
Alterations in SLC4A2, SLC26A7 and SLC26A9 Drive Acid–Base Imbalance in Gastric Neuroendocrine Tumors and Uncover a Novel Mechanism for a Co-Occurring Polyautoimmune Scenario

 , ,
, ,  , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Studied Series
2.2. Patients
2.3. Quantitative RT-PCR
2.4. Statistics
2.5. Discovery Series WES1
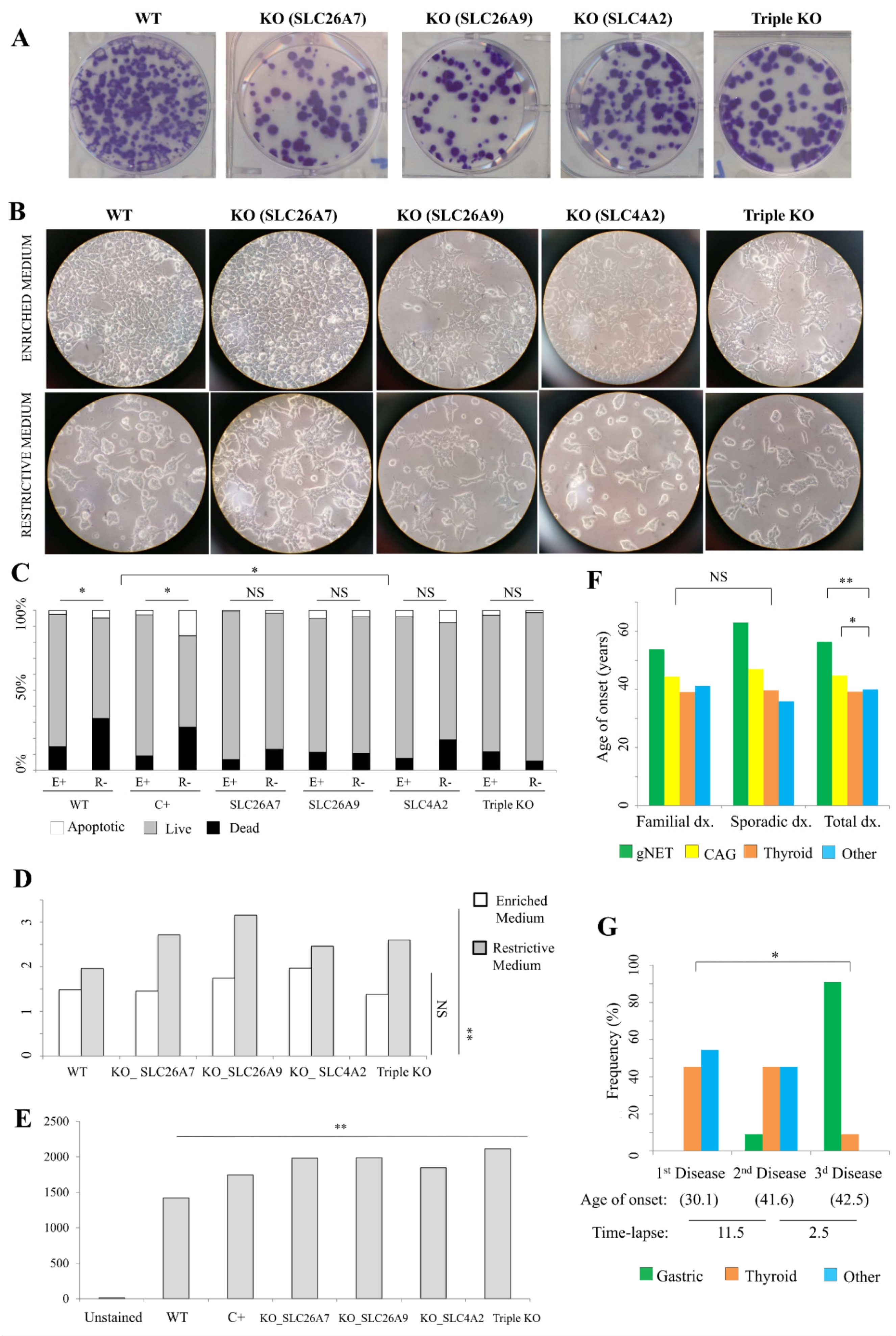
2.6. In Vitro Studies
2.7. Targeted Next Generation Sequencing (tNGS)
3. Results
3.1. Thyrogastric Syndrome
3.2. Autoimmune Polyendocrine Syndrome (APS) Series
3.2.1. Pathology Associations
3.2.2. Genetic Studies
- Efficacy per patient group.
