
Mapping the Metabolic Networks of Tumor Cells and Cancer-Associated Fibroblasts


Abstract
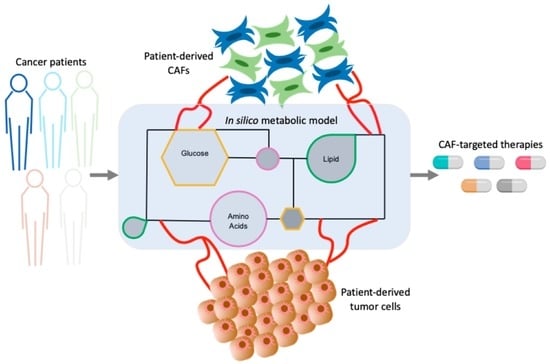
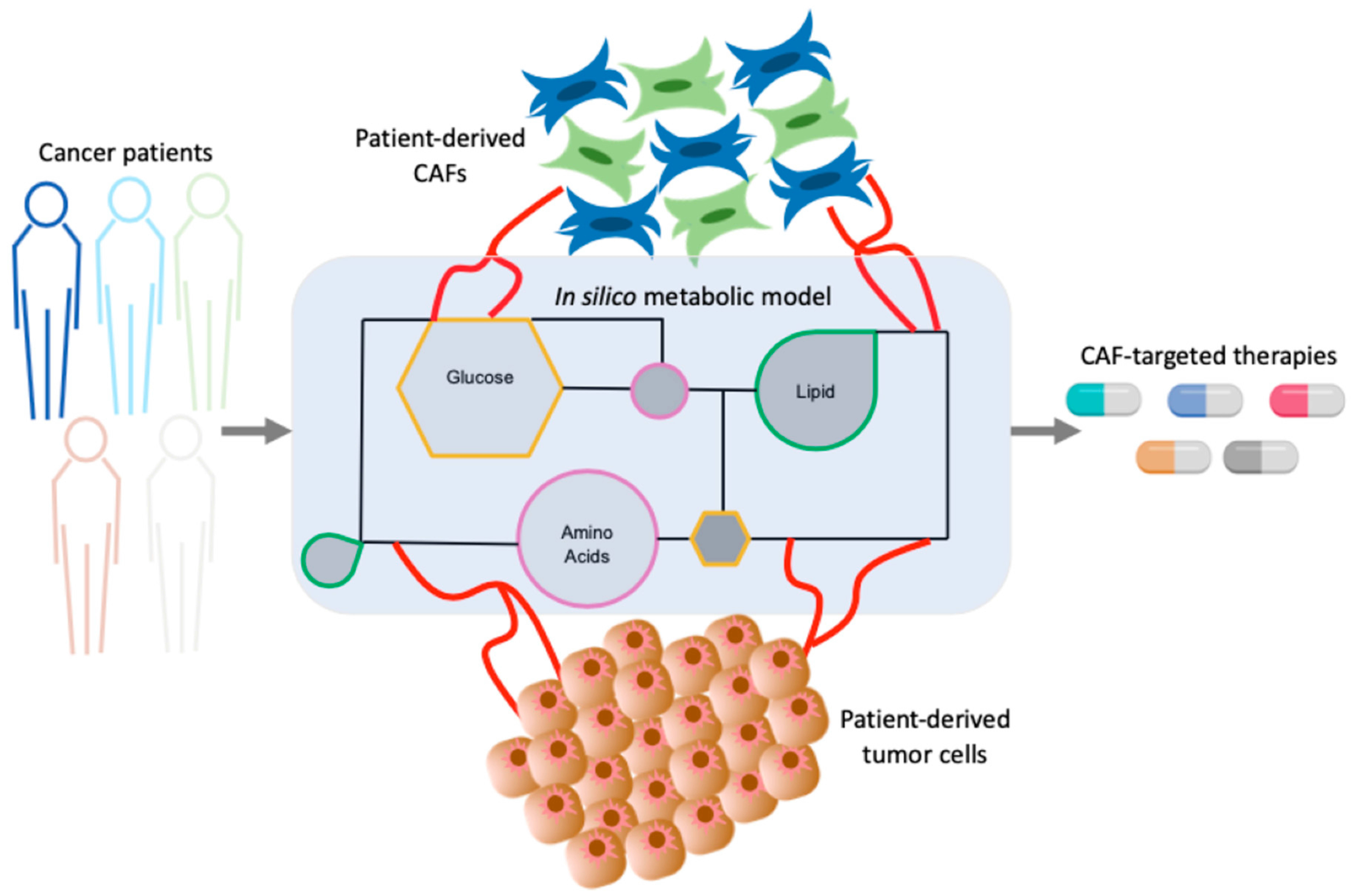
1. Introduction
1.1. CAFs as the Epitome of Tumor Metabolism
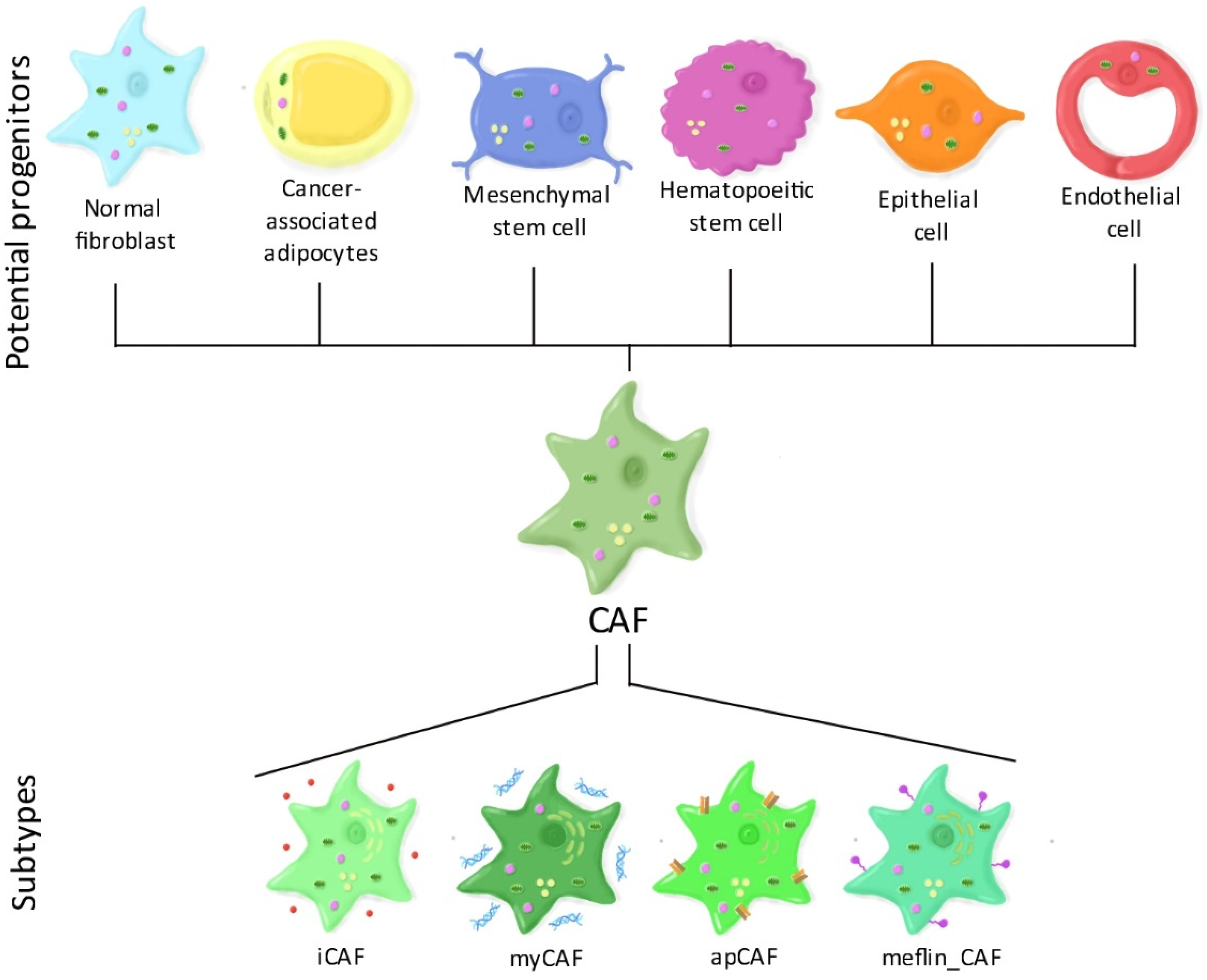
1.2. CAF Heterogeneity: The Different CAF Subtypes
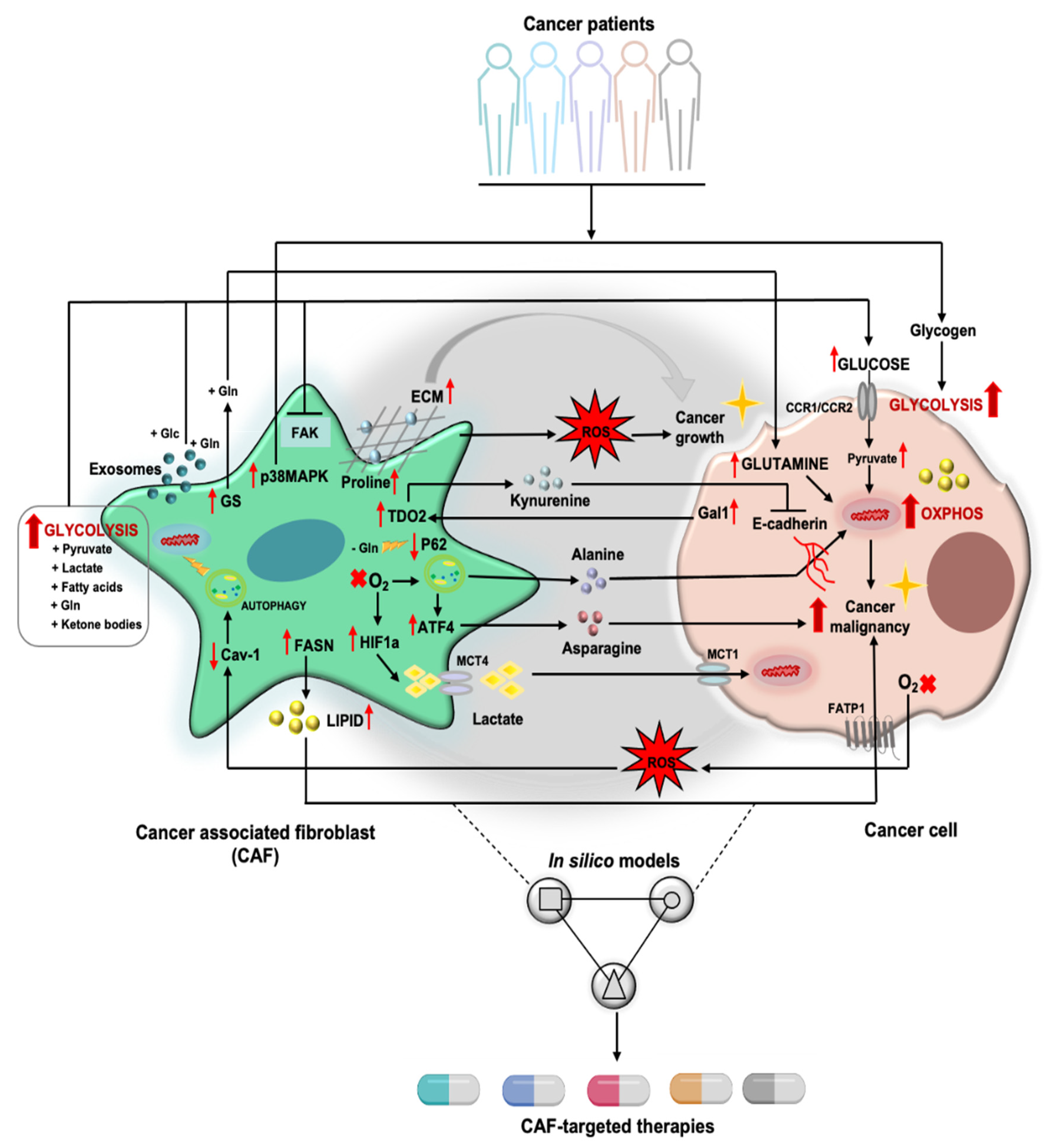
2. Tumor Cell/CAF Interaction-Driven Metabolic Rewiring in Cancer
2.1. Glucose Metabolism and Other Sugar Metabolism
2.2. Amino Acid Metabolism
2.3. Lipid Metabolism
2.4. Immune Modulation by CAF-Derived Metabolism
3. CAFs and Reactive Oxygen Species
4. Metabolic Approaches to Study the Cross-Talks between CAFs and Tumor Cells
4.1. Mass Spectrometry-Based Metabolomics
4.2. Metabolic Flux Analysis
4.3. Seahorse Extracellular Flux
4.4. Computational Approaches to Unravel Tumor-CAF Metabolic Reprogramming
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Glossary
| Abbreviation | Full Text |
| α-KG | Alpha-ketoglutarate |
| apCAF | Antigen-presenting fibroblasts |
| CAFs | Cancer-associated fibroblasts |
| CRC | Colorectal cancer |
| CDEs | CAF-derived exosomes |
| EAAs | Essential amino acids |
| ECAR | Extracellular acidification rate |
| FA | Fatty acids |
| FASN | Fatty acid synthase |
| FCCP | (trifluoromethoxy)phenylhydrazone |
| GC | Gas chromatography |
| Glc | Glucose |
| Gln | Glutamine |
| GS | Glutamine synthetase |
| HPLC | High performance liquid chromatography |
| iCAF | Inflammatory fibroblasts |
| LC | Liquid chromatography |
| LPC | Lysophophatidylcholine |
| LPA | Lysophosphatidic acid |
| MCTs | Mono-carboxylate transporters |
| Meflin_CAF | Meflin-positive fibroblasts |
| MFA | Metabolite flux analysis |
| MS | Mass spectrometry |
| mt(ROS) | Mitochondrial reactive oxygen species |
| myCAF | Myofibroblasts |
| NEAAs | Non-essential amino acids |
| NFs | Normal fibroblasts |
| OCR | Oxygen consumption rate |
| OXPHOS | Oxidative phosphorylation |
| PDAC | Pancreatic ductal adenocarcinoma |
| PSCs | Pancreatic stellate cells |
| ROS | Reactive oxygen species |
| TCA | Tricarboxylic acid cycle |
| TME | Tumor microenvironment |
| UPLC | Ultra-performance liquid chromatography |
References
- Dhom, G. The cancer cell and the connective tissue: A historical review. Pathologe 1994, 15, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.L.; Mukherjee, A.; Kenny, H.A.; Litchfield, L.M.; Lengyel, E. Molecular pathways: Trafficking of metabolic resources in the tumor microenvironment. Clin. Cancer Res. 2015, 21, 680–686. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. DMM Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.; Zhou, J.; Lu, L.; Wang, H.; Zhang, G.; Wan, G.; Cai, S.; Du, J. Nodal Facilitates Differentiation of Fibroblasts to Cancer-Associated Fibroblasts that Support Tumor Growth in Melanoma and Colorectal Cancer. Cells 2019, 8, 538. [Google Scholar] [CrossRef]
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells Toward Breast Tumor Progression. Cells 2019, 8, 223. [Google Scholar] [CrossRef]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast cancer tumor stroma: Cellular components, phenotypic heterogeneity, intercellular communication, prognostic implications and therapeutic opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Porada, G.; Atala, A.J.; Porada, C.D. Therapeutic Mesenchymal Stromal Cells for Immunotherapy and for Gene and Drug Delivery. Mol. Ther. Methods Clin. Dev. 2020, 16, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.K.; Thakrar, R.M.; Janes, S.M. Genetically modified mesenchymal stromal cells in cancer therapy. Cytotherapy 2016, 18, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic Potential of Mesenchymal Stem Cells for Cancer Therapy. Front. Bioeng. Biotechnol. 2020, 8, 1–13. [Google Scholar] [CrossRef]
