
LIM Kinases in Osteosarcoma Development



Abstract
:
1. Introduction
2. LIMKs and Cancer
2.1. The LIMK Family
2.1.1. Protein Structure
2.1.2. LIMKs and Actin Cytoskeleton or MT Rearrangement
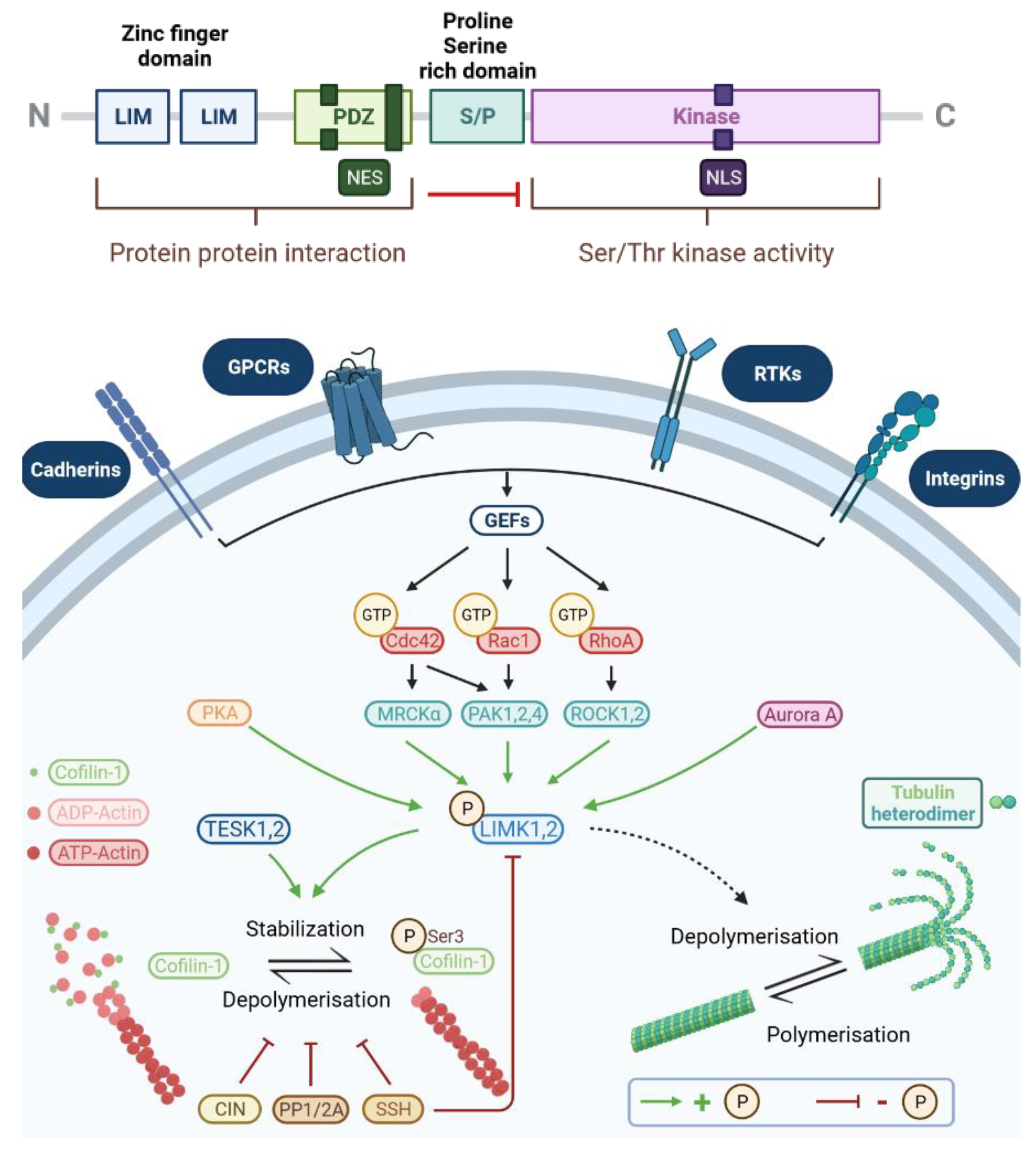
2.1.3. Activation of LIMKs
2.2. LIMKs and Cancer
2.2.1. LIMKs and Cell Cycle
2.2.2. LIMKs and Cell Migration
3. LIMKs and Primary Bone Tumour
3.1. LIMKs and Bone Remodeling
3.1.1. Brief Overview of Bone Remodelling
- (a)
- Osteoblastogenesis
- (b)
- Osteoclastogenesis
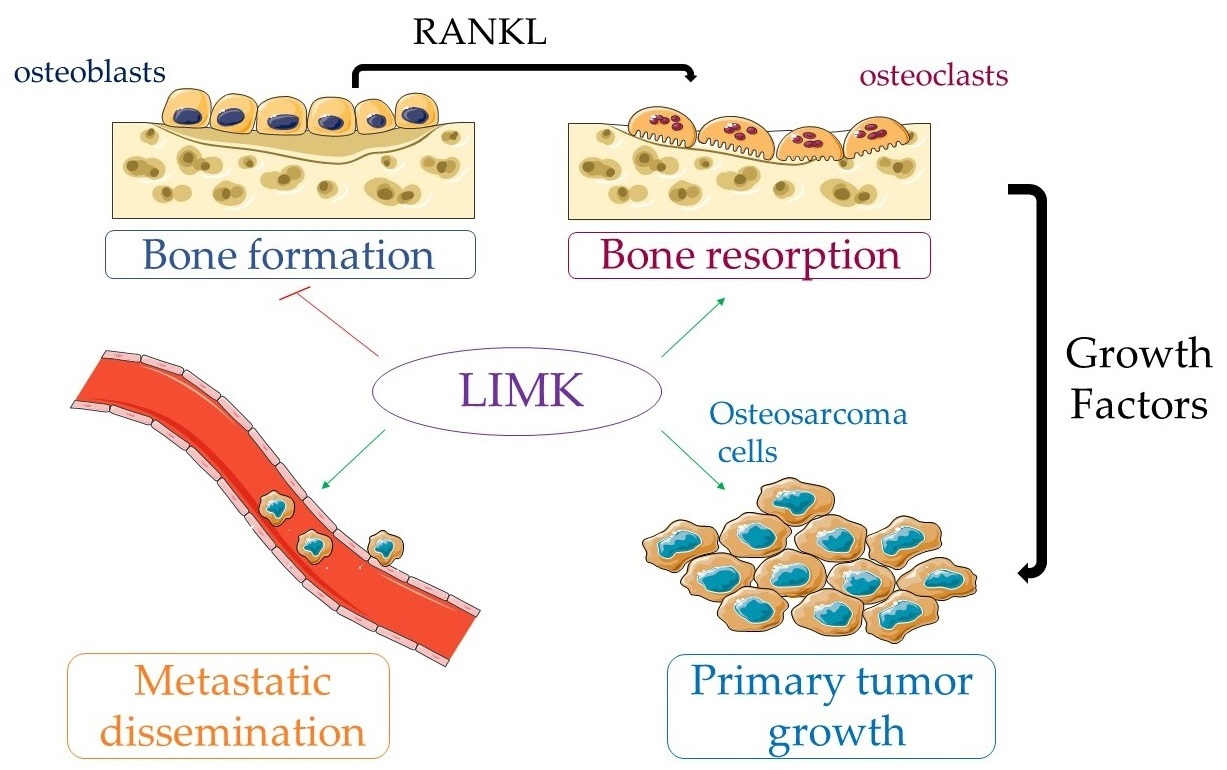
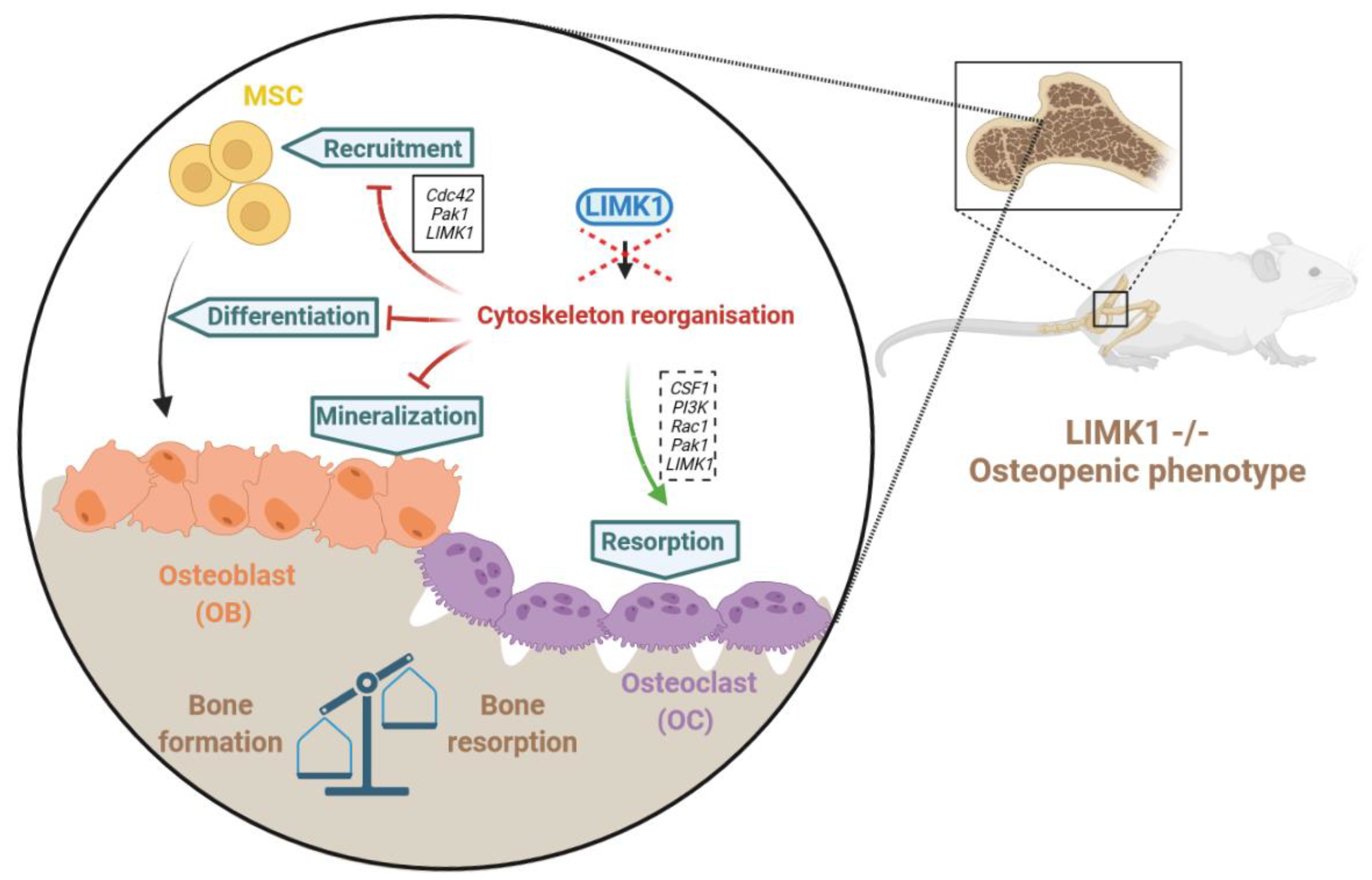
3.1.2. LIMKs and Bone Remodelling
3.2. LIMKs and Pediatric Bone Tumors
3.2.1. Osteosarcoma (OS)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Osteosarcoma | |
|---|---|
| Paediatric bone tumor ranking | 1st |
| Incidence (worldwide) | 3.4 per million per year |
| Age of patients | 18 |
| Preferential localization | Long bones |
| Cellular origin | Mesenchymal cells or osteoblast |
| Histology | Osteoblastic/chondroblastic/fibroblastic |
| Main metastases localization | lungs |
| Treatment | Chemotherapies/chirurgical resection/chemotherapies |
| 5-year survival | 70/75% (localised form) or 20/25% (metastatic form) |
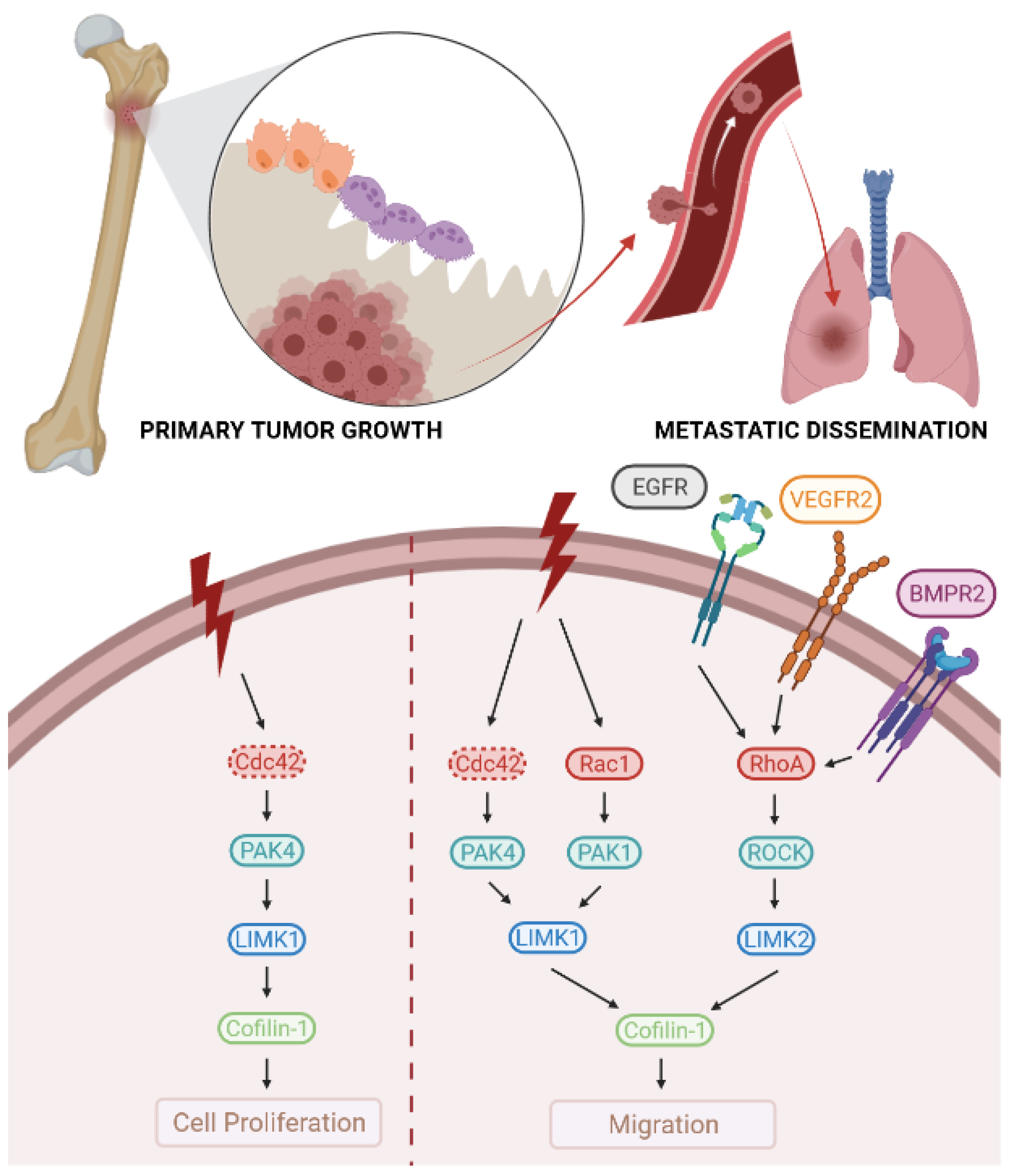
3.2.2. LIMKs and OS
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gaspar, N.; Occean, B.-V.; Pacquement, H.; Bompas, E.; Bouvier, C.; Brisse, H.J.; Castex, M.-P.; Cheurfa, N.; Corradini, N.; Delaye, J.; et al. Results of Methotrexate-Etoposide-Ifosfamide Based Regimen (M-EI) in Osteosarcoma Patients Included in the French OS2006/Sarcome-09 Study. Eur. J. Cancer 2018, 88, 57–66. [Google Scholar] [CrossRef]
- Heare, T.; Hensley, M.A.; Dell’Orfano, S. Bone Tumors: Osteosarcoma and Ewing’s Sarcoma. Curr. Opin. Pediatr. 2009, 21, 365–372. [Google Scholar] [CrossRef]
- Moore, D.D.; Luu, H.H. Osteosarcoma. Cancer Treat. Res. 2014, 162, 65–92. [Google Scholar] [CrossRef]
- Kempf-Bielack, B.; Bielack, S.S.; Jürgens, H.; Branscheid, D.; Berdel, W.E.; Exner, G.U.; Göbel, U.; Helmke, K.; Jundt, G.; Kabisch, H.; et al. Osteosarcoma Relapse after Combined Modality Therapy: An Analysis of Unselected Patients in the Cooperative Osteosarcoma Study Group (COSS). J. Clin. Oncol. 2005, 23, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.C.; Gill, G.N. Structural Features of LIM Kinase That Control Effects on the Actin Cytoskeleton. J. Biol. Chem. 1999, 274, 11352–11361. [Google Scholar] [CrossRef] [Green Version]
- Stanyon, C.A.; Bernard, O. LIM-Kinase1. Int. J. Biochem. Cell Biol. 1999, 31, 389–394. [Google Scholar] [CrossRef]
- Bernard, O. Lim Kinases, Regulators of Actin Dynamics. Int. J. Biochem. Cell Biol. 2007, 39, 1071–1076. [Google Scholar] [CrossRef]
- Vallée, B.; Cuberos, H.; Doudeau, M.; Godin, F.; Gosset, D.; Vourc’h, P.; Andres, C.R.; Bénédetti, H. LIMK2-1, a New Isoform of Human LIMK2, Regulates Actin Cytoskeleton Remodeling via a Different Signaling Pathway than That of Its Two Homologs, LIMK2a and LIMK2b. Biochem. J. 2018, 475, 3745–3761. [Google Scholar] [CrossRef]
- Takahashi, H.; Koshimizu, U.; Nakamura, T. A Novel Transcript Encoding Truncated LIM Kinase 2 Is Specifically Expressed in Male Germ Cells Undergoing Meiosis. Biochem. Biophys. Res. Commun. 1998, 249, 138–145. [Google Scholar] [CrossRef]
- Røsok, O.; Pedeutour, F.; Ree, A.H.; Aasheim, H.C. Identification and Characterization of TESK2, a Novel Member of the LIMK/TESK Family of Protein Kinases, Predominantly Expressed in Testis. Genomics 1999, 61, 44–54. [Google Scholar] [CrossRef]
- Nagata, K.; Ohashi, K.; Yang, N.; Mizuno, K. The N-Terminal LIM Domain Negatively Regulates the Kinase Activity of LIM-Kinase 1. Biochem. J. 1999, 343 Pt 1, 99–105. [Google Scholar] [CrossRef]
- Tomiyoshi, G.; Horita, Y.; Nishita, M.; Ohashi, K.; Mizuno, K. Caspase-Mediated Cleavage and Activation of LIM-Kinase 1 and Its Role in Apoptotic Membrane Blebbing. Genes Cells 2004, 9, 591–600. [Google Scholar] [CrossRef]
- Hiraoka, J.; Okano, I.; Higuchi, O.; Yang, N.; Mizuno, K. Self-Association of LIM-Kinase 1 Mediated by the Interaction between an N-Terminal LIM Domain and a C-Terminal Kinase Domain. FEBS Lett. 1996, 399, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Mizuno, K. Nuclear Export of LIM-Kinase 1, Mediated by Two Leucine-Rich Nuclear-Export Signals within the PDZ Domain. Biochem. J. 1999, 338 Pt 3, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Higuchi, O.; Mizuno, K. Cytoplasmic Localization of LIM-Kinase 1 Is Directed by a Short Sequence within the PDZ Domain. Exp. Cell Res. 1998, 241, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Pandey, D.; Siess, W. Phosphorylation-Dependent Regulation of Unique Nuclear and Nucleolar Localization Signals of LIM Kinase 2 in Endothelial Cells. J. Biol. Chem. 2006, 281, 25223–25230. [Google Scholar] [CrossRef] [Green Version]