- Efficacy per gene.
- Efficacy per gene in co-occurring pathologies.
- Correlation of the efficacy with the number of pathologies per patient.
4. Discussion
4.1. Achlorhydria and Gastric Disease
4.2. Autoimmune Thyrogastric Syndrome
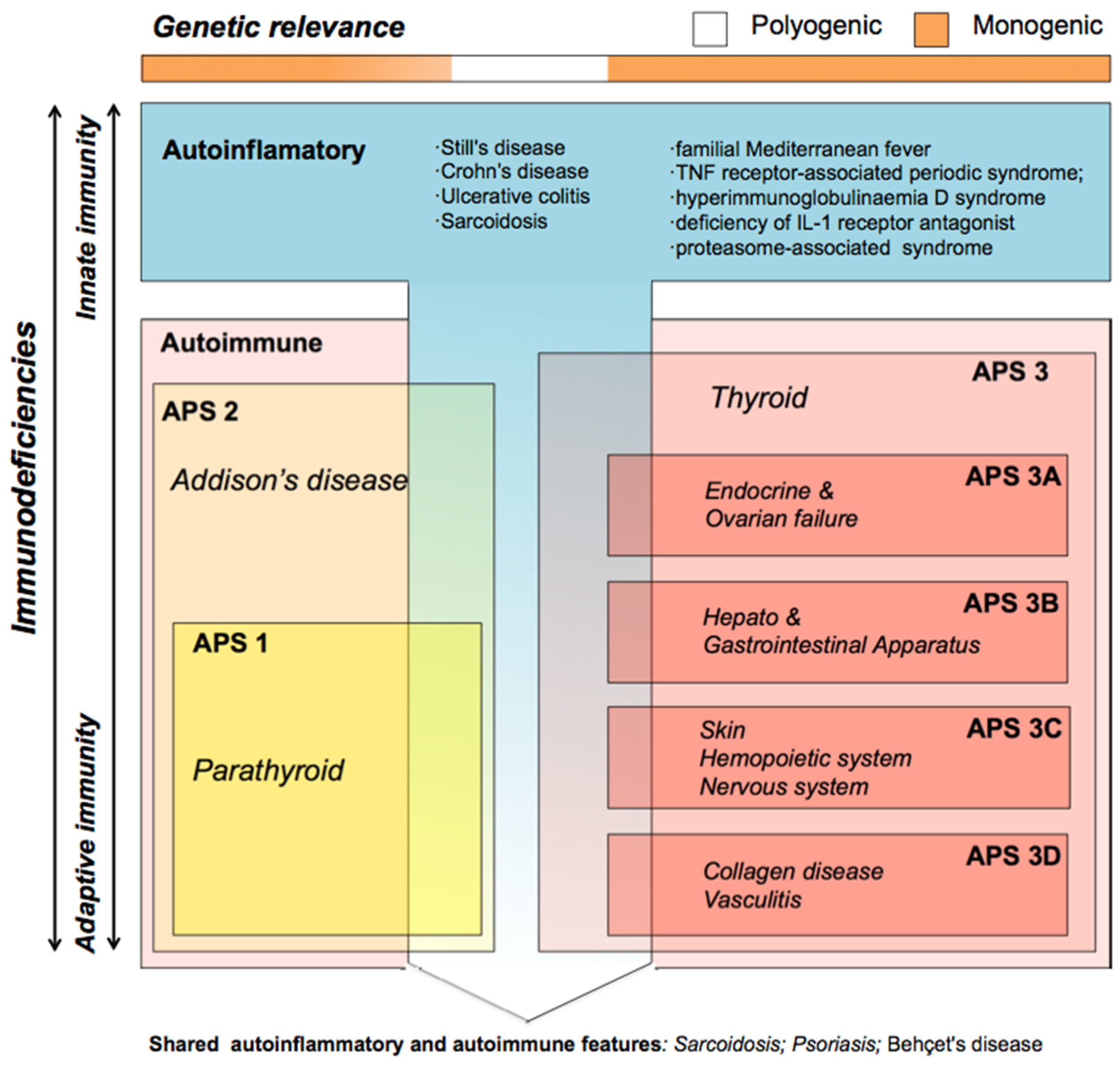
4.3. Co-Occurring Immunopathies (Polyautoimmune Syndrome)
4.3.1. Pathogenic Mechanisms
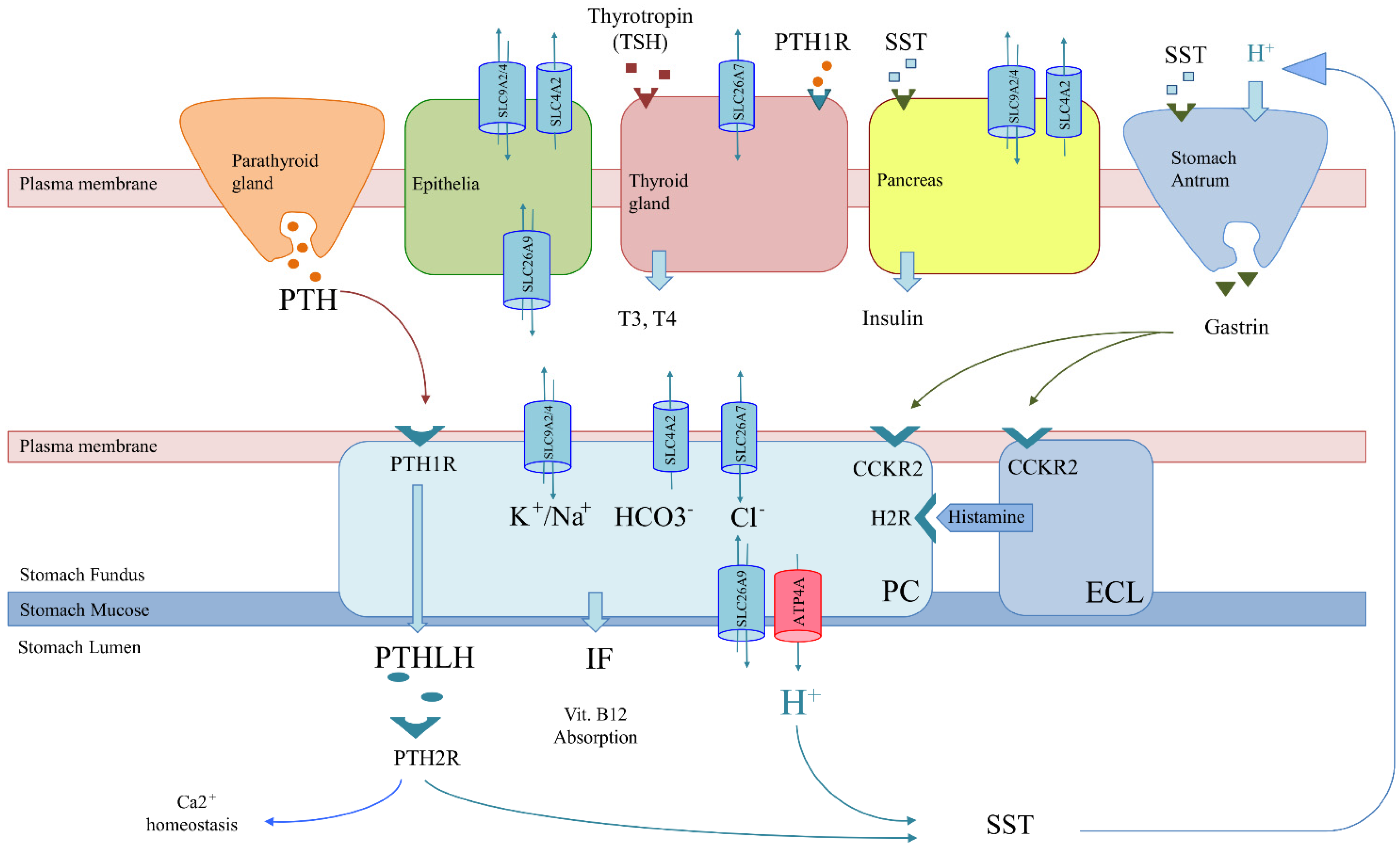
- Co-expression scenario: Genes involved in achlorhydria are also expressed in tissues other than gastric tissues (SLC26A7, SLC26A9, SLC4A2, SLC9A2 and SLC9A4) (Figure 3). Thus, like the thyrogastric syndrome, APS might be explained by monogenic models with variants in genes from the achlorhydria panel, since high positivity was observed in several associated immunodeficiencies (Table 3). Similarly, the co-expression scenario comprises not only SLC genes but also other genes, such as AIRE [7] and CTLA-4 [8], which were previously described to be involved in APS disorders.
