuan, J.; Nusse, R.; Kuo, C.J. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc. Natl. Acad. Sci. USA 2004, 101, 266–271. [Google Scholar] [CrossRef]
- Ireland, H.; Kemp, R.; Houghton, C.; Howard, L.; Clarke, A.R.; Sansom, O.J.; Winton, D.J. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: Effect of loss of beta-catenin. Gastroenterology 2004, 126, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; Mao, J.; Taketo, M.M.; Shivdasani, R.A. Phases of canonical Wnt signaling during the development of mouse intestinal epithelium. Gastroenterology 2007, 133, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Bettess, M.D.; Dubois, N.; Murphy, M.J.; Dubey, C.; Roger, C.; Robine, S.; Trumpp, A. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol. Cell Biol. 2005, 25, 7868–7878. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Kakitani, M.; Zhao, J.; Oshima, T.; Tang, T.; Binnerts, M.; Liu, Y.; Boyle, B.; Park, E.; Emtage, P.; et al. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science 2005, 309, 1256–1259. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Gregorieff, A.; Begthel, H.; Clevers, H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003, 17, 1709–1713. [Google Scholar] [CrossRef]
- Batlle, E.; Henderson, J.T.; Beghtel, H.; van den Born, M.M.; Sancho, E.; Huls, G.; Meeldijk, J.; Robertson, J.; van de Wetering, M.; Pawson, T.; et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 2002, 111, 251–263. [Google Scholar] [CrossRef]
- Demitrack, E.S.; Samuelson, L.C. Notch regulation of gastrointestinal stem cells. J. Physiol. 2016, 594, 4791–4803. [Google Scholar] [CrossRef]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a Notch. Development 2013, 140, 689–704. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Van Es, J.H.; van Gijn, M.E.; Riccio, O.; van den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef]
- Riccio, O.; van Gijn, M.E.; Bezdek, A.C.; Pellegrinet, L.; van Es, J.H.; Zimber-Strobl, U.; Strobl, L.J.; Honjo, T.; Clevers, H.; Radtke, F. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. Embo Rep. 2008, 9, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinet, L.; Rodilla, V.; Liu, Z.; Chen, S.; Koch, U.; Espinosa, L.; Kaestner, K.H.; Kopan, R.; Lewis, J.; Radtke, F. Dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology 2011, 140, 1230–1240.e7. [Google Scholar] [CrossRef] [PubMed]
- Milano, J.; McKay, J.; Dagenais, C.; Foster-Brown, L.; Pognan, F.; Gadient, R.; Jacobs, R.T.; Zacco, A.; Greenberg, B.; Ciaccio, P.J. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol. Sci. 2004, 82, 341–358. [Google Scholar] [CrossRef]
- Fre, S.; Huyghe, M.; Mourikis, P.; Robine, S.; Louvard, D.; Artavanis-Tsakonas, S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005, 435, 964–968. [Google Scholar] [CrossRef]
- Stanger, B.Z.; Datar, R.; Murtaugh, L.C.; Melton, D.A. Direct regulation of intestinal fate by Notch. Proc. Natl. Acad. Sci. USA 2005, 102, 12443–12448. [Google Scholar] [CrossRef]
- Fre, S.; Pallavi, S.K.; Huyghe, M.; Lae, M.; Janssen, K.P.; Robine, S.; Artavanis-Tsakonas, S.; Louvard, D. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 6309–6314. [Google Scholar] [CrossRef]
- Ishizuya-Oka, A.; Shi, Y.B. Evolutionary insights into postembryonic development of adult intestinal stem cells. Cell BioSci. 2011, 1, 37. [Google Scholar] [CrossRef] [PubMed]
