
Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration

Abstract

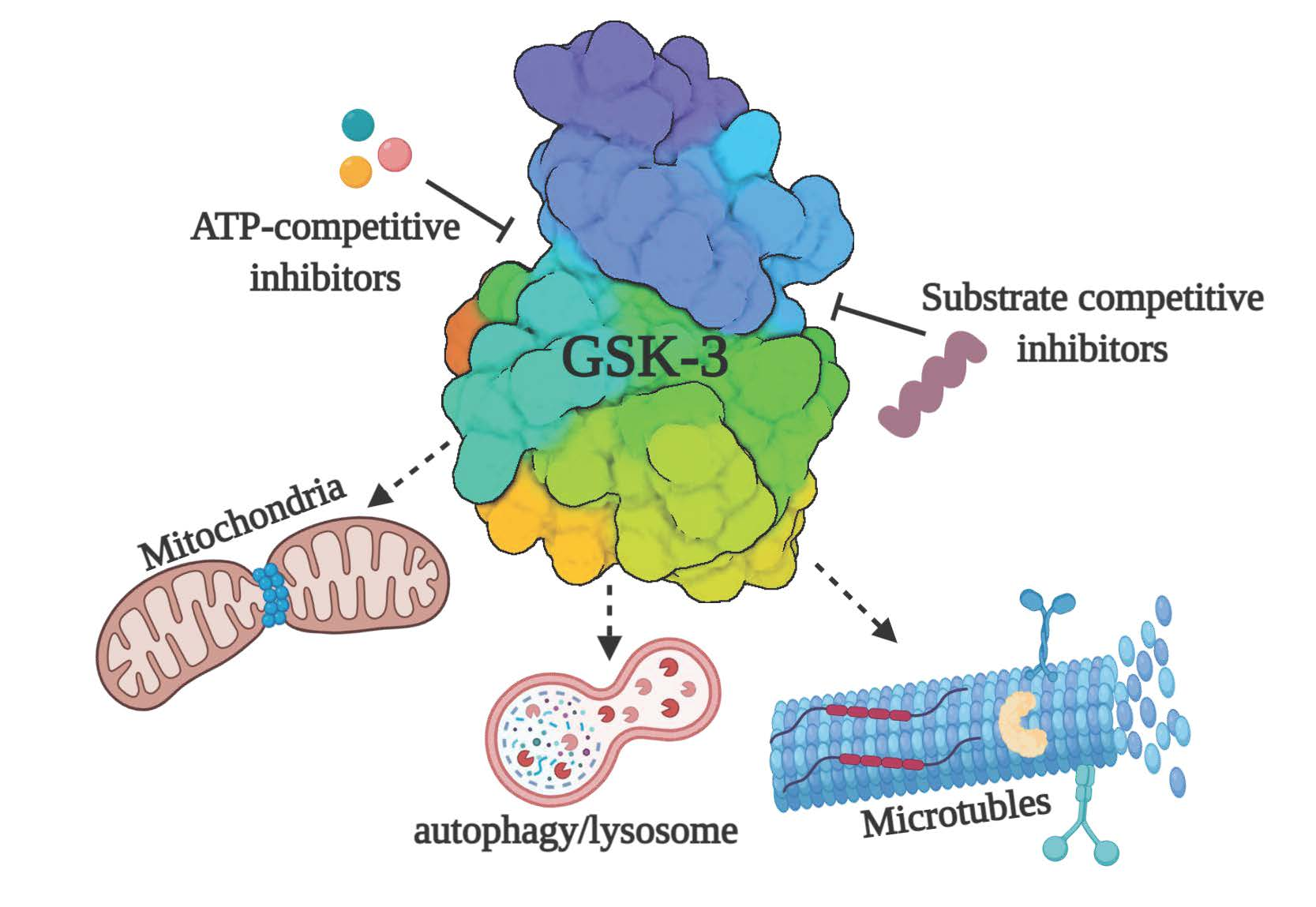{kind=link}
{kind=link}
{kind=link}
1. GSK-3—A Story of Two Isozymes
2. GSK-3 in Neurodegeneration
3. GSK-3—A Regulator of Microtubule Network and Axonal Transport
4. GSK-3—An Upstream Regulator of mTORC1/Autophagy Axis
5. GSK-3 and Mitochondria—Energetic Regulation and Cell Death
6. GSK-3–A Target for Inhibition
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woodgett, J.R.; Cohen, P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase- II (glycogen synthase kinase-5). Biochim. Biophys. Acta 1984, 788, 339–347. [Google Scholar] [CrossRef]
- Fiol, C.J.; Mahrenholz, A.M.; Wang, Y.; Roeske, R.W.; Roach, P.J. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J. Biol. Chem. 1987, 262, 14042–14048. [Google Scholar] [CrossRef]
- Liberman, Z.; Eldar-Finkelman, H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 2005, 280, 4422–4428. [Google Scholar] [CrossRef] [PubMed]
- Sharfi, H.; Eldar-Finkelman, H. Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E307–E315. [Google Scholar] [CrossRef] [PubMed]
- Yost, C.; Torres, M.; Miller, J.; Huang, E.; Kimelman, D.; Moon, R. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes. Dev. 1996, 10, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998, 17, 1371–1384. [Google Scholar] [CrossRef]
- Beals, C.R.; Sheridan, C.M.; Turck, C.W.; Gardner, P.; Crabtree, G.R. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 1997, 275, 1930–1934. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Soncin, F.; Price, B.D.; Stevenson, M.A.; Calderwood, S.K. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J. Biol. Chem. 1996, 271, 30847–30857. [Google Scholar] [CrossRef]
- Grimes, C.A.; Jope, R.S. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J. Neurochem. 2001, 78, 1219–1232. [Google Scholar] [CrossRef]
- Sanchez, J.F.; Sniderhan, L.F.; Williamson, A.L.; Fan, S.; Chakraborty-Sett, S.; Maggirwar, S.B. Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol. Cell. Biol. 2003, 23, 4649–4662. [Google Scholar] [CrossRef]
- Fiol, C.J.; Williams, J.S.; Chou, C.H.; Wang, Q.M.; Roach, P.J.; Andrisani, O.M. A secondary phosphorylation of CREB341 at Ser129 is required for the cAMP-mediated control of gene expression. A role for glycogen synthase kinase-3 in the control of gene expression. J. Biol. Chem. 1994, 269, 32187–32193. [Google Scholar] [CrossRef]
- Twomey, C.; McCarthy, J.V. Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett. 2006, 580, 4015–4020. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P.; Biondi, R.M. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Hagen, T.; Di Daniel, E.; Culbert, A.A.; Reith, A.D. Expression and characterization of GSK-3 mutants and their effect on beta-catenin phosphorylation in intact cells. J. Biol. Chem. 2002, 277, 23330–23335. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jorgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Alabed, Y.Z.; Pool, M.; Ong Tone, S.; Sutherland, C.; Fournier, A.E. GSK3 beta regulates myelin-dependent axon outgrowth inhibition through CRMP4. J. Neurosci. 2011, 30, 5635–5643. [Google Scholar] [CrossRef]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factorA. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef]
- Mukai, F.; Ishiguro, K.; Sano, Y.; Fujita, S.C. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J. Neurochem. 2002, 81, 1073–1083. [Google Scholar] [CrossRef]
- Soutar, M.P.; Kim, W.Y.; Williamson, R.; Peggie, M.; Hastie, C.J.; McLauchlan, H.; Snider, W.D.; Gordon-Weeks, P.R.; Sutherland, C. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 2011, 115, 974–983. [Google Scholar] [CrossRef]
- Alon, L.T.; Pietrokovski, S.; Barkan, S.; Avrahami, L.; Kaidanovich-Beilin, O.; Woodgett, J.R.; Barnea, A.; Eldar-Finkelman, H. Selective loss of glycogen synthase kinase-3alpha in birds reveals distinct roles for GSK-3 isozymes in tau phosphorylation. FEBS Lett. 2011, 585, 1158–1162. [Google Scholar] [CrossRef]
- Yao, H.B.; Shaw, P.C.; Wong, C.C.; Wan, D.C. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J. Chem. Neuroanat. 2002, 23, 291–297. [Google Scholar] [CrossRef]
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Force, T.; Woodgett, J.R. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J. Biol. Chem. 2009, 284, 9643–9647. [Google Scholar] [CrossRef]
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.W.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.M.; Woodgett, J.R.; et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618. [Google Scholar]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Lipina, T.V.; Takao, K.; van Eede, M.; Hattori, S.; Laliberté, C.; Khan, M.; Okamoto, K.; Chambers, J.W.; Fletcher, P.J.; et al. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2009, 2, 1–23. [Google Scholar] [CrossRef]
- Zhou, J.; Freeman, T.A.; Ahmad, F.; Shang, X.; Mangano, E.; Gao, E.; Farber, J.; Wang, Y.; Ma, X.L.; Woodgett, J.; et al. GSK-3alpha is a central regulator of age-related pathologies in mice. J. Clin. Investig. 2013, 123, 1821–1832. [Google Scholar] [CrossRef]