- Polygenic model: The cumulative effect of variants in tissue-specifically expressed genes mainly involved in the acid–base balance process might compose a digenic/polygenic scenario together with the ATP4A gene. On the other hand, skin pathologies as well as the set of autoinflammatory diseases had the lowest percentage of variants (positivity) in the genes included in the panel (Table 1), which suggests a polygenic model or other mechanisms different from an acid–base deregulation for APS patients when they co-occur with APS3C or autoinflammatory diseases. APS including skin diseases might be better explained by variants in genes of the immune system, as previously reported [9]. In addition, previously described susceptibility genes to explain individual pathologies, such as NLRP1 and XBP1 (vitiligo) [35], or TCF7L2, ABCC8 and CAPN10 (DM2) [36], were included in this model. However, we cannot rule out a different genetic behavior for a pathology when it is co-occurring or when it is diagnosed alone. Likewise, a polygenic model also describes a syndrome that mimic APS (APS-like syndrome) caused by an accumulation of independent pathologies with no shared genetic origin.
- Genes involved in achlorhydria that are not expressed in other tissues might interfere with downstream pathways or acid-dependent absorption. Thus, an alteration in the H+/somatostatin (SST) pathway, which is involved in thyroid regulation and gastrin and insulin secretion, may trigger not only achlorhydria but also hypothyroidism and diabetes (Figure 3). The inflammatory bowel disease described in the F1 family affected with gNETs caused by ATP4A mutations fits in this scenario [15]. Similarly, an alteration in the PTH/PTHLH pathway, which depends on PTH secreted by the parathyroid gland, would affect both gastric and Ca2+ homeostasis (Figure 3). The rheumatoid arthritis and gNET described in the F2 family is in agreement with this scenario [12].