- Plateroti, M.; Chassande, O.; Fraichard, A.; Gauthier, K.; Freund, J.N.; Samarut, J.; Kedinger, M. Involvement of T3Ralpha- and beta-receptor subtypes in mediation of T3 functions during postnatal murine intestinal development. Gastroenterology 1999, 116, 1367–1378. [Google Scholar] [CrossRef]
- Harper, J.; Mould, A.; Andrews, R.M.; Bikoff, E.K.; Robertson, E.J. The transcriptional repressor Blimp1/Prdm1 regulates postnatal reprogramming of intestinal enterocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10585–10590. [Google Scholar] [CrossRef]
- Muncan, V.; Heijmans, J.; Krasinski, S.D.; Buller, N.V.; Wildenberg, M.E.; Meisner, S.; Radonjic, M.; Stapleton, K.A.; Lamers, W.H.; Biemond, I.; et al. Blimp1 regulates the transition of neonatal to adult intestinal epithelium. Nat. Commun. 2011, 2, 452. [Google Scholar] [CrossRef]
- Matsuda, H.; Shi, Y.B. An essential and evolutionarily conserved role of protein arginine methyltransferase 1 for adult intestinal stem cells during postembryonic development. Stem Cells 2010, 28, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Blache, P.; van de Wetering, M.; Duluc, I.; Domon, C.; Berta, P.; Freund, J.N.; Clevers, H.; Jay, P. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J. Cell Biol. 2004, 166, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Mori-Akiyama, Y.; van den Born, M.; van Es, J.H.; Hamilton, S.R.; Adams, H.P.; Zhang, J.; Clevers, H.; de Crombrugghe, B. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology 2007, 133, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Neu, J.; Walker, W.A. Necrotizing enterocolitis. N. Engl. J. Med. 2011, 364, 255–264. [Google Scholar] [CrossRef]
- Frost, B.L.; Modi, B.P.; Jaksic, T.; Caplan, M.S. New Medical and Surgical Insights Into Neonatal Necrotizing Enterocolitis: A Review. JAMA Pediatr. 2017, 171, 83–88. [Google Scholar] [CrossRef]
- Walsh, M.C.; Kliegman, R.M. Necrotizing enterocolitis: Treatment based on staging criteria. Pediatr. Clin. N. Am. 1986, 33, 179–201. [Google Scholar] [CrossRef]
- Bell, M.J.; Ternberg, J.L.; Feigin, R.D.; Keating, J.P.; Marshall, R.; Barton, L.; Brotherton, T. Neonatal necrotizing enterocolitis. Therapeutic decisions based upon clinical staging. Ann. Surg. 1978, 187, 1–7. [Google Scholar] [CrossRef]
- Nino, D.F.; Sodhi, C.P.; Hackam, D.J. Necrotizing enterocolitis: New insights into pathogenesis and mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 590–600. [Google Scholar] [CrossRef]
- Afrazi, A.; Sodhi, C.P.; Richardson, W.; Neal, M.; Good, M.; Siggers, R.; Hackam, D.J. New insights into the pathogenesis and treatment of necrotizing enterocolitis: Toll-like receptors and beyond. Pediatr. Res. 2011, 69, 183–188. [Google Scholar] [CrossRef]
- Caplan, M.S.; Underwood, M.A.; Modi, N.; Patel, R.; Gordon, P.V.; Sylvester, K.G.; McElroy, S.; Manzoni, P.; Gephart, S.; Chwals, W.J.; et al. Necrotizing Enterocolitis: Using Regulatory Science and Drug Development to Improve Outcomes. J. Pediatr. 2019, 212, 208–215.e201. [Google Scholar] [CrossRef]
- Caplan, M.; Frost, B. Myth: Necrotizing enterocolitis: Probiotics will end the disease, and surgical intervention improves the outcome. Semin. Fetal Neonatal Med. 2011, 16, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Neal, M.D.; Siggers, R.; Sho, S.; Ma, C.; Branca, M.F.; Prindle, T., Jr.; Russo, A.M.; Afrazi, A.; Good, M.; et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 2012, 143, 708–718.e705. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.; Gutle, D.; Walther, S.; Menard, S.; Bogdan, C.; Hornef, M.W. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J. Exp. Med. 2006, 203, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Cario, E.; Podolsky, D.K. Intestinal epithelial TOLLerance versus inTOLLerance of commensals. Mol. Immunol. 2005, 42, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Renz, H.; Brandtzaeg, P.; Hornef, M. The impact of perinatal immune development on mucosal homeostasis and chronic inflammation. Nat. Rev. Immunol. 2011, 12, 9–23. [Google Scholar] [CrossRef]