- Barrett, R.; Puré, E. Cancer-associated fibroblasts: Key determinants of tumor immunity and immunotherapy. Curr. Opin. Immunol. 2020, 64, 80–87. [Google Scholar] [CrossRef]
- Salimifard, S.; Masjedi, A.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Irandoust, M.; Azizi, G.; Mohammadi, H.; Keramati, M.R.; Jadidi-Niaragh, F. Cancer associated fibroblasts as novel promising therapeutic targets in breast cancer. Pathol. Res. Pract. 2020, 216, 152915. [Google Scholar] [CrossRef]
- Bhattacharya, S.D.; Mi, Z.; Talbot, L.J.; Guo, H.; Kuo, P.C. Human mesenchymal stem cell and epithelial hepatic carcinoma cell lines in admixture: Concurrent stimulation of cancer-associated fibroblasts and epithelial-to-mesenchymal transition markers. Surgery 2012, 152, 449–454. [Google Scholar] [CrossRef][Green Version]
- Hossen, M.N.; Rao, G.; Dey, A.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Gold Nanoparticle Transforms Activated Cancer-Associated Fibroblasts to Quiescence. ACS Appl. Mater. Interfaces 2019, 11, 26060–26068. [Google Scholar] [CrossRef]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A stromal lysolipid–autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef]
- Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Jotzu, C. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal. Cell. Pathol. 2010, 33, 61–79. [Google Scholar] [CrossRef]
- McDonald, L.T.; LaRue, A.C. Hematopoietic stem cell derived carcinoma-associated fibroblasts: A novel origin. Int. J. Clin. Exp. Pathol. 2012, 5, 863–873. [Google Scholar] [PubMed]
- Augsten, M. Cancer-Associated Fibroblasts as Another Polarized Cell Type of the Tumor Microenvironment. Front. Oncol. 2014, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. Il1-induced Jak/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef]
- Ocaña, M.C.; Martínez-Poveda, B.; Quesada, A.R.; Medina, M.Á. Metabolism within the tumor microenvironment and its implication on cancer progression: An ongoing therapeutic target. Med. Res. Rev. 2019, 39, 70–113. [Google Scholar] [CrossRef]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef]
- Sun, K.; Tang, S.; Hou, Y.; Xi, L.; Chen, Y.; Yin, J.; Peng, M.; Zhao, M.; Cui, X.; Liu, M. Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine 2019, 41, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141–155.e9. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Kunou, S.; Shimada, K.; Tsunoda, M.; Aoki, T.; Iriyama, C.; Tomita, A.; Nakamura, S.; Hayakawa, F.; Kiyoi, H. Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci. 2019, 110, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Balaji, U.; Freinkman, E.; McCue, P.; Witkiewicz, A.K. Unique metabolic features of pancreatic cancer stroma: Relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget 2016, 7, 78396–78411. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5. [Google Scholar] [CrossRef]
- Mestre-Farrera, A.; Bruch-Oms, M.; Peña, R.; Rodríguez-Morató, J.; Alba-Castellón, L.; Comerma, L.; Quintela-Fandino, M.; Duñach, M.; Baulida, J.; Pozo, Ó.J.; et al. Glutamine-directed migration of cancer-activated fibroblasts facilitates epithelial tumor invasion. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hung, J.Y.; Chiang, S.Y.; Jian, S.F.; Wu, C.Y.; Lin, Y.S.; Tsai, Y.M.; Chou, S.H.; Tsai, M.J.; Kuo, P.L. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 2016, 7, 27584–27598. [Google Scholar] [CrossRef]