4.3.2. Clinical Associations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frizinsky, S.; Haj-Yahia, S.; MacHnes Maayan, D.; Lifshitz, Y.; Maoz-Segal, R.; Offengenden, I.; Kidon, M.; Agmon-Levin, N. The innate immune perspective of autoimmune and autoinflammatory conditions. Rheumatology 2019, 58 (Suppl. 6), vi1–vi8. [Google Scholar] [CrossRef]
- Schmidt, R.E.; Grimbacher, B.; Witte, T. Autoimmunity and primary immunodeficiency: Two sides of the same coin? Nat. Rev. Rheumatol. 2018, 14, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Betterle, C.; Dal Pra, C.; Mantero, F.; Zanchetta, R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: Autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr. Rev. 2002, 23, 327–364. [Google Scholar] [CrossRef]
- Husebye, E.S.; Anderson, M.S.; Kampe, O. Autoimmune polyendocrine syndromes. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef]
- Cutolo, M. Autoimmune polyendocrine syndromes. Autoimmun. Rev. 2014, 13, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Manthiram, K.; Zhou, Q.; Manthiram, K.; Zhou, Q.; Aksentijevich, I.; Kastner, D.L. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat. Immunol. 2017, 18, 832–842. [Google Scholar] [CrossRef]
- Anderson, M.S.; Su, M.A. AIRE expands: New roles in immune tolerance and beyond. Nat. Rev. Immunol. 2016, 16, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, B.; Imrie, H.; Geatch, D.R.; Perros, P.; Ball, S.G.; Baylis, P.H.; Carr, D.; Hurel, S.J.; James, R.A.; Kelly, W.F.; et al. Association analysis of the cytotoxic T lymphocyte antigen-4 (CTLA-4) and autoimmune regulator-1 (AIRE-1) genes in sporadic autoimmune Addison’s disease. J. Clin. Endocrinol. Metab. 2000, 85, 688–691. [Google Scholar] [CrossRef]
- Fischer, A.; Provot, J.; Jais, J.P.; Alcais, A.; Mahlaoui, N.; Adoue, D.; Aladjidi, N.; Amoura, Z.; Arlet, P.; Armari-Alla, C.; et al. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J. Allergy Clin. Immunol. 2017, 140, 1388–1393. [Google Scholar] [CrossRef]
- Massironi, S.; Zilli, A.; Elvevi, A.; Invernizzi, P. The changing face of chronic autoimmune atrophic gastritis: An updated comprehensive perspective. Autoimmun. Rev. 2019, 18, 215–222. [Google Scholar] [CrossRef]
- Calvete, O.; Reyes, J.; Zuñiga, S.; Paumard-Hernández, B.; Fernández, V.; Bujanda, L.; Rodriguez-Pinilla, M.S.; Palacios, J.; Heine-Suñer, D.; Banka, S.; et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastricneuroendocrine tumour. Hum. Mol. Genet. 2015, 24, 2914–2922. [Google Scholar] [CrossRef]
- Calvete, O.; Herraiz, M.; Reyes, J.; Patiño, A.; Benitez, J. A cumulative effect involving malfunction of the PTH1R and ATP4A genes explains a familial gastric neuroendocrine tumor with hypothyroidism and arthritis. Gastric Cancer 2017, 21, 998–1003. [Google Scholar] [CrossRef][Green Version]
- Calvete, O.; Varro, A.; Pritchard, D.M.; Barroso, A.; Oteo, M.; Morcillo, M.Á.; Vargiu, P.; Dodd, S.; Garcia, M.; Reyes, J.; et al. A knockin mouse model for human ATP4aR703C mutation identified in familial gastric neuroendocrine tumors recapitulates the premalignant condition of the human disease and suggests new therapeutic strategies. Dis. Model. Mech. 2016, 9, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Benítez, J.; Marra, R.; Reyes, J.; Calvete, O. A genetic origin for acid–base imbalance triggers the mitochondrial damage that explains the autoimmune response and drives to gastric neuroendocrine tumours. Gastric Cancer 2019, 23, 52–63. [Google Scholar] [CrossRef]