- Prunier, C.; Prudent, R.; Kapur, R.; Sadoul, K.; Lafanechère, L. LIM Kinases: Cofilin and Beyond. Oncotarget 2017, 8, 41749–41763. [Google Scholar] [CrossRef] [Green Version]
- Bamburg, J.R. Proteins of the ADF/Cofilin Family: Essential Regulators of Actin Dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230. [Google Scholar] [CrossRef]
- Bamburg, J.R.; Wiggan, O.P. ADF/Cofilin and Actin Dynamics in Disease. Trends Cell Biol. 2002, 12, 598–605. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Bamburg, J.R. ADF/Cofilin: A Functional Node in Cell Biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Moon, A.; Drubin, D.G. The ADF/Cofilin Proteins: Stimulus-Responsive Modulators of Actin Dynamics. Mol. Biol. Cell 1995, 6, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Van Troys, M.; Huyck, L.; Leyman, S.; Dhaese, S.; Vandekerkhove, J.; Ampe, C. Ins and Outs of ADF/Cofilin Activity and Regulation. Eur. J. Cell Biol. 2008, 87, 649–667. [Google Scholar] [CrossRef]
- Yang, X.; Yu, K.; Hao, Y.; Li, D.; Stewart, R.; Insogna, K.L.; Xu, T. LATS1 Tumour Suppressor Affects Cytokinesis by Inhibiting LIMK1. Nat. Cell Biol. 2004, 6, 609–617. [Google Scholar] [CrossRef]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of Actin Dynamics through Phosphorylation of Cofilin by LIM-Kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Mizuno, K. Signaling Mechanisms and Functional Roles of Cofilin Phosphorylation and Dephosphorylation. Cell. Signal. 2013, 25, 457–469. [Google Scholar] [CrossRef]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of Actin Reorganization by Slingshot, a Family of Phosphatases That Dephosphorylate ADF/Cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Gohla, A.; Birkenfeld, J.; Bokoch, G.M. Chronophin, a Novel HAD-Type Serine Protein Phosphatase, Regulates Cofilin-Dependent Actin Dynamics. Nat. Cell Biol. 2005, 7, 21–29. [Google Scholar] [CrossRef]
- Meberg, P.J.; Ono, S.; Minamide, L.S.; Takahashi, M.; Bamburg, J.R. Actin Depolymerizing Factor and Cofilin Phosphorylation Dynamics: Response to Signals That Regulate Neurite Extension. Cell Motil. Cytoskelet. 1998, 39, 172–190. [Google Scholar] [CrossRef]
- Soosairajah, J.; Maiti, S.; Wiggan, O.; Sarmiere, P.; Moussi, N.; Sarcevic, B.; Sampath, R.; Bamburg, J.R.; Bernard, O. Interplay between Components of a Novel LIM Kinase-Slingshot Phosphatase Complex Regulates Cofilin. EMBO J. 2005, 24, 473–486. [Google Scholar] [CrossRef]
- Shishkin, S.; Eremina, L.; Pashintseva, N.; Kovalev, L.; Kovaleva, M. Cofilin-1 and Other ADF/Cofilin Superfamily Members in Human Malignant Cells. Int. J. Mol. Sci. 2016, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Prudent, R.; Vassal-Stermann, E.; Nguyen, C.-H.; Pillet, C.; Martinez, A.; Prunier, C.; Barette, C.; Soleilhac, E.; Filhol, O.; Beghin, A.; et al. Pharmacological Inhibition of LIM Kinase Stabilizes Microtubules and Inhibits Neoplastic Growth. Cancer Res. 2012, 72, 4429–4439. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, K.; Li, R.; Soo, P.; Suryadinata, R.; Sarcevic, B.; Valova, V.A.; Graham, M.E.; Robinson, P.J.; Bernard, O. The Phosphorylation of P25/TPPP by LIM Kinase 1 Inhibits Its Ability to Assemble Microtubules. Exp. Cell Res. 2007, 313, 4091–4106. [Google Scholar] [CrossRef]
- Heng, Y.-W.; Lim, H.-H.; Mina, T.; Utomo, P.; Zhong, S.; Lim, C.-T.; Koh, C.-G. TPPP Acts Downstream of RhoA-ROCK-LIMK2 to Regulate Astral Microtubule Organization and Spindle Orientation. J. Cell Sci. 2012, 125, 1579–1590. [Google Scholar] [CrossRef] [Green Version]
- Schofield, A.V.; Steel, R.; Bernard, O. Rho-Associated Coiled-Coil Kinase (ROCK) Protein Controls Microtubule Dynamics in a Novel Signaling Pathway That Regulates Cell Migration. J. Biol. Chem. 2012, 287, 43620–43629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhu, Y.; Cao, Y.; Wang, Q.; Du, J.; Tian, J.; Liang, Y.; Ma, W. LIM Kinase Activity Is Required for Microtubule Organising Centre Positioning in Mouse Oocyte Meiosis. Reprod. Fertil. Dev. 2017, 29, 791–804. [Google Scholar] [CrossRef]
- Bishop, A.L.; Hall, A. Rho GTPases and Their Effector Proteins. Biochem. J. 2000, 348 Pt 2, 241–255. [Google Scholar] [CrossRef]
- Amano, T.; Tanabe, K.; Eto, T.; Narumiya, S.; Mizuno, K. LIM-Kinase 2 Induces Formation of Stress Fibres, Focal Adhesions and Membrane Blebs, Dependent on Its Activation by Rho-Associated Kinase-Catalysed Phosphorylation at Threonine-505. Biochem. J. 2001, 354, 149–159. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Shibuya, A.; Nakamura, T. Activation of LIM Kinases by Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinase Alpha. J. Biol. Chem. 2001, 276, 23092–23096. [Google Scholar] [CrossRef] [Green Version]