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsai, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014, 16, 1–14. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Kay, E.; Paterson, K.; Sumpton, D.; Stepanova, E.; Boldrini, C.; Hernandez-Fernaud, J.; Dhayade, S.; Gjerga, E.; Shaw, R.; Neilson, L.; et al. Metabolic control of tumour extracellular matrix production in cancer-associated fibroblasts. BioRxiv 2020, 1–46. [Google Scholar] [CrossRef]
- Lopes-Coelho, F.; André, S.; Félix, A.; Serpa, J. Breast cancer metabolic cross-talk: Fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol. Cell. Endocrinol. 2018, 462, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Courtney, K.D.; Bezwada, D.; Mashimo, T.; Pichumani, K.; Vemireddy, V.; Funk, A.M.; Wimberly, J.; McNeil, S.S.; Kapur, P.; Lotan, Y.; et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab. 2018, 28, 793–800.e2. [Google Scholar] [CrossRef]
- Sanderson, S.M.; Locasale, J.W. Revisiting the Warburg Effect: Some Tumors Hold Their Breath. Cell Metab. 2018, 28, 669–670. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Blomme, A.; Van Simaeys, G.; Doumont, G.; Costanza, B.; Bellier, J.; Otaka, Y.; Sherer, F.; Lovinfosse, P.; Boutry, S.; Palacios, A.P.; et al. Murine stroma adopts a human-like metabolic phenotype in the PDX model of colorectal cancer and liver metastases. Oncogene 2018, 37, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-P.; Luo, Q.; Deng, B.; Ju, Y.; Song, G.-B. Stiffer Matrix Accelerates Migration of Hepatocellular Carcinoma Cells through Enhanced Aerobic Glycolysis Via the MAPK-YAP Signaling. Cancers 2020, 12, 490. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Brian, I.; Pocaterra, A.; Romani, P.; Franzolin, E.; Rampazzo, C.; Bicciato, S.; Dupont, S. d NTP metabolism links mechanical cues and YAP / TAZ to cell growth and oncogene-induced senescence. EMBO J. 2018, 37, 1–16. [Google Scholar] [CrossRef]
- Mah, E.J.; Lefebvre, A.E.Y.T.; McGahey, G.E.; Yee, A.F.; Digman, M.A. Collagen density modulates triple-negative breast cancer cell metabolism through adhesion-mediated contractility. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Choi, B.H.; Coloff, J.L. The diverse functions of non-essential amino acids in cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef]
- Sanford-Crane, H.; Abrego, J.; Sherman, M.H. Fibroblasts as modulators of local and systemic cancer metabolism. Cancers 2019, 11, 619. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.W.; Qu, J.; Black, D.D.; Tso, P. Regulation of intestinal lipid metabolism: Current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; De Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef]
- Romani, P.; Brian, I.; Santinon, G.; Pocaterra, A.; Audano, M.; Pedretti, S.; Mathieu, S.; Forcato, M.; Bicciato, S.; Manneville, J.B.; et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Biol. 2019, 21, 338–347. [Google Scholar] [CrossRef]
- Boulter, E.; Estrach, S.; Tissot, F.S.; Hennrich, M.L.; Tosello, L.; Cailleteau, L.; de la Ballina, L.R.; Pisano, S.; Gavin, A.C.; Féral, C.C. Cell metabolism regulates integrin mechanosensing via an SLC3A2-dependent sphingolipid biosynthesis pathway. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- De Jaeghere, E.A.; Denys, H.G.; De Wever, O. Fibroblasts Fuel Immune Escape in the Tumor Microenvironment. Trends Cancer 2019, 5, 704–723. [Google Scholar] [CrossRef]