- Calvete, O.; Reyes, J.; Benítez, J. Case Report: CMV Infection and Same Mechanism-Originated Intestinal Inflammation Compatible With Bowel/Crohn’s Disease Is Suggested in ATP4A Mutated-Driven Gastric Neuroendocrine Tumors. Front. Med. 2021, 8, 553110. [Google Scholar] [CrossRef]
- Cellini, M.; Santaguida, M.G.; Virili, C.; Capriello, S.; Brusca, N.; Gargano, L.; Centanni, M. Hashimoto’s thyroiditis and autoimmune gastritis. Front. Front. Endocrinol. 2017, 8, 92. [Google Scholar] [CrossRef]
- Xu, J.; Song, P.; Nakamura, S.; Miller, M.; Barone, S.; Alper, S.L.; Riederer, B.; Bonhagen, J.; Arend, L.J.; Amlal, H.; et al. Deletion of the chloride transporter Slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J. Biol. Chem. 2009, 284, 29470–29479. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Song, P.; Miller, M.L.; Borgese, F.; Barone, S.; Riederer, B.; Wang, Z.; Alper, S.L.; Forte, J.G.; Shull, G.E.; et al. Deletion of the chloride transporter Slc26a9 causes loss of tubulovesicles in parietal cells and impairs acid secretion in the stomach. Proc. Natl. Acad. Sci. USA 2008, 105, 17955–17960. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Trauner, M. When lightning strikes twice: The plot thickens for a dual role of the anion exchanger 2 (AE2/SLC4A2) in the pathogenesis and treatment of primary biliary cirrhosis. J. Hepatol. 2008, 50, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, M.; Maclaren, N.; Blizzard, R. Autoimmune polyglandular syndromes. Pediatr. Ann. 1980, 9, 43–53. [Google Scholar] [CrossRef]
- Minalyan, A.; Benhammou, J.N.; Artashesyan, A.; Lewis, M.S.; Pisegna, J.R. Autoimmune atrophic gastritis: Current perspectives. Clin. Exp. Gastroenterol. 2017, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Neumann, W.L.; Coss, E.; Rugge, M.; Genta, R.M. Autoimmune atrophic gastritis-pathogenesis, pathology and management. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; Ryan, K.; Thomson, W.; Newman, W.G. Pernicious anemia-Genetic insights. Autoimmun. Rev. 2011, 10, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Seidler, U.; Nikolovska, K. Slc26 family of anion transporters in the gastrointestinal tract: Expression, function, regulation, and role in disease. Compr. Physiol. 2019, 9, 839–872. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Corrado, A.; Di Domenicantonio, A.; Fallahi, P. Autoimmune thyroid disorders. Autoimmun. Rev. 2015, 14, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Alzahrani, A.S.; Al-Odaib, A.; Alqahtani, M.A.; Babiker, O.; Al-Rijjal, R.A.; Binessa, H.A.; Kattan, W.E.; Al-Enezi, A.F.; Al Qarni, A.; et al. Molecular Analysis of Congenital Hypothyroidism in Saudi Arabia: SLC26A7 Mutation Is a Novel Defect in Thyroid Dyshormonogenesis. J. Clin. Endocrinol. Metab. 2018, 103, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Cangul, H.; Liao, X.H.; Schoenmakers, E.; Kero, J.; Barone, S.; Srichomkwun, P.; Iwayama, H.; Serra, E.G.; Saglam, H.; Eren, E.; et al. Homozygous loss-of-function mutations in SLC26A7 cause goitrous congenital hypothyroidism. JCI Insight 2018, 3, e99631. [Google Scholar] [CrossRef]