- Maurin, H.; Lechat, B.; Dewachter, I.; Ris, L.; Louis, J.V.; Borghgraef, P.; Devijver, H.; Jaworski, T.; Van Leuven, F. Neurological characterization of mice deficient in GSK3alpha highlight pleiotropic physiological functions in cognition and pathological activity as Tau kinase. Mol. Brain 2013, 6, 27. [Google Scholar] [CrossRef]
- Hurtado, D.E.; Molina-Porcel, L.; Carroll, J.C.; Macdonald, C.; Aboagye, A.K.; Trojanowski, J.Q.; Lee, V.M. Selectively Silencing GSK-3 Isoforms Reduces Plaques and Tangles in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2012, 32, 7392–7402. [Google Scholar] [CrossRef]
- Latapy, C.; Rioux, V.; Guitton, M.J.; Beaulieu, J.M. Selective deletion of forebrain glycogen synthase kinase 3beta reveals a central role in serotonin-sensitive anxiety and social behaviour. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2460–2474. [Google Scholar] [CrossRef] [PubMed]
- Nakao, K.; Singh, M.; Sapkota, K.; Hagler, B.C.; Hunter, R.N.; Raman, C.; Hablitz, J.J.; Nakazawa, K. GSK3beta inhibition restores cortical gamma oscillation and cognitive behavior in a mouse model of NMDA receptor hypofunction relevant to schizophrenia. Neuropsychopharmacology 2020, 45, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, W.T.; Harper, A.D.; Jove, F.; Woodgett, J.R.; Maretto, S.; Piccolo, S.; Klein, P.S. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 2004, 24, 6791–6798. [Google Scholar] [CrossRef] [PubMed]
- Bersudsky, Y.; Shaldubina, A.; Kozlovsky, N.; Woodgett, J.R.; Agam, G.; Belmaker, R.H. Glycogen synthase kinase-3beta heterozygote knockout mice as a model of findings in postmortem schizophrenia brain or as a model of behaviors mimicking lithium action: Negative results. Behav. Pharmacol. 2008, 19, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Ochs, S.M.; Dorostkar, M.M.; Aramuni, G.; Schon, C.; Filser, S.; Poschl, J.; Kremer, A.; Van Leuven, F.; Ovsepian, S.V.; Herms, J. Loss of neuronal GSK3beta reduces dendritic spine stability and attenuates excitatory synaptic transmission via beta-catenin. Mol. Psychiatry 2015, 20, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Spittaels, K.; Van den Haute, C.; Van Dorpe, J.; Terwel, D.; Vandezande, K.; Lasrado, R.; Bruynseels, K.; Irizarry, M.; Verhoye, M.; Van Lint, J.; et al. Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience 2002, 113, 797–808. [Google Scholar] [CrossRef]
- Kim, W.Y.; Zhou, F.Q.; Zhou, J.; Yokota, Y.; Wang, Y.M.; Yoshimura, T.; Kaibuchi, K.; Woodgett, J.R.; Anton, E.S.; Snider, W.D. Essential Roles for GSK-3s and GSK-3-Primed Substrates in Neurotrophin-Induced and Hippocampal Axon Growth. Neuron 2006, 52, 981–996. [Google Scholar] [CrossRef]
- Jung, E.M.; Ka, M.; Kim, W.Y. Loss of GSK-3 Causes Abnormal Astrogenesis and Behavior in Mice. Mol. Neurobiol. 2016, 53, 3954–3966. [Google Scholar] [CrossRef]
- Morgan-Smith, M.; Wu, Y.; Zhu, X.; Pringle, J.; Snider, W.D. GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. eLife 2014, 3, e02663. [Google Scholar] [CrossRef]
- Kim, W.Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef]
- Aloni, E.; Shapira, M.; Eldar-Finkelman, H.; Barnea, A. GSK-3beta Inhibition Affects Singing Behavior and Neurogenesis in Adult Songbirds. Brain Behav. Evol. 2015, 85, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.J.; Hernandez, F.; Gomez-Ramos, P.; Moran, M.A.; Hen, R.; Avila, J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001, 20, 27–39. [Google Scholar] [CrossRef] [PubMed]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Prickaerts, J.; Moechars, D.; Cryns, K.; Lenaerts, I.; van Craenendonck, H.; Goris, I.; Daneels, G.; Bouwknecht, J.A.; Steckler, T. Transgenic mice overexpressing glycogen synthase kinase 3beta: A putative model of hyperactivity and mania. J. Neurosci. 2006, 26, 9022–9029. [Google Scholar] [CrossRef]
- Eom, T.Y.; Jope, R.S. Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol. Psychiatry 2009, 66, 494–502. [Google Scholar] [CrossRef]
- Wagner, F.F.; Benajiba, L.; Campbell, A.J.; Weiwer, M.; Sacher, J.R.; Gale, J.P.; Ross, L.; Puissant, A.; Alexe, G.; Conway, A.; et al. Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci. Transl. Med. 2018, 10, eaam8460. [Google Scholar] [CrossRef]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Selective inhibition of glycogen synthase kinase 3alpha corrects pathophysiology in a mouse model of fragile X syndrome. Sci. Transl. Med. 2020, 12, eaam8572. [Google Scholar] [CrossRef]
- Klein, P.S.; Melton, D.A. A Molecular Mechanism for the Effect of Lithium on Development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef]
- O’Brien, W.T.; Klein, P.S. Validating GSK3 as an in vivo target of lithium action. Biochem. Soc. Trans. 2009, 37 Pt 5, 1133–1138. [Google Scholar]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Glycogen synthase kinase-3 is increased in white cells early in Alzheimer’s disease. Neurosci. Lett. 2005, 373, 1–4. [Google Scholar]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; Tanaka, T.; Tung, Y.C.; Braak, E.; Iqbal, K.; Grundke-Iqbal, I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 1997, 56, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Borrell, J.; Guaza, C.; Avila, J.; Lucas, J.J. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 2002, 83, 1529–1533. [Google Scholar] [CrossRef]