- Cheng, J.; Deng, Y.; Yi, H.; Wang, G.; Fu, B.; Chen, W.; Liu, W.; Tai, Y.; Peng, Y.; Zhang, Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 2016, 5, e198. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II Expressed in Cancer-Associated Fibroblasts Indicates Tissue Hypoxia and Predicts Poor Outcome in Patients with Pancreatic Cancer. PLoS ONE 2013, 8, e55146. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, X.; Zhang, L.; Li, W.; Wu, H.; Yuan, X.; Mao, F.; Wang, M.; Zhu, W.; Qian, H.; et al. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 2014, 5, e1295. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing ROS-Generating Monocytic MDSCs in Lung Squamous Cell Carcinoma. Cancer Immunol. Res. 2020, 8, 436–450. [Google Scholar] [CrossRef]
- Sampson, N.; Brunner, E.; Weber, A.; Puhr, M.; Schäfer, G.; Szyndralewiez, C.; Klocker, H. Inhibition of Nox4-dependent ROS signaling attenuates prostate fibroblast activation and abrogates stromal-mediated protumorigenic interactions. Int. J. Cancer 2018, 143, 383–395. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Nietzel, T.; Mostertz, J.; Hochgräfe, F.; Schwarzländer, M. Redox regulation of mitochondrial proteins and proteomes by cysteine thiol switches. Mitochondrion 2017, 33, 72–83. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Rajamanickam, S.; Loranc, E.; Masamsetti, V.P.; Gorthi, A.; Romero, J.C.; Tonapi, S.; Gonçalves, R.M.; Reddick, R.L.; Benavides, R.; et al. Sorafenib improves alkylating therapy by blocking induced inflammation, invasion and angiogenesis in breast cancer cells. Cancer Lett. 2018, 425, 101–115. [Google Scholar] [CrossRef]
- Zhao, T.; Zhu, Y.; Morinibu, A.; Kobayashi, M.; Shinomiya, K.; Itasaka, S.; Yoshimura, M.; Guo, G.; Hiraoka, M.; Harada, H. HIF-1-mediated metabolic reprogramming reduces ROS levels and facilitates the metastatic colonization of cancers in lungs. Sci. Rep. 2014, 4, 3793. [Google Scholar] [CrossRef]
- Shan, T.; Lu, H.; Ji, H.; Li, Y.; Guo, J.; Chen, X.; Wu, T. Loss of stromal caveolin-1 expression: A novel tumor microenvironment biomarker that can predict poor clinical outcomes for pancreatic cancer. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Dasgupta, A.; Sotgia, F.; Mercier, I.; Pestell, R.G.; Sabel, M.; Kleer, C.G.; Brody, J.R.; Lisanti, M.P. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am. J. Pathol. 2009, 174, 2023–2034. [Google Scholar] [CrossRef]
- Chen, F.; Barman, S.; Yu, Y.; Haigh, S.; Wang, Y.; Dou, H.; Bagi, Z.; Han, W.; Su, Y.; Fulton, D.J.R. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2014, 73, 201–213. [Google Scholar] [CrossRef]
- Arcucci, A.; Ruocco, M.R.; Granato, G.; Sacco, A.M.; Montagnani, S. Cancer: An Oxidative Crosstalk between Solid Tumor Cells and Cancer Associated Fibroblasts. BioMed Res. Int. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment—Accomplices in tumor malignancy. Cell. Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.C.; Franzen, C.A.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015, 34, 4821–4833. [Google Scholar] [CrossRef]
- Liu, F.L.; Mo, E.P.; Yang, L.; Du, J.; Wang, H.S.; Zhang, H.; Kurihara, H.; Xu, J.; Cai, S.H. Autophagy is involved in TGF-β1-induced protective mechanisms and formation of cancer-associated fibroblasts phenotype in tumor microenvironment. Oncotarget 2016, 7, 4122–4141. [Google Scholar] [CrossRef]