- Ravi, N.; Yang, M.; Mylona, N.; Wennerberg, J.; Paulsson, K. Global RNA expression and DNA methylation patterns in primary anaplastic thyroid cancer. Cancers 2020, 12, 680. [Google Scholar] [CrossRef]
- Valdes Socin, H.; Lutteri, L.; Cavalier, E.; Polus, M.; Geenen, V.; Louis, E.; Beckers, A. The thyrogastric syndrome: Its effects on micronutriments and gastric tumorigenesis | Le syndrome auto-immun thyro-gastrique: Ses effets sur les micronutriments et la tumorigenèse gastrique. Rev. Med. De Liege 2013, 68, 579–584. [Google Scholar]
- Saitoh, O.; Takiuchi, H.; Egashira, Y.; Sakane, S.; Sasaki, I.; Takamatsu, J.; Hirata, I.; Katsu, K.I. Gastric carcinoid tumor associated with Graves’ disease: A case report. Dig. Endosc. 1998, 10, 223–226. [Google Scholar] [CrossRef]
- De Block, C.E.M.; De Leeuw, I.H.; Van Gaal, L.F. Autoimmune gastritis in type 1 diabetes: A clinically oriented review. J. Clin. Endocrinol. Metab. 2008, 93, 363–371. [Google Scholar] [CrossRef]
- Amerio, P.; Tracanna, M.; De Remigis, P.; Betterle, C.; Vianale, L.; Marra, M.E.; Di Rollo, D.; Capizzi, R.; Feliciani, C.; Tulli, A. Vitiligo associated with other autoimmune diseases: Polyglandular autoimmune syndrome types 3B + C and 4. Clin. Exp. Dermatol. 2006, 31, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Zelissen, P.M.J.; Bast, E.J.E.G.; Croughs, R.J.M. Associated autoimmunity in addison’s disease. J. Autoimmun. 1995, 8, 121–130. [Google Scholar] [CrossRef]
- Chang, K.H.; Lyu, R.K.; Ro, L.S.; Wu, Y.R.; Chen, C.M. Coexistence of pernicious anemia and myasthenia gravis-A rare combination of autoimmune diseases in Taiwan. J. Formos. Med Assoc. 2006, 105, 946–949. [Google Scholar] [CrossRef]
- Spritz, R.A.; Andersen, G.H.L. Genetics of Vitiligo. Dermatol. Clin. 2017, 35, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. Genomic screening for monogenic forms of diabetes. BMC Med. 2018, 16, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Batterham, R.L.; Cummings, D.E. Mechanisms of diabetes improvement following bariatric/metabolic surgery. Diabetes Care 2016, 39, 893–901. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Information | tNGS Study | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients | Gastric Disease | Autoimmune Polyendocrine Syndrome (APS) | NON-APS | ||||||||||||||||||
| N | APS1 | APS2 | APS3 (Graves) | APS3 (Hashimoto) | APS3A (DM1) | APS3B (gastric) | APS3C (skin) | APS3D (collagen) | DM2 | Autoinfl. | ATP4A | SLC26A9 | SLC9A4 | SLC9A2 | PTH2R | PTH1R | SLC4A2 | SLC26A7 | KCNQ1 | ||
| Previously | gNET | F1 | p.Arg703Cys | ||||||||||||||||||
| studied | F2 | p.Gln680Leu | p.Glu546Lys | ||||||||||||||||||
| Discovery WES1 | gNET | F3 | p.Ile194Met | p.Arg25Gln | |||||||||||||||||
| CAG | F4 | p.Val172Met/p.Arg849Gln | |||||||||||||||||||
| F5 | p.Ile215Val | ||||||||||||||||||||
| F6 | p.Pro240His | p.Ile215Val | |||||||||||||||||||
| F7 | p.Arg849Gln | ||||||||||||||||||||
| FAMILIAL | gNET | F8 | p.Gln680Leu/c.1500+5G>T | ||||||||||||||||||
| F9 | p.Thr628Lys | p.Pro102Leu | |||||||||||||||||||
| F10 | p.Pro240His | ||||||||||||||||||||
| F11 | p.Thr312Ile | ||||||||||||||||||||