- Engel, T.; Hernandez, F.; Avila, J.; Lucas, J.J. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 2006, 26, 5083–5090. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Milman, A.; Weizman, A.; Pick, C.; Eldar-Finkelman, H. Rapid anti-depressive like activity of specific GSK-3 inhibitor, and its effect on beta-catenin in the mouse hippocampus. Biol. Psychiatry 2004, 55, 781–784. [Google Scholar] [CrossRef]
- Polter, A.; Beurel, E.; Yang, S.; Garner, R.; Song, L.; Miller, C.A.; Sweatt, J.D.; McMahon, L.; Bartolucci, A.A.; Li, X.; et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology 2010, 35, 1761–1774. [Google Scholar] [CrossRef]
- Mines, M.A.; Yuskaitis, C.J.; King, M.K.; Beurel, E.; Jope, R.S. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS ONE 2010, 5, e9706. [Google Scholar] [CrossRef]
- Du, J.; Wei, Y.; Liu, L.; Wang, Y.; Khairova, R.; Blumenthal, R.; Tragon, T.; Hunsberger, J.G.; Machado-Vieira, R.; Drevets, W.; et al. A kinesin signaling complex mediates the ability of GSK-3beta to affect mood-associated behaviors. Proc. Natl. Acad. Sci. USA 2010, 107, 11573–11578. [Google Scholar] [CrossRef]
- Emamian, E.S.; Hall, D.; Birnbaum, M.J.; Karayiorgou, M.; Gogos, J.A. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 2004, 36, 131–137. [Google Scholar] [CrossRef]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Roca, C.; Campillo, N.E. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2016–2019). Expert Opin. Ther. Pat. 2020, 30, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Martinez, A. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2014–2015). Expert Opin. Ther. Pat. 2017, 27, 657–666. [Google Scholar] [CrossRef] [PubMed]
- King, M.K.; Pardo, M.; Cheng, Y.; Downey, K.; Jope, R.S.; Beurel, E. Glycogen synthase kinase-3 inhibitors: Rescuers of cognitive impairments. Pharmacol. Ther. 2014, 141, 1–12. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Tolosa, E.; Litvan, I.; Hoglinger, G.U.; Burn, D.; Lees, A.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; Del Ser, T.; Investigators, T. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 2014, 29, 470–478. [Google Scholar] [CrossRef]
- Dent, E.W.; Gertler, F.B. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 2003, 40, 209–227. [Google Scholar] [CrossRef]
- Lansbergen, G.; Akhmanova, A. Microtubule plus end: A hub of cellular activities. Traffic 2006, 7, 499–507. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. From signaling pathways to microtubule dynamics: The key players. Curr. Opin. Cell Biol. 2010, 22, 104–111. [Google Scholar] [CrossRef]
- Eira, J.; Silva, C.S.; Sousa, M.M.; Liz, M.A. The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog. Neurobiol. 2016, 141, 61–82. [Google Scholar] [CrossRef] [PubMed]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, N.; Marsh, P.; Goold, R.G.; Wood-Kaczmar, A.; Gordon-Weeks, P.R. Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J. Cell Sci. 2005, 118 Pt 5, 993–1005. [Google Scholar] [CrossRef]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [PubMed]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef]
- Zhou, F.Q.; Zhou, J.; Dedhar, S.; Wu, Y.H.; Snider, W.D. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron 2004, 42, 897–912. [Google Scholar] [CrossRef]
- Shi, S.H.; Cheng, T.; Jan, L.Y.; Jan, Y.N. APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr. Biol. 2004, 14, 2025–2032. [Google Scholar] [CrossRef]
- Liu, C.M.; Hur, E.M.; Zhou, F.Q. Coordinating Gene Expression and Axon Assembly to Control Axon Growth: Potential Role of GSK3 Signaling. Front. Mol. Neurosci. 2012, 5, 3. [Google Scholar] [CrossRef]
- Zhou, F.Q.; Snider, W.D. Cell biology. GSK-3beta and microtubule assembly in axons. Science 2005, 308, 211–214. [Google Scholar] [CrossRef]
- Morfini, G.; Szebenyi, G.; Elluru, R.; Ratner, N.; Brady, S.T. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002, 21, 281–293. [Google Scholar] [CrossRef]
- Pigino, G.; Morfini, G.; Pelsman, A.; Mattson, M.P.; Brady, S.T.; Busciglio, J. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J. Neurosci. 2003, 23, 4499–4508. [Google Scholar] [CrossRef] [PubMed]
- Dolma, K.; Iacobucci, G.J.; Hong Zheng, K.; Shandilya, J.; Toska, E.; White, J.A., 2nd; Spina, E.; Gunawardena, S. Presenilin influences glycogen synthase kinase-3 beta (GSK-3beta) for kinesin-1 and dynein function during axonal transport. Hum. Mol. Genet. 2014, 23, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.M.; Zhou, F.Q. GSK3 signalling in neural development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Reynolds, C.H.; Latimer, D.; Davis, D.R.; Anderton, B.H.; Gallo, J.M.; Hanger, D.; Mulot, S.; Marquardt, B.; Stabel, S.; et al. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 1994, 4, 1077–1086. [Google Scholar] [CrossRef]