- Pierro, M.; Ionescu, L.; Montemurro, T.; Vadivel, A.; Weissmann, G.; Oudit, G.; Emery, D.; Bodiga, S.; Eaton, F.; Péault, B.; et al. Short-term, long-term and paracrine effect of human umbilical cord-derived stem cells in lung injury prevention and repair in experimental bronchopulmonary dysplasia. Thorax 2013, 68, 475–484. [Google Scholar] [CrossRef]
- Claud, E.C.; Lu, L.; Anton, P.M.; Savidge, T.; Walker, W.A.; Cherayil, B.J. Developmentally regulated IkappaB expression in intestinal epithelium and susceptibility to flagellin-induced inflammation. Proc. Natl. Acad. Sci. USA 2004, 101, 7404–7408. [Google Scholar] [CrossRef]
- Nanthakumar, N.; Meng, D.; Goldstein, A.M.; Zhu, W.; Lu, L.; Uauy, R.; Llanos, A.; Claud, E.C.; Walker, W.A. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: An immature innate immune response. PLoS ONE 2011, 6, e17776. [Google Scholar] [CrossRef]
- Cuna, A.; Yu, W.; Menden, H.L.; Feng, L.; Srinivasan, P.; Chavez-Bueno, S.; Ahmed, I.; Umar, S.; Sampath, V. NEC-like intestinal injury is ameliorated by Lactobacillus rhamnosus GG in parallel with SIGIRR and A20 induction in neonatal mice. Pediatr. Res. 2020, 88, 546–555. [Google Scholar] [CrossRef]
- Ganguli, K.; Meng, D.; Rautava, S.; Lu, L.; Walker, W.A.; Nanthakumar, N. Probiotics prevent necrotizing enterocolitis by modulating enterocyte genes that regulate innate immune-mediated inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G132–G141. [Google Scholar] [CrossRef]
- Minekawa, R.; Takeda, T.; Sakata, M.; Hayashi, M.; Isobe, A.; Yamamoto, T.; Tasaka, K.; Murata, Y. Human breast milk suppresses the transcriptional regulation of IL-1beta-induced NF-kappaB signaling in human intestinal cells. Am. J. Physiol. Cell Physiol. 2004, 287, C1404–C1411. [Google Scholar] [CrossRef] [PubMed]
- Nanthakumar, N.N.; Fusunyan, R.D.; Sanderson, I.; Walker, W.A. Inflammation in the developing human intestine: A possible pathophysiologic contribution to necrotizing enterocolitis. Proc. Natl. Acad. Sci. USA 2000, 97, 6043–6048. [Google Scholar] [CrossRef] [PubMed]
- Pammi, M.; Cope, J.; Tarr, P.I.; Warner, B.B.; Morrow, A.L.; Mai, V.; Gregory, K.E.; Kroll, J.S.; McMurtry, V.; Ferris, M.J.; et al. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: A systematic review and meta-analysis. Microbiome 2017, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Bhattacharya, N.; Crits-Christoph, A.; Firek, B.A.; Baker, R.; Song, Y.S.; Morowitz, M.J.; Banfield, J.F. Necrotizing enterocolitis is preceded by increased gut bacterial replication, Klebsiella, and fimbriae-encoding bacteria. Sci. Adv. 2019, 5, eaax5727. [Google Scholar] [CrossRef] [PubMed]
- Jilling, T.; Simon, D.; Lu, J.; Meng, F.J.; Li, D.; Schy, R.; Thomson, R.B.; Soliman, A.; Arditi, M.; Caplan, M.S. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J. Immunol. 2006, 177, 3273–3282. [Google Scholar] [CrossRef]