- Cheng, N.; Bhowmick, N.A.; Chytil, A.; Gorksa, A.E.; Brown, K.A.; Muraoka, R.; Arteaga, C.L.; Neilson, E.G.; Hayward, S.W.; Moses, H.L. Loss of TGf-β type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-α-, MSP- and HGF-mediated signaling networks. Oncogene 2005, 24, 5053–5068. [Google Scholar] [CrossRef]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S.; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-β signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef]
- Chan, J.S.K.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; Li, L.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2017, 8, e2562. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Augsten, M.; Rundqvist, H.; Bianchi, J.; Sarne, V.; Egevad, L.; Bykov, V.J.; Östman, A.; Wiman, K.G. Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis. 2017, 8, e2848. [Google Scholar] [CrossRef]
- Kim, Y.M.; Heyman, H.M. Mass spectrometry-based metabolomics. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1775, pp. 107–118. [Google Scholar]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef]
- Parri, M.; Ippolito, L.; Cirri, P.; Ramazzotti, M.; Chiarugi, P. Metabolic cell communication within tumour microenvironment: Models, methods and perspectives. Curr. Opin. Biotechnol. 2020, 63, 210–219. [Google Scholar] [CrossRef]
- Lagziel, S.; Lee, W.D.; Shlomi, T. Studying metabolic flux adaptations in cancer through integrated experimental-computational approaches. BMC Biol. 2019, 17, 1–11. [Google Scholar] [CrossRef]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med. 2018, 50, 19. [Google Scholar] [CrossRef]
- Faubert, B.; Deberardinis, R.J. Analyzing tumor metabolism in vivo. Annu. Rev. Cancer Biol. 2017, 1, 99–117. [Google Scholar] [CrossRef]
- Elia, I.; Fendt, S.M. In vivo cancer metabolism is defined by the nutrient microenvironment. Transl. Cancer Res. 2016, 5, S1284–S1287. [Google Scholar] [CrossRef]
- Fernández-García, J.; Altea-Manzano, P.; Pranzini, E.; Fendt, S.M. Stable Isotopes for Tracing Mammalian-Cell Metabolism In Vivo. Trends Biochem. Sci. 2020, 45, 185–201. [Google Scholar] [CrossRef]
- Application Brief Agilent Technologies. Measuring Glycolysis and Oxidative Metabolism in Cancer Cells. Available online: https://www.agilent.com (accessed on 19 August 2020).
- Plitzko, B.; Loesgen, S. Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism. Bio-Protocol 2018, 8. [Google Scholar] [CrossRef]
- Qureshi-Baig, K.; Ullmann, P.; Haan, S.; Letellier, E. Tumor-Initiating Cells: A criTICal review of isolation approaches and new challenges in targeting strategies. Mol. Cancer 2017, 16, 1–16. [Google Scholar] [CrossRef]
- Russell, S.; Wojtkowiak, J.; Neilson, A.; Gillies, R.J. Metabolic Profiling of healthy and cancerous tissues in 2D and 3D. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Sauer, U. Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2006, 2, 62. [Google Scholar] [CrossRef]
- Ji, Z.; Yan, K.; Li, W.; Hu, H.; Zhu, X. Mathematical and Computational Modeling in Complex Biological Systems. BioMed Res. Int. 2017, 2017, 5958321. [Google Scholar] [CrossRef]
- Alm, E.; Arkin, A.P. Biological networks. Curr. Opin. Struct. Biol. 2003, 13, 193–202. [Google Scholar] [CrossRef]
- Hadjicharalambous, M.; Wijeratne, P.A.; Vavourakis, V. From tumour perfusion to drug delivery and clinical translation of in silico cancer models. Methods 2020. [Google Scholar] [CrossRef]
- Werner, H.M.J.; Mills, G.B.; Ram, P.T. Cancer systems biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef]
- Thiele, I.; Swainston, N.; Fleming, R.M.T.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef]