- Hanger, D.P.; Noble, W. Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation. Int. J. Alzheimers Dis. 2011, 2011, 352805. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef]
- Kosik, K.S.; Caceres, A. Tau protein and the establishment of an axonal morphology. J. Cell Sci. Suppl. 1991, 15, 69–74. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 2006, 45, 3134–3145. [Google Scholar] [CrossRef]
- Wagner, U.; Utton, M.; Gallo, J.M.; Miller, C.C. Cellular phosphorylation of tau by GSK-3 beta influences tau binding to microtubules and microtubule organisation. J. Cell Sci. 1996, 109 Pt 6, 1537–1543. [Google Scholar]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Plattner, F.; Angelo, M.; Giese, K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef]
- Baumann, K.; Mandelkow, E.M.; Biernat, J.; Piwnica-Worms, H.; Mandelkow, E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993, 336, 417–424. [Google Scholar] [CrossRef]
- Su, S.C.; Tsai, L.H. Cyclin-dependent kinases in brain development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 465–491. [Google Scholar] [CrossRef] [PubMed]
- Gomez de Barreda, E.; Perez, M.; Gomez Ramos, P.; de Cristobal, J.; Martin-Maestro, P.; Moran, A.; Dawson, H.N.; Vitek, M.P.; Lucas, J.J.; Hernandez, F.; et al. Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol. Dis. 2011, 37, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Selenica, M.L.; Jensen, H.S.; Larsen, A.K.; Pedersen, M.L.; Helboe, L.; Leist, M.; Lotharius, J. Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br. J. Pharmacol. 2007, 152, 959–979. [Google Scholar] [CrossRef] [PubMed]
- Culbert, A.A.; Brown, M.J.; Frame, S.; Hagen, T.; Cross, D.A.; Bax, B.; Reith, A.D. GSK-3 inhibition by adenoviral FRAT1 overexpression is neuroprotective and induces Tau dephosphorylation and beta-catenin stabilisation without elevation of glycogen synthase activity. FEBS Lett. 2001, 507, 288–294. [Google Scholar] [CrossRef]
- Dickson, D.W.; Farrer, M.J. Tau kinases and Parkinson’s disease: Guilt by association? Ann. Neurol. 2005, 58, 819–820. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau and Parkinson disease. JAMA 2001, 286, 2324–2326. [Google Scholar] [CrossRef]
- Wood, H. Neurodegenerative disease: Tau is linked to cognitive decline in Huntington disease. Nat. Rev. Neurol. 2015, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Gaisina, I.N.; Petukhov, P.A.; Sridhar, J.; King, L.T.; Blond, S.Y.; Duka, T.; Rusnak, M.; Sidhu, A. Highly potent and specific GSK-3beta inhibitors that block tau phosphorylation and decrease alpha-synuclein protein expression in a cellular model of Parkinson’s disease. ChemMedChem 2006, 1, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Albert, I.; Porfiri, E.; Fiol, C.; Munemitsu, S.; Polakis, P. Binding of GSK-3 beta to the APC-beta-catenin complex and regulation of complex assembly. Science 1996, 272, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Farr, G.H., 3rd; Ferkey, D.M.; Yost, C.; Pierce, S.B.; Weaver, C.; Kimelman, D. Interaction among GSK-3, GBP, axin, and APC in Xenopus axis specification. J. Cell Biol. 2000, 148, 691–702. [Google Scholar] [CrossRef]
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef]
- Akhmanova, A.; Hoogenraad, C.C. Microtubule plus-end-tracking proteins: Mechanisms and functions. Curr. Opin. Cell Biol. 2005, 17, 47–54. [Google Scholar] [CrossRef]
- Reilein, A.; Nelson, W.J. APC is a component of an organizing template for cortical microtubule networks. Nat. Cell Biol. 2005, 7, 463–473. [Google Scholar] [CrossRef]
- Barth, A.I.; Caro-Gonzalez, H.Y.; Nelson, W.J. Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin Cell Dev. Biol. 2008, 19, 245–251. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Boeda, B.; Etienne-Manneville, S. APC binds intermediate filaments and is required for their reorganization during cell migration. J. Cell Biol. 2013, 200, 249–258. [Google Scholar] [CrossRef]
- Hart, M.J.; de los Santos, R.; Albert, I.N.; Rubinfeld, B.; Polakis, P. Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr. Biol. 1998, 8, 573–581. [Google Scholar] [CrossRef]
- Zumbrunn, J.; Kinoshita, K.; Hyman, A.A.; Nathke, I.S. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr. Biol. 2001, 11, 44–49. [Google Scholar] [CrossRef]
- Asada, N.; Sanada, K. LKB1-mediated spatial control of GSK3beta and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing neocortex. J. Neurosci. 2010, 30, 8852–8865. [Google Scholar] [CrossRef]
- Wang, L.H.; Strittmatter, S.M. A family of rat CRMP genes is differentially expressed in the nervous system. J. Neurosci. 1996, 16, 6197–6207. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Yoshimura, T.; Tsuboi, D.; Kawabata, S.; Kaneko-Kawano, T.; Shirataki, H.; Takenawa, T.; Kaibuchi, K. CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Mol. Cell. Biol. 2005, 25, 9920–9935. [Google Scholar] [CrossRef] [PubMed]
- Charrier, E.; Reibel, S.; Rogemond, V.; Aguera, M.; Thomasset, N.; Honnorat, J. Collapsin response mediator proteins (CRMPs): Involvement in nervous system development and adult neurodegenerative disorders. Mol. Neurobiol. 2003, 28, 51–64. [Google Scholar] [CrossRef]