- Sumi, T.; Matsumoto, K.; Nakamura, T. Specific Activation of LIM Kinase 2 via Phosphorylation of Threonine 505 by ROCK, a Rho-Dependent Protein Kinase. J. Biol. Chem. 2001, 276, 670–676. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D.C.; Sanders, L.C.; Bokoch, G.M.; Gill, G.N. Activation of LIM-Kinase by Pak1 Couples Rac/Cdc42 GTPase Signalling to Actin Cytoskeletal Dynamics. Nat. Cell Biol. 1999, 1, 253–259. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the Actin Cytoskeleton through Protein Kinases ROCK and LIM-Kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef]
- Dan, C.; Kelly, A.; Bernard, O.; Minden, A. Cytoskeletal Changes Regulated by the PAK4 Serine/Threonine Kinase Are Mediated by LIM Kinase 1 and Cofilin. J. Biol. Chem. 2001, 276, 32115–32121. [Google Scholar] [CrossRef] [Green Version]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Binding of Activated Alpha2-Macroglobulin to Its Cell Surface Receptor GRP78 in 1-LN Prostate Cancer Cells Regulates PAK-2-Dependent Activation of LIMK. J. Biol. Chem. 2005, 280, 26278–26286. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Zheng, Y.; Wang, Z.-X. Evaluation of the Catalytic Mechanism of the P21-Activated Protein Kinase PAK2. Biochemistry 2003, 42, 1129–1139. [Google Scholar] [CrossRef]
- Ohashi, K.; Nagata, K.; Maekawa, M.; Ishizaki, T.; Narumiya, S.; Mizuno, K. Rho-Associated Kinase ROCK Activates LIM-Kinase 1 by Phosphorylation at Threonine 508 within the Activation Loop. J. Biol. Chem. 2000, 275, 3577–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorovoy, M.; Niu, J.; Bernard, O.; Profirovic, J.; Minshall, R.; Neamu, R.; Voyno-Yasenetskaya, T. LIM Kinase 1 Coordinates Microtubule Stability and Actin Polymerization in Human Endothelial Cells. J. Biol. Chem. 2005, 280, 26533–26542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadella, K.S.; Saji, M.; Jacob, N.K.; Pavel, E.; Ringel, M.D.; Kirschner, L.S. Regulation of Actin Function by Protein Kinase A-Mediated Phosphorylation of Limk1. EMBO Rep. 2009, 10, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nishita, M.; Mishima, T.; Ohashi, K.; Mizuno, K. MAPKAPK-2-Mediated LIM-Kinase Activation Is Critical for VEGF-Induced Actin Remodeling and Cell Migration. EMBO J. 2006, 25, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Ritchey, L.; Ottman, R.; Roumanos, M.; Chakrabarti, R. A Functional Cooperativity between Aurora A Kinase and LIM Kinase1: Implication in the Mitotic Process. Cell Cycle 2012, 11, 296–309. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.O.; Chang, K.-H.; Ghosh, S.; Venkatesh, C.; Giger, K.; Low, P.S.; Shah, K. LIMK2 Is a Crucial Regulator and Effector of Aurora-A-Kinase-Mediated Malignancy. J. Cell Sci. 2012, 125, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagheri-Yarmand, R.; Mazumdar, A.; Sahin, A.A.; Kumar, R. LIM Kinase 1 Increases Tumor Metastasis of Human Breast Cancer Cells via Regulation of the Urokinase-Type Plasminogen Activator System. Int. J. Cancer 2006, 118, 2703–2710. [Google Scholar] [CrossRef]
- Zhao, J.; Li, D.; Fang, L. MiR-128-3p Suppresses Breast Cancer Cellular Progression via Targeting LIMK1. Biomed. Pharmacother. 2019, 115, 108947. [Google Scholar] [CrossRef]
- Prunier, C.; Josserand, V.; Vollaire, J.; Beerling, E.; Petropoulos, C.; Destaing, O.; Montemagno, C.; Hurbin, A.; Prudent, R.; de Koning, L.; et al. LIM Kinase Inhibitor Pyr1 Reduces the Growth and Metastatic Load of Breast Cancers. Cancer Res. 2016, 76, 3541–3552. [Google Scholar] [CrossRef] [Green Version]
- Aggelou, H.; Chadla, P.; Nikou, S.; Karteri, S.; Maroulis, I.; Kalofonos, H.P.; Papadaki, H.; Bravou, V. LIMK/Cofilin Pathway and Slingshot Are Implicated in Human Colorectal Cancer Progression and Chemoresistance. Virchows Arch. 2018, 472, 727–737. [Google Scholar] [CrossRef]
- Su, J.; Zhou, Y.; Pan, Z.; Shi, L.; Yang, J.; Liao, A.; Liao, Q.; Su, Q. Downregulation of LIMK1-ADF/Cofilin by DADS Inhibits the Migration and Invasion of Colon Cancer. Sci. Rep. 2017, 7, 45624. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.; Frost, A.R.; Grizzle, W.E.; Chakrabarti, R. LIM Kinase 1 Is Essential for the Invasive Growth of Prostate Epithelial Cells: Implications in Prostate Cancer. J. Biol. Chem. 2003, 278, 36868–36875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Chen, R.; Li, X.; Cai, Y.; Ye, Z.; Li, S.; Li, J.; Huang, H.; Peng, S.; Wang, J.; et al. Downregulation of MicroRNA-23a Suppresses Prostate Cancer Metastasis by Targeting the PAK6-LIMK1 Signaling Pathway. Oncotarget 2015, 6, 3904–3917. [Google Scholar] [CrossRef] [Green Version]