- Blais, E.M.; Rawls, K.D.; Dougherty, B.V.; Li, Z.I.; Kolling, G.L.; Ye, P.; Wallqvist, A.; Papin, J.A. Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Angione, C. Human Systems Biology and Metabolic Modelling: A Review-From Disease Metabolism to Precision Medicine. BioMed Res. Int. 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Patil, K.R.; Åkesson, M.; Nielsen, J. Use of genome-scale microbial models for metabolic engineering. Curr. Opin. Biotechnol. 2004, 15, 64–69. [Google Scholar] [CrossRef]
- Granata, I.; Troiano, E.; Sangiovanni, M.; Guarracino, M.R. Integration of transcriptomic data in a genome-scale metabolic model to investigate the link between obesity and breast cancer. BMC Bioinform. 2019, 20, 162. [Google Scholar] [CrossRef]
- Schultz, A.; Mehta, S.; Hu, C.W.; Hoff, F.W.; Horton, T.M.; Kornblau, S.M.; Qutub, A.A. Identifying Cancer Specific Metabolic Signatures Using Constraint-Based Models. In Proceedings of the Biocomputing 2017, Kohala Coast, HI, USA, 4–8 January 2017; World Scientific: Singapore, 2017; Volume 22, pp. 485–496. [Google Scholar]
- Aurich, M.K.; Fleming, R.M.T.; Thiele, I. MetaboTools: A comprehensive toolbox for analysis of genome-scale metabolic models. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, A.B.; Herzog, K.; Gerding, A.; Kwiatkowski, M.; Wolters, J.C.; Dolga, A.M.; van Lint, A.E.M.; Wanders, R.J.A.; Waterham, H.R.; Bakker, B.M. Fibroblast-specific genome-scale modelling predicts an imbalance in amino acid metabolism in Refsum disease. FEBS J. 2020, 287, 5096–5113. [Google Scholar] [CrossRef] [PubMed]
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst. Biol. 2012, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Vlassis, N.; Pacheco, M.P.; Sauter, T. Fast Reconstruction of Compact Context-Specific Metabolic Network Models. PLoS Comput. Biol. 2014, 10, e1003424. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.P.; John, E.; Kaoma, T.; Heinäniemi, M.; Nicot, N.; Vallar, L.; Bueb, J.L.; Sinkkonen, L.; Sauter, T. Integrated metabolic modelling reveals cell-type specific epigenetic control points of the macrophage metabolic network. BMC Genom. 2015, 16, 809. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.P.; Bintener, T.; Ternes, D.; Kulms, D.; Haan, S.; Letellier, E.; Sauter, T. Identifying and targeting cancer-specific metabolism with network-based drug target prediction. EBioMedicine 2019, 43, 98–106. [Google Scholar] [CrossRef]
- Bintener, T.; Pacheco, M.P.; Sauter, T. Towards the routine use of in silico screenings for drug discovery using metabolic modelling. Biochem. Soc. Trans. 2020, 48, 955–969. [Google Scholar] [CrossRef]
- Heinken, A.; Thiele, I. Systems biology of host-microbe metabolomics. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 195–219. [Google Scholar] [CrossRef]
- Lewis, N.E.; Schramm, G.; Bordbar, A.; Schellenberger, J.; Andersen, M.P.; Cheng, J.K.; Patel, N.; Yee, A.; Lewis, R.A.; Eils, R.; et al. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010, 28, 1279–1285. [Google Scholar] [CrossRef]
- Capuani, F.; De Martino, D.; Marinari, E.; De Martino, A. Quantitative constraint-based computational model of tumor-to-stroma coupling via lactate shuttle. Sci. Rep. 2015, 5, 11880. [Google Scholar] [CrossRef]
- Shan, M.; Dai, D.; Vudem, A.; Varner, J.D.; Stroock, A.D. Multi-scale computational study of the Warburg effect, reverse Warburg effect and glutamine addiction in solid tumors. PLoS Comput. Biol. 2018, 14, e1006584. [Google Scholar] [CrossRef]