- Niwa, S.; Nakamura, F.; Tomabechi, Y.; Aoki, M.; Shigematsu, H.; Matsumoto, T.; Yamagata, A.; Fukai, S.; Hirokawa, N.; Goshima, Y.; et al. Structural basis for CRMP2-induced axonal microtubule formation. Sci. Rep. 2017, 7, 10681. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Itoh, T.J.; Kimura, T.; Menager, C.; Nishimura, T.; Shiromizu, T.; Watanabe, H.; Inagaki, N.; Iwamatsu, A.; Hotani, H.; et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 2002, 4, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ohshima, T.; Sasaki, Y.; Suzuki, H.; Yanai, S.; Yamashita, N.; Nakamura, F.; Takei, K.; Ihara, Y.; Mikoshiba, K.; et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 2005, 10, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Eickholt, B.J.; Walsh, F.S.; Doherty, P. An inactive pool of GSK-3 at the leading edge of growth cones is implicated in Semaphorin 3A signaling. J. Cell Biol. 2002, 157, 211–217. [Google Scholar] [CrossRef]
- Cole, A.R.; Noble, W.; van Aalten, L.; Plattner, F.; Meimaridou, R.; Hogan, D.; Taylor, M.; LaFrancois, J.; Gunn-Moore, F.; Verkhratsky, A.; et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem. 2007, 103, 1132–1144. [Google Scholar] [CrossRef]
- Fang, W.; Gao, G.; Zhao, H.; Xia, Y.; Guo, X.; Li, N.; Li, Y.; Yang, Y.; Chen, L.; Wang, Q.; et al. Role of the Akt/GSK-3beta/CRMP-2 pathway in axon degeneration of dopaminergic neurons resulting from MPP+ toxicity. Brain Res. 2015, 1602, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lim, N.K.; Hung, L.W.; Pang, T.Y.; McLean, C.A.; Liddell, J.R.; Hilton, J.B.; Li, Q.X.; White, A.R.; Hannan, A.J.; Crouch, P.J. Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington’s disease affected brain. Hum. Mol. Genet. 2014, 23, 4051–4063. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.; Leidel, C.; Szpankowski, L.; Farley, N.M.; Shubeita, G.T.; Goldstein, L.S. Endogenous GSK-3/shaggy regulates bidirectional axonal transport of the amyloid precursor protein. Traffic 2013, 14, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Torroja, L.; Packard, M.; Gorczyca, M.; White, K.; Budnik, V. The Drosophila beta-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J. Neurosci. 1999, 19, 7793–7803. [Google Scholar] [CrossRef] [PubMed]
- Ally, S.; Larson, A.G.; Barlan, K.; Rice, S.E.; Gelfand, V.I. Opposite-polarity motors activate one another to trigger cargo transport in live cells. J Cell Biol. 2009, 187, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Levi, V.; Serpinskaya, A.S.; Gratton, E.; Gelfand, V. Organelle transport along microtubules in Xenopus melanophores: Evidence for cooperation between multiple motors. Biophys. J. 2006, 90, 318–327. [Google Scholar] [CrossRef]
- Kimura, T.; Watanabe, H.; Iwamatsu, A.; Kaibuchi, K. Tubulin and CRMP-2 complex is transported via Kinesin-1. J. Neurochem. 2005, 93, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Liz, M.A.; Mar, F.M.; Santos, T.E.; Pimentel, H.I.; Marques, A.M.; Morgado, M.M.; Vieira, S.; Sousa, V.F.; Pemble, H.; Wittmann, T.; et al. Neuronal deletion of GSK3beta increases microtubule speed in the growth cone and enhances axon regeneration via CRMP-2 and independently of MAP1B and CLASP2. BMC Biol. 2014, 12, 47. [Google Scholar] [CrossRef]
- Leibinger, M.; Hilla, A.M.; Andreadaki, A.; Fischer, D. GSK3-CRMP2 signaling mediates axonal regeneration induced by Pten knockout. Commun. Biol. 2019, 2, 318. [Google Scholar] [CrossRef]
- Petratos, S.; Ozturk, E.; Azari, M.F.; Kenny, R.; Lee, J.Y.; Magee, K.A.; Harvey, A.R.; McDonald, C.; Taghian, K.; Moussa, L.; et al. Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation. Brain 2012, 135 Pt 6, 1794–1818. [Google Scholar] [CrossRef]
- Dupree, J.L.; Polak, P.E.; Hensley, K.; Pelligrino, D.; Feinstein, D.L. Lanthionine ketimine ester provides benefit in a mouse model of multiple sclerosis. J. Neurochem. 2015, 134, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, H.; Li, J.; Zhang, Y.; Han, B.; Zeng, Z.; Qiao, N.; Cui, X.; Lou, J.; Li, J. Amelioration of beta-amyloid-induced cognitive dysfunction and hippocampal axon degeneration by curcumin is associated with suppression of CRMP-2 hyperphosphorylation. Neurosci. Lett. 2013, 557 Pt B, 112–117. [Google Scholar] [CrossRef]
- Khazaei, M.R.; Girouard, M.P.; Alchini, R.; Ong Tone, S.; Shimada, T.; Bechstedt, S.; Cowan, M.; Guillet, D.; Wiseman, P.W.; Brouhard, G.; et al. Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J. Biol. Chem. 2014, 289, 30133–30143. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Bernard-Marissal, N.; Vourc’h, P.; Guettard, Y.O.; Sunyach, C.; Augereau, O.; Khederchah, J.; Mouzat, K.; Antar, C.; Gordon, P.H.; et al. A rare motor neuron deleterious missense mutation in the DPYSL3 (CRMP4) gene is associated with ALS. Hum. Mutat. 2013, 34, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Duplan, L.; Bernard, N.; Casseron, W.; Dudley, K.; Thouvenot, E.; Honnorat, J.; Rogemond, V.; De Bovis, B.; Aebischer, P.; Marin, P.; et al. Collapsin response mediator protein 4a (CRMP4a) is upregulated in motoneurons of mutant SOD1 mice and can trigger motoneuron axonal degeneration and cell death. J. Neurosci. 2010, 30, 785–796. [Google Scholar] [CrossRef]