- Park, G.B.; Kim, D. PI3K Catalytic Isoform Alteration Promotes the LIMK1-Related Metastasis Through the PAK1 or ROCK1/2 Activation in Cigarette Smoke-Exposed Ovarian Cancer Cells. Anticancer Res. 2017, 37, 1805–1818. [Google Scholar] [CrossRef]
- Vlecken, D.H.; Bagowski, C.P. LIMK1 and LIMK2 Are Important for Metastatic Behavior and Tumor Cell-Induced Angiogenesis of Pancreatic Cancer Cells. Zebrafish 2009, 6, 433–439. [Google Scholar] [CrossRef]
- Wan, L.; Zhang, L.; Fan, K.; Wang, J. MiR-27b Targets LIMK1 to Inhibit Growth and Invasion of NSCLC Cells. Mol. Cell. Biochem. 2014, 390, 85–91. [Google Scholar] [CrossRef]
- Chen, Q.; Jiao, D.; Hu, H.; Song, J.; Yan, J.; Wu, L.; Xu, L.-Q. Downregulation of LIMK1 Level Inhibits Migration of Lung Cancer Cells and Enhances Sensitivity to Chemotherapy Drugs. Oncol. Res. 2013, 20, 491–498. [Google Scholar] [CrossRef]
- Manetti, F. Recent Findings Confirm LIM Domain Kinases as Emerging Target Candidates for Cancer Therapy. Curr. Cancer Drug Targets 2012, 12, 543–560. [Google Scholar] [CrossRef]
- Lee, M.-H.; Kundu, J.K.; Chae, J.-I.; Shim, J.-H. Targeting ROCK/LIMK/Cofilin Signaling Pathway in Cancer. Arch. Pharm. Res. 2019, 42, 481–491. [Google Scholar] [CrossRef]
- Sumi, T.; Hashigasako, A.; Matsumoto, K.; Nakamura, T. Different Activity Regulation and Subcellular Localization of LIMK1 and LIMK2 during Cell Cycle Transition. Exp. Cell Res. 2006, 312, 1021–1030. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Nakamura, T. Mitosis-Dependent Phosphorylation and Activation of LIM-Kinase 1. Biochem. Biophys. Res. Commun. 2002, 290, 1315–1320. [Google Scholar] [CrossRef]
- Kaji, N.; Ohashi, K.; Shuin, M.; Niwa, R.; Uemura, T.; Mizuno, K. Cell Cycle-Associated Changes in Slingshot Phosphatase Activity and Roles in Cytokinesis in Animal Cells. J. Biol. Chem. 2003, 278, 33450–33455. [Google Scholar] [CrossRef] [Green Version]
- Amano, T.; Kaji, N.; Ohashi, K.; Mizuno, K. Mitosis-Specific Activation of LIM Motif-Containing Protein Kinase and Roles of Cofilin Phosphorylation and Dephosphorylation in Mitosis. J. Biol. Chem. 2002, 277, 22093–22102. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-E.; Ryu, H.J.; Kim, M.J.; Kang, T.-C. LIM Kinase-2 Induces Programmed Necrotic Neuronal Death via Dysfunction of DRP1-Mediated Mitochondrial Fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Goyal, P.; Pandey, D.; Behring, A.; Siess, W. Inhibition of Nuclear Import of LIMK2 in Endothelial Cells by Protein Kinase C-Dependent Phosphorylation at Ser-283. J. Biol. Chem. 2005, 280, 27569–27577. [Google Scholar] [CrossRef] [Green Version]
- Croft, D.R.; Olson, M.F. The Rho GTPase Effector ROCK Regulates Cyclin A, Cyclin D1, and P27Kip1 Levels by Distinct Mechanisms. Mol. Cell. Biol. 2006, 26, 4612–4627. [Google Scholar] [CrossRef] [Green Version]
- Galkin, V.E.; Orlova, A.; Kudryashov, D.S.; Solodukhin, A.; Reisler, E.; Schröder, G.F.; Egelman, E.H. Remodeling of Actin Filaments by ADF/Cofilin Proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 20568–20572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.; Song, X.; Mouneimne, G.; Sidani, M.; Lawrence, D.S.; Condeelis, J.S. Cofilin Promotes Actin Polymerization and Defines the Direction of Cell Motility. Science 2004, 304, 743–746. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, E.S.; Higgs, H.N. The Many Faces of Actin: Matching Assembly Factors with Cellular Structures. Nat. Cell Biol. 2007, 9, 1110–1121. [Google Scholar] [CrossRef]
- Pellegrin, S.; Mellor, H. Actin Stress Fibres. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawe, H.R.; Minamide, L.S.; Bamburg, J.R.; Cramer, L.P. ADF/Cofilin Controls Cell Polarity during Fibroblast Migration. Curr. Biol. 2003, 13, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Nagata-Ohashi, K.; Ohta, Y.; Goto, K.; Chiba, S.; Mori, R.; Nishita, M.; Ohashi, K.; Kousaka, K.; Iwamatsu, A.; Niwa, R.; et al. A Pathway of Neuregulin-Induced Activation of Cofilin-Phosphatase Slingshot and Cofilin in Lamellipodia. J. Cell Biol. 2004, 165, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishita, M.; Tomizawa, C.; Yamamoto, M.; Horita, Y.; Ohashi, K.; Mizuno, K. Spatial and Temporal Regulation of Cofilin Activity by LIM Kinase and Slingshot Is Critical for Directional Cell Migration. J. Cell Biol. 2005, 171, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, K.; Fujiwara, S.; Watanabe, T.; Kondo, H.; Kiuchi, T.; Sato, M.; Mizuno, K. LIM Kinase Has a Dual Role in Regulating Lamellipodium Extension by Decelerating the Rate of Actin Retrograde Flow and the Rate of Actin Polymerization. J. Biol. Chem. 2011, 286, 36340–36351. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho GTPases and the Actin Cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex But Targetable Ecosystem. Cells 2020, 9, 976. [Google Scholar] [CrossRef] [Green Version]