- Fawley, J.; Cuna, A.; Menden, H.L.; McElroy, S.; Umar, S.; Welak, S.R.; Gourlay, D.M.; Li, X.; Sampath, V. Single-Immunoglobulin Interleukin-1-Related Receptor regulates vulnerability to TLR4-mediated necrotizing enterocolitis in a mouse model. Pediatr. Res. 2018, 83, 164–174. [Google Scholar] [CrossRef]
- Sampath, V.; Menden, H.; Helbling, D.; Li, K.; Gastonguay, A.; Ramchandran, R.; Dimmock, D.P. SIGIRR genetic variants in premature infants with necrotizing enterocolitis. Pediatrics 2015, 135, e1530–e1534. [Google Scholar] [CrossRef]
- White, J.R.; Gong, H.; Pope, B.; Schlievert, P.; McElroy, S.J. Paneth-cell-disruption-induced necrotizing enterocolitis in mice requires live bacteria and occurs independently of TLR4 signaling. Dis. Model. Mech. 2017, 10, 727–736. [Google Scholar] [CrossRef]
- Lueschow, S.R.; McElroy, S.J. The Paneth Cell: The Curator and Defender of the Immature Small Intestine. Front. Immunol. 2020, 11, 587. [Google Scholar] [CrossRef]
- Caplan, M.S.; Simon, D.; Jilling, T. The role of PAF, TLR, and the inflammatory response in neonatal necrotizing enterocolitis. Semin. Pediatr. Surg. 2005, 14, 145–151. [Google Scholar] [CrossRef]
- Soliman, A.; Michelsen, K.S.; Karahashi, H.; Lu, J.; Meng, F.J.; Qu, X.; Crother, T.R.; Rabizadeh, S.; Chen, S.; Caplan, M.S.; et al. Platelet-activating factor induces TLR4 expression in intestinal epithelial cells: Implication for the pathogenesis of necrotizing enterocolitis. PLoS ONE 2010, 5, e15044. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.L.; Caplan, M.S. Necrotizing enterocolitis: Pathophysiology, platelet-activating factor, and probiotics. Semin. Pediatr. Surg. 2013, 22, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, S.S.; Dzakpasu, P.; Piecuch, S.; Leblanc, P.; Valencia, G.; Kornecki, E. Platelet-activating factor in infants at risk for necrotizing enterocolitis. J. Pediatr. 2001, 138, 81–86. [Google Scholar] [CrossRef]
- Stewart, C.J.; Embleton, N.D.; Marrs, E.C.L.; Smith, D.P.; Fofanova, T.; Nelson, A.; Skeath, T.; Perry, J.D.; Petrosino, J.F.; Berrington, J.E.; et al. Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome 2017, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The microbiome in early life: Implications for health outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef]
- Nogacka, A.; Salazar, N.; Suarez, M.; Milani, C.; Arboleya, S.; Solis, G.; Fernandez, N.; Alaez, L.; Hernandez-Barranco, A.M.; de Los Reyes-Gavilan, C.G.; et al. Impact of intrapartum antimicrobial prophylaxis upon the intestinal microbiota and the prevalence of antibiotic resistance genes in vaginally delivered full-term neonates. Microbiome 2017, 5, 93. [Google Scholar] [CrossRef]
- Chu, D.M.; Antony, K.M.; Ma, J.; Prince, A.L.; Showalter, L.; Moller, M.; Aagaard, K.M. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Med. 2016, 8, 77. [Google Scholar] [CrossRef]
- Warner, B.B.; Deych, E.; Zhou, Y.; Hall-Moore, C.; Weinstock, G.M.; Sodergren, E.; Shaikh, N.; Hoffmann, J.A.; Linneman, L.A.; Hamvas, A.; et al. Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: A prospective case-control study. Lancet 2016, 387, 1928–1936. [Google Scholar] [CrossRef]
- Claud, E.C.; Keegan, K.P.; Brulc, J.M.; Lu, L.; Bartels, D.; Glass, E.; Chang, E.B.; Meyer, F.; Antonopoulos, D.A. Bacterial community structure and functional contributions to emergence of health or necrotizing enterocolitis in preterm infants. Microbiome 2013, 1, 20. [Google Scholar] [CrossRef]