- Damiani, C.; Maspero, D.; Di Filippo, M.; Colombo, R.; Pescini, D.; Graudenzi, A.; Westerhoff, H.V.; Alberghina, L.; Vanoni, M.; Mauri, G. Integration of single-cell RNA-seq data into population models to characterize cancer metabolism. PLoS Comput. Biol. 2019, 15, e1006733. [Google Scholar] [CrossRef]
- Huang, L.; Xu, A.M.; Liu, S.; Liu, W.; Li, T.J. Cancer-associated fibroblasts in digestive tumors. World J. Gastroenterol. 2014, 20, 17804–17818. [Google Scholar] [CrossRef]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. BioMed Res. Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Cancer Type | In Vitro Analysis | In Vivo Analysis | Ex Vivo Analysis | Technical Advantage | Technical Limitation | Ref. |
|---|---|---|---|---|---|---|
| Glucose metabolism | ||||||
| Breast and pancreatic | NA |
|
|
|
| Demircioglu et al. 2020 [29] |
| Mechanistic highlight: FAK-deletion in CAFs induced malignant cell glycolysis and tumor growth via CCR1/CCR2 | ||||||
| Breast |
| NA |
|
|
| L.M. Becker et al. 2020 [30] |
| Mechanistic highlight: Chronic hypoxia induced NFs to adopt a pro-glycolytic CAF phenotype via epigenetic reprogramming, which fuelled cancer cells’ metabolism and their growth. | ||||||
| Breast |
| NA | NA |
|
| Sun et al. 2019 [31] |
| Mechanistic highlight: Lactate secreted by hypoxic and pro-glycolytic CAFs was driven by GLUT1 phosphorylation and PKM2 upregulation, and this lactate promoted cancer cell invasion via activated TGFB1/p38 MAPK/MMP2/9 signaling and increased cancer cells mitochondrial activity. | ||||||
| Ovarian |
| NA | NA |
|
| Curtis et al. 2019 [32] |
| Mechanistic highlight: Glycogen utilization in cancer cells was dependent on p38-alpha MAPK activation in CAFs and this supported their proliferation, invasion and metastasis | ||||||
| Lymphoma |
| NA | NA |
|
| Sakamoto et al. 2019 [33] |
| Mechanistic highlight: CAF-secreted pyruvate supported citric acid cycle while inhibited redox regulation to promote cancer cells survival | ||||||
| Pancreatic |
| NA | NA |
|
| Knudsen et al. 2016 [34] |
| Mechanistic highlight: HIF1alpha-driven hypoxic and pro-glycolytic CAFs upregulated carbonic anhydrase IX (CAIX) and MCT4 expressions, secreted lactate and supported cancer invasion | ||||||
| Glucose and glutamine metabolism | ||||||
| Breast |
|
| NA |
|
| Li et al. 2018 [35] |
| Mechanistic highlight: Breast cancer-secreted extracellular vesicles (EVs) containing miR-105 induced a MYC-dependent pro-glycolysis and pro-glutaminolysis in CAFs under sufficient nutrients. However, in nutrient-deprived conditions, these mir-105-reprogrammed CAFs converted metabolic wastes (i.e., lactic acid and ammonium) into energy-rich metabolites to sustain tumor growth. | ||||||
| Prostate and pancreatic |
| NA | NA |
|
| Zhao et al. 2016 [36] |
| Mechanistic highlight: Under starvation, CAFs-derived exosomes (CDEs) were smuggled in by cancer cells as the required building blocks. This caused a decrease in mitochondrial OXPHOS, while increase in glucose and glutamine tumor cell metabolism, enhancing cancer cells survival via a Kras-independent mechanism. | ||||||
| Breast and CRC | Invasion and migration Organotypic invasion Directional migration in chemotaxis μ-slides. | Migration | NA | Exploring the roles of metabolite gradient on CAFs and tumor cell migration | Mestre-Farrera et al. 2020 [37] | |
| Mechanistic insight: CAFs are more sensitive to low glutamine levels than their cancer counterparts. As the tumor core is depleted in glutamine, CAFs move towards glutamine rich areas in an ATK2 dependent mechanism. This movement along with the racks that CAFs create allow tumor cells to invade tissues and escape their original site. | ||||||