- Amarasekara, D.S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int. J. Mol. Sci. 2021, 22, 2851. [Google Scholar] [CrossRef]
- Owen, M. Marrow Stromal Stem Cells. J. Cell Sci. 1988, 1988 (Suppl. 10), 63–76. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Yang, S.; Shao, J.; Li, Y.-P. Signaling and Transcriptional Regulation in Osteoblast Commitment and Differentiation. Front. Biosci. 2007, 12, 3068–3092. [Google Scholar] [CrossRef] [Green Version]
- Pontikoglou, C.; Deschaseaux, F.; Sensebé, L.; Papadaki, H.A. Bone Marrow Mesenchymal Stem Cells: Biological Properties and Their Role in Hematopoiesis and Hematopoietic Stem Cell Transplantation. Stem Cell Rev. Rep. 2011, 7, 569–589. [Google Scholar] [CrossRef]
- Augello, A.; De Bari, C. The Regulation of Differentiation in Mesenchymal Stem Cells. Hum. Gene Ther. 2010, 21, 1226–1238. [Google Scholar] [CrossRef]
- Neve, A.; Corrado, A.; Cantatore, F.P. Osteoblast Physiology in Normal and Pathological Conditions. Cell Tissue Res. 2011, 343, 289–302. [Google Scholar] [CrossRef]
- Väänänen, H.K. Mesenchymal Stem Cells. Ann. Med. 2005, 37, 469–479. [Google Scholar] [CrossRef]
- Karsenty, G. Principles of Bone Biology, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Schroeder, T.M.; Jensen, E.D.; Westendorf, J.J. Runx2: A Master Organizer of Gene Transcription in Developing and Maturing Osteoblasts. Birth Defects Res. Part C Embryo Today Rev. 2005, 75, 213–225. [Google Scholar] [CrossRef]
- Marie, P.J. Transcription Factors Controlling Osteoblastogenesis. Arch. Biochem. Biophys. 2008, 473, 98–105. [Google Scholar] [CrossRef]
- Wagner, E.F.; Karsenty, G. Genetic Control of Skeletal Development. Curr. Opin. Genet. Dev. 2001, 11, 527–532. [Google Scholar] [CrossRef]
- Komori, T. Signaling Networks in RUNX2-Dependent Bone Development. J. Cell. Biochem. 2011, 112, 750–755. [Google Scholar] [CrossRef]
- Guasto, A.; Cormier-Daire, V. Signaling Pathways in Bone Development and Their Related Skeletal Dysplasia. Int. J. Mol. Sci. 2021, 22, 4321. [Google Scholar] [CrossRef]
- Komori, T. Regulation of Bone Development and Extracellular Matrix Protein Genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Kim, H.J.; Li, Q.L.; Chi, X.Z.; Ueta, C.; Komori, T.; Wozney, J.M.; Kim, E.G.; Choi, J.Y.; Ryoo, H.M.; et al. Runx2 Is a Common Target of Transforming Growth Factor Beta1 and Bone Morphogenetic Protein 2, and Cooperation between Runx2 and Smad5 Induces Osteoblast-Specific Gene Expression in the Pluripotent Mesenchymal Precursor Cell Line C2C12. Mol. Cell. Biol. 2000, 20, 8783–8792. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, A.; Srinaath, N.; Rohini, M.; Selvamurugan, N. Regulation of Runx2 by MicroRNAs in Osteoblast Differentiation. Life Sci. 2019, 232, 116676. [Google Scholar] [CrossRef]
- Vimalraj, S.; Arumugam, B.; Miranda, P.J.; Selvamurugan, N. Runx2: Structure, Function, and Phosphorylation in Osteoblast Differentiation. Int. J. Biol. Macromol. 2015, 78, 202–208. [Google Scholar] [CrossRef]
- Talbot, J.; Brion, R.; Lamora, A.; Mullard, M.; Morice, S.; Heymann, D.; Verrecchia, F. Connexin43 Intercellular Communication Drives the Early Differentiation of Human Bone Marrow Stromal Cells into Osteoblasts. J. Cell. Physiol. 2018, 233, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A Transcriptional Activator of Osteoblast Differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Takarada, T.; Hinoi, E.; Nakazato, R.; Ochi, H.; Xu, C.; Tsuchikane, A.; Takeda, S.; Karsenty, G.; Abe, T.; Kiyonari, H.; et al. An Analysis of Skeletal Development in Osteoblast-Specific and Chondrocyte-Specific Runt-Related Transcription Factor-2 (Runx2) Knockout Mice. J. Bone Miner. Res. 2013, 28, 2064–2069. [Google Scholar] [CrossRef]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted Disruption of Cbfa1 Results in a Complete Lack of Bone Formation Owing to Maturational Arrest of Osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Mundlos, S. Cleidocranial Dysplasia: Clinical and Molecular Genetics. J. Med. Genet. 1999, 36, 177–182. [Google Scholar] [PubMed]
- Guo, Y.-W.; Chiu, C.-Y.; Liu, C.-L.; Jap, T.-S.; Lin, L.-Y. Novel Mutation of RUNX2 Gene in a Patient with Cleidocranial Dysplasia. Int. J. Clin. Exp. Pathol. 2015, 8, 1057–1062. [Google Scholar] [PubMed]
- Zhou, X.; Zhang, Z.; Feng, J.Q.; Dusevich, V.M.; Sinha, K.; Zhang, H.; Darnay, B.G.; de Crombrugghe, B. Multiple Functions of Osterix Are Required for Bone Growth and Homeostasis in Postnatal Mice. Proc. Natl. Acad. Sci. USA 2010, 107, 12919–12924. [Google Scholar] [CrossRef] [Green Version]