- Salinas, S.; Bilsland, L.G.; Schiavo, G. Molecular landmarks along the axonal route: Axonal transport in health and disease. Curr. Opin. Cell Biol. 2008, 20, 445–453. [Google Scholar] [CrossRef]
- Encalada, S.E.; Goldstein, L.S. Biophysical challenges to axonal transport: Motor-cargo deficiencies and neurodegeneration. Annu. Rev. Biophys. 2014, 43, 141–169. [Google Scholar] [CrossRef]
- Stokin, G.B.; Goldstein, L.S. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 2006, 75, 607–627. [Google Scholar] [CrossRef]
- Kieran, D.; Hafezparast, M.; Bohnert, S.; Dick, J.R.; Martin, J.; Schiavo, G.; Fisher, E.M.; Greensmith, L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J. Cell Biol. 2005, 169, 561–567. [Google Scholar] [CrossRef]
- McGuire, J.R.; Rong, J.; Li, S.H.; Li, X.J. Interaction of Huntingtin-associated protein-1 with kinesin light chain: Implications in intracellular trafficking in neurons. J. Biol. Chem. 2006, 281, 3552–3559. [Google Scholar] [CrossRef]
- Goldstein, L.S. Molecular motors: From one motor many tails to one motor many tales. Trends Cell Biol. 2001, 11, 477–482. [Google Scholar] [CrossRef]
- Mokhtar, S.H.; Kim, M.J.; Magee, K.A.; Aui, P.M.; Thomas, S.; Bakhuraysah, M.M.; Alrehaili, A.A.; Lee, J.Y.; Steer, D.L.; Kenny, R.; et al. Amyloid-beta-dependent phosphorylation of collapsin response mediator protein-2 dissociates kinesin in Alzheimer’s disease. Neural Regen. Res. 2018, 13, 1066–1080. [Google Scholar] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.; Pollak, M.; Sonenberg, N. Current status and challenges associated with targeting mTOR for cancer therapy. BioDrugs 2009, 23, 77–91. [Google Scholar] [CrossRef]
- Proud, C.G. mTOR-mediated regulation of translation factors by amino acids. Biochem. Biophys. Res. Commun. 2004, 313, 429–436. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Klionsky, D. An overview of autophagy: Morphology, mechanism and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Walker, S.A.; Ktistakis, N.T. Autophagosome Biogenesis Machinery. J. Mol. Biol. 2020, 432, 2449–2461. [Google Scholar] [CrossRef]
- Tang, B.L. mTOR, autophagy, and reprogramming. Front. Cell Dev. Biol. 2013, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Boya, P. Lysosomal Function and Dysfunction: Mechanism and Disease. Antioxid. Redox Signal. 2012, 17, 766–774. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Skalecka, A.; Liszewska, E.; Bilinski, R.; Gkogkas, C.; Khoutorsky, A.; Malik, A.R.; Sonenberg, N.; Jaworski, J. mTOR kinase is needed for the development and stabilization of dendritic arbors in newly born olfactory bulb neurons. Dev. Neurobiol. 2016, 76, 1308–1327. [Google Scholar] [CrossRef]
- Lipton, J.O.; Sahin, M. The neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [PubMed]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134 Pt 1, 258–277. [Google Scholar] [CrossRef]
- Lee, J.A.; Gao, F.B. Roles of ESCRT in autophagy-associated neurodegeneration. Autophagy 2008, 4, 230–232. [Google Scholar] [CrossRef]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef]
- Tsvetkov, A.S.; Miller, J.; Arrasate, M.; Wong, J.S.; Pleiss, M.A.; Finkbeiner, S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc. Natl. Acad. Sci. USA 2010, 107, 16982–16987. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Bove, J.; Martinez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26C, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.L.; Matus, S.; Bargsted, L.; Hetz, C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 2014, 35, 583–591. [Google Scholar] [CrossRef]
- Welsh, G.I.; Proud, C.G. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem. J. 1993, 294, 625–629. [Google Scholar] [CrossRef]
- Karyo, R.; Eskira, Y.; Pinhasov, A.; Belmaker, R.; Agam, G.; Eldar-Finkelman, H. Identification of eukaryotic elongation factor-2 as a novel cellular target of lithium and glycogen synthase kinase-3. Mol. Cell. Neurosci. 2010, 45, 449–455. [Google Scholar] [CrossRef]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Azoulay-Alfaguter, I.; Elya, R.; Avrahami, L.; Katz, A.; Eldar-Finkelman, H. Combined regulation of mTORC1 and lysosomal acidification by GSK-3 suppresses autophagy and contributes to cancer cells growth. Oncogene 2015, 34, 4613–4623. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, L.; Paz, R.; Dominko, K.; Hecimovic, S.; Bucci, C.; Eldar-Finkelman, H. GSK-3-TSC axis governs lysosomal acidification through autophagy and endocytic pathways. Cell. Signal. 2020, 71, 109597. [Google Scholar] [CrossRef]
- Shin, S.; Wolgamott, L.; Yu, Y.; Blenis, J.; Yoon, S.O. Glycogen synthase kinase (GSK)-3 promotes p70 ribosomal protein S6 kinase (p70S6K) activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2011, 108, E1204–E1213. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Cao, L. Starvation signals in yeast are integrated to coordinate metabolic reprogramming and stress response to ensure longevity. Curr. Genet. 2017, 63, 839–843. [Google Scholar] [CrossRef]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of GSK-3 Ameliorates beta-Amyloid (A-beta) Pathology and Restores Lysosomal Acidification and mTOR Activity in the Alzheimer’s Disease Mouse Model. In vivo and In vitro Studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef]