| Amino acids metabolism | ||||||
| Ovarian |
| NA |
|
|
| Yang et al. 2016 [38] |
| Mechanistic highlight: Under glutamine deprived conditions, CAFs harnessed atypical carbon and nitrogen sources to boost their glutamine production, and support cancer cells proliferation. This relied on the expression of glutamine synthetase (GLUL) in CAFs and glutaminase (GLS) in cancer cells. | ||||||
| Lung |
| NA | NA |
|
| Hsu et al. 2016 [39] |
| Mechanistic highlight: Tumor-fibroblast interaction induced galactin-1 overexpression in lung cancer which led to TDO2-dependent kynurenine secretion by fibroblasts. Fibroblasts-secreted kynurenine promoted cancer growth, invasion and immunosuppression through the AKT/CREB/WNK1 axis. | ||||||
| Breast |
| NA | NA |
|
| Chen et al. 2014 [40] |
| Mechanistic highlight: IDO-dependent kynurenine secretion by CAFs promoted E-cadherin degradation and increased invasion of cancer cells. PGE2 released by cancer cells promoted the expression of stromal IDO via STAT3/COX2 activation. | ||||||
| Pancreatic |
| NA | NA |
|
| Sousa et al. 2016 [41] |
| Mechanistic highlight: Autophagy dependent-alanine secretion by PSCs became an alternative carbon source for cancer cells. This led to an increase in the OCR of PDAC cells. | ||||||
| Breast |
| NA | NA |
|
| Kay et al. 2020 [42] |
| Mechanistic highlight: Proline synthesis in CAFs caused tumor epigenetic reprogramming, which enhanced ECM production and supported tumor growth. | ||||||
| Lipid metabolism | ||||||
| Pancreatic |
| NA |
|
|
| Auciello et al. 2019 [19] |
| Mechanistic highlight: PSC secreted lysophospatidylcholines (LPC) promoted the secretion of oncogenic autotaxin-lysophospatidic acid (LPA), which supported proliferation, migration and AKT activation in PDAC | ||||||
| Breast |
| NA | NA |
|
| Coelho et al. 2018 [43] |
| Mechanistic highlight: Lipids were transferred from CAFs to tumor cells, which was dependent on fatty acid transporter-1 (FATP1), and promoted tumor growth. | ||||||
| Breast |
| NA | NA |
|
| Radhakrishnan et al. 2018 [44] |
| Mechanistic highlight: Under normoxia and hypoxia, the secreted LPA by ovarian cancer cells (OCC) induced pro-glycolytic phenotypes in both ovarian NFs and CAFs. This was due to LPA triggered HIF1 alpha-dependent pseudohypoxic oxidative stress in OCC. | ||||||
| Colorectal |
|
|
| Gong et al. 2020 [45] | ||
| Mechanistic highlight: FASN-dependent CAFs-secreted lipids were taken up by tumor cells and induced tumor migration capacity. | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karta, J.; Bossicard, Y.; Kotzamanis, K.; Dolznig, H.; Letellier, E. Mapping the Metabolic Networks of Tumor Cells and Cancer-Associated Fibroblasts. Cells 2021, 10, 304. https://doi.org/10.3390/cells10020304
Karta J, Bossicard Y, Kotzamanis K, Dolznig H, Letellier E. Mapping the Metabolic Networks of Tumor Cells and Cancer-Associated Fibroblasts. Cells. 2021; 10(2):304. https://doi.org/10.3390/cells10020304
Chicago/Turabian StyleKarta, Jessica, Ysaline Bossicard, Konstantinos Kotzamanis, Helmut Dolznig, and Elisabeth Letellier. 2021. "Mapping the Metabolic Networks of Tumor Cells and Cancer-Associated Fibroblasts" Cells 10, no. 2: 304. https://doi.org/10.3390/cells10020304
APA StyleKarta, J., Bossicard, Y., Kotzamanis, K., Dolznig, H., & Letellier, E. (2021). Mapping the Metabolic Networks of Tumor Cells and Cancer-Associated Fibroblasts. Cells, 10(2), 304. https://doi.org/10.3390/cells10020304