- Coker, M.O.; Hoen, A.G.; Dade, E.; Lundgren, S.; Li, Z.; Wong, A.D.; Zens, M.S.; Palys, T.J.; Morrison, H.G.; Sogin, M.L.; et al. Specific class of intrapartum antibiotics relates to maturation of the infant gut microbiota: A prospective cohort study. BJOG 2020, 127, 217–227. [Google Scholar] [CrossRef]
- Zwittink, R.D.; van Zoeren-Grobben, D.; Renes, I.B.; van Lingen, R.A.; Norbruis, O.F.; Martin, R.; Groot Jebbink, L.J.; Knol, J.; Belzer, C. Dynamics of the bacterial gut microbiota in preterm and term infants after intravenous amoxicillin/ceftazidime treatment. BMC Pediatr. 2020, 20, 195. [Google Scholar] [CrossRef] [PubMed]
- Groer, M.W.; Luciano, A.A.; Dishaw, L.J.; Ashmeade, T.L.; Miller, E.; Gilbert, J.A. Development of the preterm infant gut microbiome: A research priority. Microbiome 2014, 2, 38. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H. Paediatrics: Gut microbiota dysbiosis precedes NEC. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Cuna, A.; George, L.; Sampath, V. Genetic predisposition to necrotizing enterocolitis in premature infants: Current knowledge, challenges, and future directions. Semin. Fetal Neonatal Med. 2018, 23, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Hartel, C.; Hartz, A.; Pagel, J.; Rupp, J.; Stein, A.; Kribs, A.; Muller, A.; Haase, R.; Gille, C.; Bottger, R.; et al. NOD2 Loss-of-Function Mutations and Risks of Necrotizing Enterocolitis or Focal Intestinal Perforation in Very Low-birth-weight Infants. Inflamm. Bowel Dis. 2016, 22, 249–256. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sampath, V.; Bhandari, V.; Berger, J.; Merchant, D.; Zhang, L.; Ladd, M.; Menden, H.; Garland, J.; Ambalavanan, N.; Mulrooney, N.; et al. A functional ATG16L1 (T300A) variant is associated with necrotizing enterocolitis in premature infants. Pediatr. Res. 2017, 81, 582–588. [Google Scholar] [CrossRef]
- Garlanda, C.; Anders, H.J.; Mantovani, A. TIR8/SIGIRR: An IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol. 2009, 30, 439–446. [Google Scholar] [CrossRef]
- Moonen, R.M.; Huizing, M.J.; Gonzalez-Luis, G.E.; Cavallaro, G.; Mosca, F.; Villamor, E. Risk of Necrotizing Enterocolitis Associated With the Single Nucleotide Polymorphisms VEGF C-2578A, IL-18 C-607A, and IL-4 Receptor alpha-Chain A-1902G: A Validation Study in a Prospective Multicenter Cohort. Front. Pediatr. 2020, 8, 45. [Google Scholar] [CrossRef]
- Chan, K.Y.; Leung, K.T.; Tam, Y.H.; Lam, H.S.; Cheung, H.M.; Ma, T.P.; Lee, K.H.; To, K.F.; Li, K.; Ng, P.C. Genome-wide expression profiles of necrotizing enterocolitis versus spontaneous intestinal perforation in human intestinal tissues: Dysregulation of functional pathways. Ann. Surg. 2014, 260, 1128–1137. [Google Scholar] [CrossRef]
- Neal, M.D.; Sodhi, C.P.; Jia, H.; Dyer, M.; Egan, C.E.; Yazji, I.; Good, M.; Afrazi, A.; Marino, R.; Slagle, D.; et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J. Biol. Chem. 2012, 287, 37296–37308. [Google Scholar] [CrossRef] [PubMed]
- Leaphart, C.L.; Cavallo, J.; Gribar, S.C.; Cetin, S.; Li, J.; Branca, M.F.; Dubowski, T.D.; Sodhi, C.P.; Hackam, D.J. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 2007, 179, 4808–4820. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.D.; Richardson, W.M.; Sodhi, C.P.; Russo, A.; Hackam, D.J. Intestinal stem cells and their roles during mucosal injury and repair. J. Surg. Res. 2011, 167, 1–8. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Shi, X.H.; Richardson, W.M.; Grant, Z.S.; Shapiro, R.A.; Prindle, T., Jr.; Branca, M.; Russo, A.; Gribar, S.C.; Ma, C.; et al. Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis. Gastroenterology 2010, 138, 185–196. [Google Scholar] [CrossRef]