- Koga, T.; Matsui, Y.; Asagiri, M.; Kodama, T.; de Crombrugghe, B.; Nakashima, K.; Takayanagi, H. NFAT and Osterix Cooperatively Regulate Bone Formation. Nat. Med. 2005, 11, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Lapunzina, P.; Aglan, M.; Temtamy, S.; Caparrós-Martín, J.A.; Valencia, M.; Letón, R.; Martínez-Glez, V.; Elhossini, R.; Amr, K.; Vilaboa, N.; et al. Identification of a Frameshift Mutation in Osterix in a Patient with Recessive Osteogenesis Imperfecta. Am. J. Hum. Genet. 2010, 87, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Chambers, T.J. The Regulation of Osteoclastic Development and Function. Cell Mol. Biol. Vertebr. Hard Tissues 1988, 136, 92–107. [Google Scholar] [CrossRef]
- Ueki, Y.; Lin, C.-Y.; Senoo, M.; Ebihara, T.; Agata, N.; Onji, M.; Saheki, Y.; Kawai, T.; Mukherjee, P.M.; Reichenberger, E.; et al. Increased Myeloid Cell Responses to M-CSF and RANKL Cause Bone Loss and Inflammation in SH3BP2 “Cherubism” Mice. Cell 2007, 128, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK Is the Intrinsic Hematopoietic Cell Surface Receptor That Controls Osteoclastogenesis and Regulation of Bone Mass and Calcium Metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef] [Green Version]
- Casimiro, S.; Vilhais, G.; Gomes, I.; Costa, L. The Roadmap of RANKL/RANK Pathway in Cancer. Cells 2021, 10, 1978. [Google Scholar] [CrossRef] [PubMed]
- Rashed, F.; Kamijyo, S.; Shimizu, Y.; Hirohashi, Y.; Khan, M.; Sugamori, Y.; Murali, R.; Aoki, K. The Effects of Receptor Activator of NF-ΚB Ligand-Binding Peptides on Bone Resorption and Bone Formation. Front. Cell Dev. Biol. 2021, 9, 648084. [Google Scholar] [CrossRef]
- Elango, J.; Bao, B.; Wu, W. The Hidden Secrets of Soluble RANKL in Bone Biology. Cytokine 2021, 144, 155559. [Google Scholar] [CrossRef]
- Grigoriadis, A.E.; Wang, Z.Q.; Cecchini, M.G.; Hofstetter, W.; Felix, R.; Fleisch, H.A.; Wagner, E.F. C-Fos: A Key Regulator of Osteoclast-Macrophage Lineage Determination and Bone Remodeling. Science 1994, 266, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Matsuo, K.; Suzuki, H.; Suzuki, T.; Sato, K.; Yokochi, T.; Oda, H.; Nakamura, K.; Ida, N.; et al. RANKL Maintains Bone Homeostasis through C-Fos-Dependent Induction of Interferon-Beta. Nature 2002, 416, 744–749. [Google Scholar] [CrossRef]
- Wu, P.-F.; Tang, J.; Li, K. RANK Pathway in Giant Cell Tumor of Bone: Pathogenesis and Therapeutic Aspects. Tumour Biol. 2015, 36, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Zhu, M.; Troiano, N.; Horowitz, M.; Bian, J.; Gundberg, C.; Kolodziejczak, K.; Insogna, K. LIM Kinase 1 Deficient Mice Have Reduced Bone Mass. Bone 2013, 52, 70–82. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Shi, K.; Frary, C.E.; Ditzel, N.; Hu, H.; Qiu, W.; Kassem, M. Inhibiting Actin Depolymerization Enhances Osteoblast Differentiation and Bone Formation in Human Stromal Stem Cells. Stem Cell Res. 2015, 15, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, Y.; Jiang, L.; Li, Z.; Lee, S.; Liu, C.; Wang, J.; Zhang, J. Recombinant Human BMP-2 Accelerates the Migration of Bone Marrow Mesenchymal Stem Cells via the CDC42/PAK1/LIMK1 Pathway in Vitro and in Vivo. Biomater. Sci. 2018, 7, 362–372. [Google Scholar] [CrossRef]
- Kristensen, L.P.; Chen, L.; Nielsen, M.O.; Qanie, D.W.; Kratchmarova, I.; Kassem, M.; Andersen, J.S. Temporal Profiling and Pulsed SILAC Labeling Identify Novel Secreted Proteins during Ex Vivo Osteoblast Differentiation of Human Stromal Stem Cells. Mol. Cell. Proteom. 2012, 11, 989–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Shi, K.; Andersen, T.L.; Qiu, W.; Kassem, M. KIAA1199 Is a Secreted Molecule That Enhances Osteoblastic Stem Cell Migration and Recruitment. Cell Death Dis. 2019, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Wu, C.; Shen, Y.; Zheng, S.; Chen, R. Effect of LIMK2 RNAi on Reorganization of the Actin Cytoskeleton in Osteoblasts Induced by Fluid Shear Stress. J. Biomech. 2008, 41, 3225–3228. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tan, S.; Shen, Y.; Chen, R.; Wu, C.; Xu, Y.; Song, Z.; Fu, Q. Inhibition of FSS-Induced Actin Cytoskeleton Reorganization by Silencing LIMK2 Gene Increases the Mechanosensitivity of Primary Osteoblasts. Bone 2015, 74, 182–190. [Google Scholar] [CrossRef]
- Klein, M.J.; Siegal, G.P. Osteosarcoma: Anatomic and Histologic Variants. Am. J. Clin. Pathol. 2006, 125, 555–581. [Google Scholar] [CrossRef]
- Ritter, J.; Bielack, S.S. Osteosarcoma. Ann. Oncol. 2010, 21 (Suppl. 7), vii320–vii325. [Google Scholar] [CrossRef] [PubMed]