- Guo, D.; Shen, Y.; Li, W.; Li, Q.; Zhao, Y.; Pan, C.; Chen, B.; Zhong, Y.; Miao, Y. 6-Bromoindirubin-3′-Oxime (6BIO) Suppresses the mTOR Pathway, Promotes Autophagy, and Exerts Anti-aging Effects in Rodent Liver. Front. Pharmacol. 2019, 10, 320. [Google Scholar] [CrossRef]
- Parr, C.; Carzaniga, R.; Gentleman, S.M.; Van Leuven, F.; Walter, J.; Sastre, M. Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-beta precursor protein. Mol. Cell. Biol. 2012, 32, 4410–4418. [Google Scholar] [CrossRef]
- Xiong, T.; Qu, Y.; Wang, H.; Chen, H.; Zhu, J.; Zhao, F.; Zou, R.; Zhang, L.; Mu, D. GSK-3beta/mTORC1 Couples Synaptogenesis and Axonal Repair to Reduce Hypoxia Ischemia-Mediated Brain Injury in Neonatal Rats. J. Neuropathol. Exp. Neurol. 2018, 77, 383–394. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, D.; Li, S.; Zhang, Y.; Wang, C. Underlying mechanisms of recombinant adeno-associated virus-mediated bicaudal C homolog 1 overexpression in the medial prefrontal cortex of mice with induced depressive-like behaviors. Brain Res. Bull. 2019, 150, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Zhang, J.X.; Chan, C.C.; Yuen, K.S.; Chu, L.W.; Cheung, R.T.; Chan, Y.S.; Fox, P.T.; Gao, J.H. Age-related differences in response regulation as revealed by functional MRI. Brain Res. 2006, 1076, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; DeMuth, T.M.; Miller, K.N.; Pugh, T.D.; Polewski, M.A.; Colman, R.J.; Eliceiri, K.W.; Beasley, T.M.; Johnson, S.C.; Anderson, R.M. Regional metabolic heterogeneity of the hippocampus is nonuniformly impacted by age and caloric restriction. Aging Cell 2016, 15, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Wu, X.; Mao, Z.; Khuri, F.R.; Sun, S.Y. Rictor Undergoes Glycogen Synthase Kinase 3 (GSK3)-dependent, FBXW7-mediated Ubiquitination and Proteasomal Degradation. J. Biol. Chem. 2015, 290, 14120–14129. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Manfredi, G. Neuronal degeneration and mitochondrial dysfunction. J. Clin. Investig. 2003, 111, 303–312. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Guardia-Laguarta, C.; Schon, E.A.; Przedborski, S. Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J. Clin. Investig. 2019, 129, 34–45. [Google Scholar] [CrossRef]
- Swerdlow, N.S.; Wilkins, H.M. Mitophagy and the Brain. Int. J. Mol. Sci. 2020, 21, 9661. [Google Scholar] [CrossRef]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef]
- Olson, B.L.; Hock, M.B.; Ekholm-Reed, S.; Wohlschlegel, J.A.; Dev, K.K.; Kralli, A.; Reed, S.I. SCFCdc4 acts antagonistically to the PGC-1alpha transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008, 22, 252–264. [Google Scholar] [CrossRef]
- Martin, S.A.; Souder, D.C.; Miller, K.N.; Clark, J.P.; Sagar, A.K.; Eliceiri, K.W.; Puglielli, L.; Beasley, T.M.; Anderson, R.M. GSK3beta Regulates Brain Energy Metabolism. Cell Rep. 2018, 23, 1922–1931.e4. [Google Scholar] [CrossRef]
- Yan, J.; Liu, X.H.; Han, M.Z.; Wang, Y.M.; Sun, X.L.; Yu, N.; Li, T.; Su, B.; Chen, Z.Y. Blockage of GSK3beta-mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 211–227. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef]
- Tanno, M.; Kuno, A.; Ishikawa, S.; Miki, T.; Kouzu, H.; Yano, T.; Murase, H.; Tobisawa, T.; Ogasawara, M.; Horio, Y.; et al. Translocation of glycogen synthase kinase-3beta (GSK-3beta), a trigger of permeability transition, is kinase activity-dependent and mediated by interaction with voltage-dependent anion channel 2 (VDAC2). J. Biol. Chem. 2014, 289, 29285–29296. [Google Scholar] [CrossRef]
- Linseman, D.A.; Butts, B.D.; Precht, T.A.; Phelps, R.A.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Florez-McClure, M.L.; Heidenreich, K.A. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004, 24, 9993–10002. [Google Scholar] [CrossRef]
- Pinton, P. Mitochondria-associated membranes (MAMs) and pathologies. Cell Death Dis. 2018, 9, 413. [Google Scholar] [CrossRef]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Llorens-Martin, M.; Lopez-Domenech, G.; Soriano, E.; Avila, J. GSK3beta is involved in the relief of mitochondria pausing in a Tau-dependent manner. PLoS ONE 2011, 6, e27686. [Google Scholar] [CrossRef]
- Morel, M.; Authelet, M.; Dedecker, R.; Brion, J.P. Glycogen synthase kinase-3beta and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience 2010, 167, 1044–1056. [Google Scholar] [CrossRef]
- Chen, S.; Owens, G.C.; Crossin, K.L.; Edelman, D.B. Serotonin stimulates mitochondrial transport in hippocampal neurons. Mol. Cell. Neurosci. 2007, 36, 472–483. [Google Scholar] [CrossRef]
- Jimenez-Mateos, E.M.; Gonzalez-Billault, C.; Dawson, H.N.; Vitek, M.P.; Avila, J. Role of MAP1B in axonal retrograde transport of mitochondria. Biochem. J. 2006, 397, 53–59. [Google Scholar] [CrossRef]