- Li, B.; Lee, C.; Cadete, M.; Zhu, H.; Koike, Y.; Hock, A.; Wu, R.Y.; Botts, S.R.; Minich, A.; Alganabi, M.; et al. Impaired Wnt/beta-catenin pathway leads to dysfunction of intestinal regeneration during necrotizing enterocolitis. Cell Death Dis. 2019, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Jakoniuk, I.; Anderson, S.M.; Li, B.; Pickel, J.; McKay, R.; Nadal-Ginard, B.; Bodine, D.M.; et al. Bone marrow cells regenerate infarcted myocardium. Nature 2001, 410, 701–705. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Kern, S.; Eichler, H.; Stoeve, J.; Klüter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Gorskaja, J.F.; Kulagina, N.N. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp. Hematol. 1976, 4, 267–274. [Google Scholar] [CrossRef]
- Miao, Z.; Jin, J.; Chen, L.; Zhu, J.; Huang, W.; Zhao, J.; Qian, H.; Zhang, X. Isolation of mesenchymal stem cells from human placenta: Comparison with human bone marrow mesenchymal stem cells. Cell Biol. Int. 2006, 30, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Wegmeyer, H.; Bröske, A.-M.; Leddin, M.; Kuentzer, K.; Nisslbeck, A.K.; Hupfeld, J.; Wiechmann, K.; Kuhlen, J.; von Schwerin, C.; Stein, C.; et al. Mesenchymal stromal cell characteristics vary depending on their origin. Stem Cells Dev. 2013, 22, 2606–2618. [Google Scholar] [CrossRef] [PubMed]
- Nitkin, C.R.; Bonfield, T.L. Concise Review: Mesenchymal Stem Cell Therapy for Pediatric Disease: Perspectives on Success and Potential Improvements. Stem Cells Transl. Med. 2017, 6, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Rager, T.M.; Olson, J.K.; Zhou, Y.; Wang, Y.; Besner, G.E. Exosomes secreted from bone marrow-derived mesenchymal stem cells protect the intestines from experimental necrotizing enterocolitis. J. Pediatr. Surg. 2016, 51, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Stavely, R.; Nurgali, K. The emerging antioxidant paradigm of mesenchymal stem cell therapy. Stem Cells Transl. Med. 2020, 9, 985–1006. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Lee, S.H.; Bae, D.K.; Yang, Y.-H.; Yang, G.; Kyung, J.; Kim, D.; Choi, E.-K.; Hong, J.T.; Shin, I.S.; et al. Transplantation of Human Adipose Tissue-Derived Mesenchymal Stem Cells Restores the Neurobehavioral Disorders of Rats with Neonatal Hypoxic-Ischemic Encephalopathy. Cell Med. 2013, 5, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.C.; Li, Y.T.; Chen, C.M. Human mesenchymal stem cells attenuate experimental bronchopulmonary dysplasia induced by perinatal inflammation and hyperoxia. Am. J. Transl. Res. 2016, 8, 342–353. [Google Scholar]
- Ding, H.; Zhang, H.; Ding, H.; Li, D.; Yi, X.; Ma, X.; Li, R.; Huang, M.; Ju, X. Transplantation of placenta-derived mesenchymal stem cells reduces hypoxic-ischemic brain damage in rats by ameliorating the inflammatory response. Cell. Mol. Immunol. 2017, 14, 693–701. [Google Scholar] [CrossRef]
- Woik, N.; Kroll, J. Regulation of lung development and regeneration by the vascular system. Cell. Mol. Life Sci. Cmls 2015, 72, 2709–2718. [Google Scholar] [CrossRef]
- Luan, Y.; Ding, W.; Ju, Z.-y.; Zhang, Z.-h.; Zhang, X.; Kong, F. Bone marrow-derived mesenchymal stem cells protect against lung injury in a mouse model of bronchopulmonary dysplasia. Mol. Med. Rep. 2015, 11, 1945–1950. [Google Scholar] [CrossRef][Green Version]