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A. IICC-3 contributors International Incidence of Childhood Cancer, 2001–2010: A Population-Based Registry Study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Henley, S.J.; Thomas, C.C.; Lewis, D.R.; Ward, E.M.; Islami, F.; Wu, M.; Weir, H.K.; Scott, S.; Sherman, R.L.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, Part II: Progress toward Healthy People 2020 Objectives for 4 Common Cancers. Cancer 2020, 126, 2250–2266. [Google Scholar] [CrossRef]
- Mirabello, L.; Pfeiffer, R.; Murphy, G.; Daw, N.C.; Patiño-Garcia, A.; Troisi, R.J.; Hoover, R.N.; Douglass, C.; Schüz, J.; Craft, A.W.; et al. Height at Diagnosis and Birth-Weight as Risk Factors for Osteosarcoma. Cancer Causes Control 2011, 22, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational Biology of Osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. International Osteosarcoma Incidence Patterns in Children and Adolescents, Middle Ages and Elderly Persons. Int. J. Cancer 2009, 125, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, A.M.; Synoradzki, K.; Firlej, W.; Bartnik, E.; Sobczuk, P.; Fiedorowicz, M.; Grieb, P.; Rutkowski, P. Molecular Biology of Osteosarcoma. Cancers 2020, 12, 2130. [Google Scholar] [CrossRef]
- de Azevedo, J.W.V.; de Medeiros Fernandes, T.A.A.; Fernandes, J.V.; de Azevedo, J.C.V.; Lanza, D.C.F.; Bezerra, C.M.; Andrade, V.S.; de Araújo, J.M.G.; Fernandes, J.V. Biology and Pathogenesis of Human Osteosarcoma. Oncol. Lett. 2020, 19, 1099–1116. [Google Scholar] [CrossRef]
- Mutsaers, A.J.; Walkley, C.R. Cells of Origin in Osteosarcoma: Mesenchymal Stem Cells or Osteoblast Committed Cells? Bone 2014, 62, 56–63. [Google Scholar] [CrossRef]
- Mohseny, A.B.; Szuhai, K.; Romeo, S.; Buddingh, E.P.; Briaire-de Bruijn, I.; de Jong, D.; van Pel, M.; Cleton-Jansen, A.-M.; Hogendoorn, P.C.W. Osteosarcoma Originates from Mesenchymal Stem Cells in Consequence of Aneuploidization and Genomic Loss of Cdkn2. J. Pathol. 2009, 219, 294–305. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent Somatic Structural Variations Contribute to Tumorigenesis in Pediatric Osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Toguchida, J.; Ishizaki, K.; Sasaki, M.S.; Nakamura, Y.; Ikenaga, M.; Kato, M.; Sugimot, M.; Kotoura, Y.; Yamamuro, T. Preferential Mutation of Paternally Derived RB Gene as the Initial Event in Sporadic Osteosarcoma. Nature 1989, 338, 156–158. [Google Scholar] [CrossRef]
- Sasaki, M.S.; Kato, M.; Toguchida, J.; Yamaguchi, T.; Ejima, Y.; Ishizaki, K.; Kaneko, A.; Tanooka, H. Somatic and Germinal Mutations of Tumor-Suppressor Genes in the Development of Cancer. J. Radiat. Res. 1991, 32 (Suppl. 2), 266–276. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Cogdell, D.; Yang, D.; Hu, L.; Li, H.; Zheng, H.; Du, X.; Pang, Y.; Trent, J.; Chen, K.; et al. Deletion of the WWOX Gene and Frequent Loss of Its Protein Expression in Human Osteosarcoma. Cancer Lett. 2010, 291, 31–38. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Z.; Huang, Z.; Chen, D.-C.; Zhu, X.-X.; Wang, Y.-Z.; Yan, Y.-W.; Tang, S.; Madhavan, S.; Ni, W.; et al. Super Enhancer Inhibitors Suppress MYC Driven Transcriptional Amplification and Tumor Progression in Osteosarcoma. Bone Res. 2018, 6, 11. [Google Scholar] [CrossRef]
- Ottaviani, G.; Jaffe, N. The Etiology of Osteosarcoma. Cancer Treat. Res. 2009, 152, 15–32. [Google Scholar] [CrossRef]
- Varley, J.M.; Evans, D.G.; Birch, J.M. Li-Fraumeni Syndrome—A Molecular and Clinical Review. Br. J. Cancer 1997, 76, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Correa, H. Li-Fraumeni Syndrome. J. Pediatr. Genet. 2016, 5, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Chauveinc, L.; Mosseri, V.; Quintana, E.; Desjardins, L.; Schlienger, P.; Doz, F.; Dutrillaux, B. Osteosarcoma Following Retinoblastoma: Age at Onset and Latency Period. Ophthalmic Genet. 2001, 22, 77–88. [Google Scholar] [CrossRef]
- Leonard, A.; Craft, A.W.; Moss, C.; Malcolm, A.J. Osteogenic Sarcoma in the Rothmund-Thomson Syndrome. Med. Pediatr. Oncol. 1996, 26, 249–253. [Google Scholar] [CrossRef]
- Hansen, M.F.; Seton, M.; Merchant, A. Osteosarcoma in Paget’s Disease of Bone. J. Bone Miner. Res. 2006, 21 (Suppl. 2), P58–P63. [Google Scholar] [CrossRef]
- Morice, S.; Danieau, G.; Rédini, F.; Brounais-Le-Royer, B.; Verrecchia, F. Hippo/YAP Signaling Pathway: A Promising Therapeutic Target in Bone Paediatric Cancers? Cancers 2020, 12, 645. [Google Scholar] [CrossRef] [Green Version]
- Morice, S.; Mullard, M.; Brion, R.; Dupuy, M.; Renault, S.; Tesfaye, R.; Brounais-Le Royer, B.; Ory, B.; Redini, F.; Verrecchia, F. The YAP/TEAD Axis as a New Therapeutic Target in Osteosarcoma: Effect of Verteporfin and CA3 on Primary Tumor Growth. Cancers 2020, 12, 3847. [Google Scholar] [CrossRef]