- Saraswati, A.P.; Ali Hussaini, S.M.; Krishna, N.H.; Babu, B.N.; Kamal, A. Glycogen synthase kinase-3 and its inhibitors: Potential target for various therapeutic conditions. Eur. J. Med. Chem. 2018, 144, 843–858. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. Protein kinases. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C.L. Research on resistance to cancer drug Gleevec. Science 2001, 294, 1834. [Google Scholar] [CrossRef]
- Lo Monte, F.; Kramer, T.; Gu, J.; Anumala, U.R.; Marinelli, L.; La Pietra, V.; Novellino, E.; Franco, B.; Demedts, D.; Van Leuven, F.; et al. Identification of glycogen synthase kinase-3 inhibitors with a selective sting for glycogen synthase kinase-3alpha. J. Med. Chem. 2012, 55, 4407–4424. [Google Scholar] [CrossRef]
- Neumann, T.; Benajiba, L.; Goring, S.; Stegmaier, K.; Schmidt, B. Evaluation of Improved Glycogen Synthase Kinase-3alpha Inhibitors in Models of Acute Myeloid Leukemia. J. Med. Chem. 2015, 58, 8907–8919. [Google Scholar] [CrossRef]
- Vignaux, P.A.; Minerali, E.; Foil, D.H.; Puhl, A.C.; Ekins, S. Machine Learning for Discovery of GSK3beta Inhibitors. ACS Omega 2020, 5, 26551–26561. [Google Scholar] [CrossRef]
- Martinez, A.; Perez, D.I. GSK-3 inhibitors: A ray of hope for the treatment of Alzheimer’s disease? J. Alzheimers Dis. 2008, 15, 181–191. [Google Scholar] [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Perez, C.; Moreno, F.J. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef]
- Balasubramaniam, M.; Mainali, N.; Bowroju, S.; Atluri, P.; Penthala, N.; Ayyadevera, S.; Crooks, P.; Reis, R. Structural modeling of GSK3β implicates the inactive (DFG-out) conformation as the target bound by TDZD analogs. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fujishiro, H.; Takechi, H. Efficacy and Safety of Glycogen Synthase Kinase 3 Inhibitors for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2019, 69, 1031–1039. [Google Scholar] [CrossRef]
- Horrigan, J.; Gomes, T.B.; Snape, M.; Nikolenko, N.; McMorn, A.; Evans, S.; Yaroshinsky, A.; Della Pasqua, O.; Oosterholt, S.; Lochmuller, H. A Phase 2 Study of AMO-02 (Tideglusib) in Congenital and Childhood-Onset Myotonic Dystrophy Type 1 (DM1). Pediatr. Neurol. 2020, 112, 84–93. [Google Scholar] [CrossRef]
- Ilouz, R.; Kowelsman, N.; Eisenstein, M.; Eldar-Finkelman, H. Identification of novel glycogen synthase kinase-3beta substrate-interacting residues suggests a common mechanism for substrate recognition. J. Biol. Chem. 2006, 281, 30621–30630. [Google Scholar] [CrossRef]
- Licht-Murava, A.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. Elucidating substrate and inhibitor binding sites on the surface of GSK-3beta and the refinement of a competitive inhibitor. J. Mol. Biol. 2011, 408, 366–378. [Google Scholar] [CrossRef]
- Licht-Murava, A.; Paz, R.; Vaks, L.; Avrahami, L.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. A unique type of GSK-3 inhibitor brings new opportunities to the clinic. Sci. Signal. 2016, 9, ra110. [Google Scholar] [CrossRef]
- Chen, G.; Bower, K.A.; Ma, C.; Fang, S.; Thiele, C.J.; Luo, J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004, 18, 1162–1164. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, J.; Han, S.; Liu, J.; Holzbeierlein, J.; Thrasher, J.B.; Li, B. Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate 2011, 71, 835–845. [Google Scholar] [CrossRef]
- Pardo, M.; Cheng, Y.; Velmeshev, D.; Magistri, M.; Eldar-Finkelman, H.; Martinez, A.; Faghihi, M.A.; Jope, R.S.; Beurel, E. Intranasal siRNA administration reveals IGF2 deficiency contributes to impaired cognition in Fragile X syndrome mice. JCI Insight 2017, 2, e91782. [Google Scholar] [CrossRef]
- Beurel, E.; Kaidanovich-Beilin, O.; Yeh, W.I.; Song, L.; Palomo, V.; Michalek, S.M.; Woodgett, J.R.; Harrington, L.E.; Eldar-Finkelman, H.; Martinez, A.; et al. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J. Immunol. 2013, 190, 5000–5011. [Google Scholar] [CrossRef]
- Rippin, I.; Khazanov, N.; Shirley Ben, J.; Kudinov, T.; Berent, E.; Arciniegas Ruiz, S.M.; Marciano, D.; Levy, L.; Gruzman, A.; Senderowitz, H.; et al. Discovery and Design of Novel Small Molecule GSK-3 Inhibitors Targeting the Substrate Binding Site. Int. J. Mol. Sci. 2020, 21, 8709. [Google Scholar] [CrossRef]
- Zhou, Y.; Peng, Z.; Seven, E.S.; Leblanc, R.M. Crossing the blood-brain barrier with nanoparticles. J. Control. Release 2018, 270, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rippin, I.; Eldar-Finkelman, H. Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration. Cells 2021, 10, 262. https://doi.org/10.3390/cells10020262
Rippin I, Eldar-Finkelman H. Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration. Cells. 2021; 10(2):262. https://doi.org/10.3390/cells10020262
Chicago/Turabian StyleRippin, Ido, and Hagit Eldar-Finkelman. 2021. "Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration" Cells 10, no. 2: 262. https://doi.org/10.3390/cells10020262
APA StyleRippin, I., & Eldar-Finkelman, H. (2021). Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration. Cells, 10(2), 262. https://doi.org/10.3390/cells10020262