- Schlosser, K.; Wang, J.-P.; Dos Santos, C.; Walley, K.R.; Marshall, J.; Fergusson, D.A.; Winston, B.W.; Granton, J.; Watpool, I.; Stewart, D.J.; et al. Effects of Mesenchymal Stem Cell Treatment on Systemic Cytokine Levels in a Phase 1 Dose Escalation Safety Trial of Septic Shock Patients. Crit. Care Med. 2019, 47, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Panés, J.; García-Olmo, D.; Van Assche, G.; Colombel, J.F.; Reinisch, W.; Baumgart, D.C.; Dignass, A.; Nachury, M.; Ferrante, M.; Kazemi-Shirazi, L.; et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn’s disease: A phase 3 randomised, double-blind controlled trial. Lancet 2016, 388, 1281–1290. [Google Scholar] [CrossRef]
- Drucker, N.A.; McCulloh, C.J.; Li, B.; Pierro, A.; Besner, G.E.; Markel, T.A. Stem cell therapy in necrotizing enterocolitis: Current state and future directions. Semin. Pediatr. Surg. 2018, 27, 57–64. [Google Scholar] [CrossRef] [PubMed]
- McCulloh, C.J.; Olson, J.K.; Zhou, Y.; Wang, Y.; Besner, G.E. Stem cells and necrotizing enterocolitis: A direct comparison of the efficacy of multiple types of stem cells. J. Pediatr. Surg. 2017, 52, 999–1005. [Google Scholar] [CrossRef]
- Pisano, C.; Besner, G.E. Potential role of stem cells in disease prevention based on a murine model of experimental necrotizing enterocolitis. J. Pediatr. Surg. 2019, 54, 413–416. [Google Scholar] [CrossRef]
- McCulloh, C.J.; Olson, J.K.; Wang, Y.; Vu, J.; Gartner, S.; Besner, G.E. Evaluating the efficacy of different types of stem cells in preserving gut barrier function in necrotizing enterocolitis. J. Surg. Res. 2017, 214, 278–285. [Google Scholar] [CrossRef]
- McCulloh, C.J.; Olson, J.K.; Wang, Y.; Zhou, Y.; Tengberg, N.H.; Deshpande, S.; Besner, G.E. Treatment of experimental necrotizing enterocolitis with stem cell-derived exosomes. J. Pediatr. Surg. 2018, 53, 1215–1220. [Google Scholar] [CrossRef]
- Chen, W.; Wang, X.; Yan, X.; Yu, Z.; Zhang, J.; Han, S. The emerging role of exosomes in the pathogenesis, prognosis and treatment of necrotizing enterocolitis. Am. J. Transl. Res. 2020, 12, 7020–7033. [Google Scholar]
- Miyake, H.; Lee, C.; Chusilp, S.; Bhalla, M.; Li, B.; Pitino, M.; Seo, S.; O’Connor, D.L.; Pierro, A. Human breast milk exosomes attenuate intestinal damage. Pediatr. Surg. Int. 2020, 36, 155–163. [Google Scholar] [CrossRef]
- O’Reilly, D.; Dorodnykh, D.; Avdeenko, N.V.; Nekliudov, N.A.; Garssen, J.; Elolimy, A.A.; Petrou, L.; Simpson, M.R.; Yeruva, L.; Munblit, D. Perspective: The Role of Human Breast-Milk Extracellular Vesicles in Child Health and Disease. Adv. Nutr. 2020. [Google Scholar] [CrossRef]
- Gao, R.; Zhang, R.; Qian, T.; Peng, X.; He, W.; Zheng, S.; Cao, Y.; Pierro, A.; Shen, C. A comparison of exosomes derived from different periods breast milk on protecting against intestinal organoid injury. Pediatr. Surg. Int. 2019, 35, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Patel, M.; Williams, S.; Arora, H.; Brawner, K.; Sims, B. Human breast milk-derived exosomes attenuate cell death in intestinal epithelial cells. Innate Immun. 2018, 24, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.; Miyake, H.; Li, B.; Lee, C.; Ermini, L.; Koike, Y.; Chen, Y.; Maattanen, P.; Zani, A.; Pierro, A. Breast milk-derived exosomes promote intestinal epithelial cell growth. J. Pediatr. Surg. 2017, 52, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Long, W.; Cao, Y.; Li, J.; You, L.; Fan, Y. Mesenchymal stem cell-derived secretomes for therapeutic potential of premature infant diseases. BioSci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Watkins, D.; Chen, C.