- Morice, S.; Danieau, G.; Tesfaye, R.; Mullard, M.; Brion, R.; Dupuy, M.; Ory, B.; Brounais-Le Royer, B.; Corre, I.; Redini, F.; et al. Involvement of the TGF-β Signaling Pathway in the Development of YAP-Driven Osteosarcoma Lung Metastasis. Front. Oncol. 2021, 11, 765711. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Fischer, R.S.; Pan, D.; Waterman, C.M. YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-Dependent, Myosin II- and Phospho-YAP-Independent Pathway during Extracellular Matrix Mechanosensing. J. Biol. Chem. 2016, 291, 6096–6110. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; Chen, J.; Lim, Y.B.; Finch-Edmondson, M.L.; Seshachalam, V.P.; Qin, L.; Jiang, T.; Low, B.C.; Singh, H.; Lim, C.T.; et al. YAP Regulates Actin Dynamics through ARHGAP29 and Promotes Metastasis. Cell Rep. 2017, 19, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Casali, P.G.; Bielack, S.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brennan, B.; et al. Bone Sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv79–iv95. [Google Scholar] [CrossRef]
- Ta, H.T.; Dass, C.R.; Choong, P.F.M.; Dunstan, D.E. Osteosarcoma Treatment: State of the Art. Cancer Metastasis Rev. 2009, 28, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-F.; Yao, Y.-D.; Zhao, Y.-Y.; Liu, Y.; Liu, Z.-H.; Hu, P.; Zhu, Z.-R. Effects of PAK4/LIMK1/Cofilin-1 Signaling Pathway on Proliferation, Invasion, and Migration of Human Osteosarcoma Cells. J. Clin. Lab. Anal. 2020, 34, e23362. [Google Scholar] [CrossRef]
- Yang, J.-Z.; Huang, L.-H.; Chen, R.; Meng, L.-J.; Gao, Y.-Y.; Ji, Q.-Y.; Wang, Y. LIM Kinase 1 Serves an Important Role in the Multidrug Resistance of Osteosarcoma Cells. Oncol. Lett. 2018, 15, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Y.; Xing, F.; Wang, J.; Wang, Y.; Wang, H.; Yang, Y.; Gao, Z. Overexpression of LIMK1 Promotes Migration Ability of Multidrug-Resistant Osteosarcoma Cells. Oncol. Res. 2011, 19, 501–509. [Google Scholar] [CrossRef]
- Zhang, H.-S.; Zhao, J.-W.; Wang, H.; Zhang, H.-Y.; Ji, Q.-Y.; Meng, L.-J.; Xing, F.-J.; Yang, S.-T.; Wang, Y. LIM Kinase 1 Is Required for Insulin—Dependent Cell Growth of Osteosarcoma Cell Lines. Mol. Med. Rep. 2014, 9, 103–108. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Nakamura, S.; Sugiyama, Y.; Tamai, S.; Ishida, Y.; Sueyoshi, M.; Toda, Y.; Hosogi, S.; Yano, Y.; Ashihara, E. 6-Hydroxythiobinupharidine Inhibits Migration of LM8 Osteosarcoma Cells by Decreasing Expression of LIM Domain Kinase 1. Anticancer Res. 2019, 39, 6507–6513. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, L.; Liu, Y.; Chen, M.; Zhang, S.; Kong, D. Sea Cucumber Cucumaria Frondosa Fucoidan Inhibits Osteosarcoma Adhesion and Migration by Regulating Cytoskeleton Remodeling. Oncol. Rep. 2020, 44, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ren, T.; Jiao, G.; Huang, Y.; Bao, X.; Zhang, F.; Liu, K.; Zheng, B.; Sun, K.; Guo, W. BMPR2 Promotes Invasion and Metastasis via the RhoA-ROCK-LIMK2 Pathway in Human Osteosarcoma Cells. Oncotarget 2017, 8, 58625–58641. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.; Zheng, B.; Huang, Y.; Wang, S.; Bao, X.; Liu, K.; Guo, W. Osteosarcoma Cell Intrinsic PD-L2 Signals Promote Invasion and Metastasis via the RhoA-ROCK-LIMK2 and Autophagy Pathways. Cell Death Dis. 2019, 10, 261. [Google Scholar] [CrossRef]
- Zheng, B.; Zhou, C.; Qu, G.; Ren, C.; Yan, P.; Guo, W.; Yue, B. VEGFR2 Promotes Metastasis and PD-L2 Expression of Human Osteosarcoma Cells by Activating the STAT3 and RhoA-ROCK-LIMK2 Pathways. Front. Oncol. 2020, 10, 543562. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Qu, R.; Zhang, L.; Huang, W. Rho A Regulates Epidermal Growth Factor-Induced Human Osteosarcoma MG63 Cell Migration. Int. J. Mol. Sci. 2018, 19, 1437. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brion, R.; Regnier, L.; Mullard, M.; Amiaud, J.; Rédini, F.; Verrecchia, F. LIM Kinases in Osteosarcoma Development. Cells 2021, 10, 3542. https://doi.org/10.3390/cells10123542
Brion R, Regnier L, Mullard M, Amiaud J, Rédini F, Verrecchia F. LIM Kinases in Osteosarcoma Development. Cells. 2021; 10(12):3542. https://doi.org/10.3390/cells10123542
Chicago/Turabian StyleBrion, Régis, Laura Regnier, Mathilde Mullard, Jérome Amiaud, Françoise Rédini, and Franck Verrecchia. 2021. "LIM Kinases in Osteosarcoma Development" Cells 10, no. 12: 3542. https://doi.org/10.3390/cells10123542
APA StyleBrion, R., Regnier, L., Mullard, M., Amiaud, J., Rédini, F., & Verrecchia, F. (2021). LIM Kinases in Osteosarcoma Development. Cells, 10(12), 3542. https://doi.org/10.3390/cells10123542