-L.; Bhushan, B.; Zhou, Y.; Besner, G.E. Heparin-binding epidermal growth factor-like growth factor and mesenchymal stem cells act synergistically to prevent experimental necrotizing enterocolitis. J. Am. Coll. Surg. 2012, 215, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lee, C.; O’Connell, J.S.; Antounians, L.; Ganji, N.; Alganabi, M.; Cadete, M.; Nascimben, F.; Koike, Y.; Hock, A.; et al. Activation of Wnt signaling by amniotic fluid stem cell-derived extracellular vesicles attenuates intestinal injury in experimental necrotizing enterocolitis. Cell Death Dis. 2020, 11, 750. [Google Scholar] [CrossRef]
- Oszvald, A.; Szvicsek, Z.; Sandor, G.O.; Kelemen, A.; Soos, A.A.; Paloczi, K.; Bursics, A.; Dede, K.; Tolgyes, T.; Buzas, E.I.; et al. Extracellular vesicles transmit epithelial growth factor activity in the intestinal stem cell niche. Stem Cells 2020, 38, 291–300. [Google Scholar] [CrossRef]
- Li, B.; Hock, A.; Wu, R.Y.; Minich, A.; Botts, S.R.; Lee, C.; Antounians, L.; Miyake, H.; Koike, Y.; Chen, Y.; et al. Bovine milk-derived exosomes enhance goblet cell activity and prevent the development of experimental necrotizing enterocolitis. PLoS ONE 2019, 14, e0211431. [Google Scholar] [CrossRef]
- Pisano, C.; Galley, J.; Elbahrawy, M.; Wang, Y.; Farrell, A.; Brigstock, D.; Besner, G.E. Human Breast Milk-Derived Extracellular Vesicles in the Protection Against Experimental Necrotizing Enterocolitis. J. Pediatr. Surg. 2020, 55, 54–58. [Google Scholar] [CrossRef]
- Sun, J.; Ding, X.; Liu, S.; Duan, X.; Liang, H.; Sun, T. Adipose-derived mesenchymal stem cells attenuate acute lung injury and improve the gut microbiota in septic rats. Stem Cell Res. Ther. 2020, 11, 384. [Google Scholar] [CrossRef]
- Akduman, H.; Dilli, D.; Ergün, E.; Çakmakçı, E.; Çelebi, S.K.; Çitli, R.; Zenciroğlu, A. Successful Mesenchymal Stem Cell Application in Supraventricular Tachycardia-Related Necrotizing Enterocolitis: A Case Report. Fetal Pediatr. Pathol. 2019, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Guillot, M.; Asad, S.; Lalu, M.M.; Lemyre, B.; Castillo, G.; Thébaud, B.; Presseau, J. So You Want to Give Stem Cells to Babies? Neonatologists and Parents’ Views to Optimize Clinical Trials. J. Pediatr. 2019, 210, 41–47.e41. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatraman, A.; Yu, W.; Nitkin, C.; Sampath, V. Intestinal Stem Cell Development in the Neonatal Gut: Pathways Regulating Development and Relevance to Necrotizing Enterocolitis. Cells 2021, 10, 312. https://doi.org/10.3390/cells10020312
Venkatraman A, Yu W, Nitkin C, Sampath V. Intestinal Stem Cell Development in the Neonatal Gut: Pathways Regulating Development and Relevance to Necrotizing Enterocolitis. Cells. 2021; 10(2):312. https://doi.org/10.3390/cells10020312
Chicago/Turabian StyleVenkatraman, Aparna, Wei Yu, Christopher Nitkin, and Venkatesh Sampath. 2021. "Intestinal Stem Cell Development in the Neonatal Gut: Pathways Regulating Development and Relevance to Necrotizing Enterocolitis" Cells 10, no. 2: 312. https://doi.org/10.3390/cells10020312
APA StyleVenkatraman, A., Yu, W., Nitkin, C., & Sampath, V. (2021). Intestinal Stem Cell Development in the Neonatal Gut: Pathways Regulating Development and Relevance to Necrotizing Enterocolitis. Cells, 10(2), 312. https://doi.org/10.3390/cells10020312