| F12 | p.Ala820Ser | ||||||||||||||||||||
| F13 | p.Gly841Glu | ||||||||||||||||||||
| F14 | |||||||||||||||||||||
| F15 | |||||||||||||||||||||
| F16 | |||||||||||||||||||||
| F17 | |||||||||||||||||||||
| CAG | F18 | p.V371A*54 | p.Glu546Lys | ||||||||||||||||||
| F19 | c.1500+5G>T | p.Arg143Cys | p.Ala459Val | ||||||||||||||||||
| F20 | c.544-6G>C | p.Val648Ile | |||||||||||||||||||
| F21 | p.Ter888GlnextTer2 | p.Gly116Ser | |||||||||||||||||||
| F22 | p.His748Arg | p.Arg143Cys | p.Glu485Val | ||||||||||||||||||
| F23 | p.Asn501Se | p.Glu546Lys | p.Glu485Val | ||||||||||||||||||
| F24 | p.Ala820Ser/p.Val622Leu | p.Ile215Val | |||||||||||||||||||
| F25 | p.Arg668Cys | c.1777-4C>T | |||||||||||||||||||
| F26 | c.1500+5G>T | ||||||||||||||||||||
| F27 | p.Ala459Val | ||||||||||||||||||||
| F28 | p.Gly562Arg | ||||||||||||||||||||
| F29 | p.Pro240His | ||||||||||||||||||||
| F30 | p.Val744Met | ||||||||||||||||||||
| F31 | p.Ala459Val | ||||||||||||||||||||
| F32 | p.Arg656Gln | ||||||||||||||||||||
| F33 | |||||||||||||||||||||
| F34 | |||||||||||||||||||||
| F35 | |||||||||||||||||||||
| F36 | |||||||||||||||||||||
| F37 | |||||||||||||||||||||
| F38 | |||||||||||||||||||||
| F39 | |||||||||||||||||||||
| F40 | |||||||||||||||||||||
| F41 | |||||||||||||||||||||
| F42 | |||||||||||||||||||||
| F43 | |||||||||||||||||||||
| F44 | |||||||||||||||||||||
| F45 | |||||||||||||||||||||
| F46 | |||||||||||||||||||||
| F47 | |||||||||||||||||||||
| F48 | |||||||||||||||||||||
| F49 | |||||||||||||||||||||
| F50 | |||||||||||||||||||||
| F51 | |||||||||||||||||||||
| Non-gastric | F52 | p.Val172Met | p.Leu349Phe | ||||||||||||||||||
| F53 | p.Asn501Ser | p.Glu546Lys | p.Glu485Val | ||||||||||||||||||
| F54 | c.1500+5G>T | ||||||||||||||||||||
| F55 | p.Arg849Gln | ||||||||||||||||||||
| F56 | |||||||||||||||||||||
| F57 | |||||||||||||||||||||
| SPORADIC | gNET | S1 | p.Ile215Val | ||||||||||||||||||
| S2 | c.1777-4C>T | ||||||||||||||||||||
| S3 | p.Val172Met | ||||||||||||||||||||
| S4 | |||||||||||||||||||||
| S5 | |||||||||||||||||||||
| S6 | |||||||||||||||||||||
| S7 | |||||||||||||||||||||
| S8 | |||||||||||||||||||||
| S9 | |||||||||||||||||||||
| S10 | |||||||||||||||||||||
| S11 | |||||||||||||||||||||
| CAG | S12 | p.Arg143Cys | p.Val455Met | ||||||||||||||||||
| S13 | p.Pro114Leu | ||||||||||||||||||||
| S14 | c.1500+5G>T | p.Val172Met | p.Glu546Lys | ||||||||||||||||||
| S15 | c.313+4C>T | ||||||||||||||||||||
| S16 | |||||||||||||||||||||
| S17 | |||||||||||||||||||||
| S18 | |||||||||||||||||||||
| S19 | |||||||||||||||||||||
| Pathology found in the proband and other family members | |||||||||||||||||||||
| Pathology found only in the proband | |||||||||||||||||||||
| Pathology not found in proband but in other members of the family | |||||||||||||||||||||
| Pathologies | Autoimmune Polyendocrine Syndrome (APS) | NON-APS | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| APS1 * | APS2 * | APS3 (Graves) | APS3 (Hashimoto) | APS3A (DM1) | APS3B (gNET) | APS3B (CAG) | APS3C (skin) | APS3D (collagen) | non-gastric ** | DM2 | Autoinflammatory | TOTAL | |||||||||||||||
| Familial | N | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) |
| APS1 * | 3 | NA | NA | 0 | 0.0 | 0 | 0.0 | 2 | 66.7 | 1 | 33.3 | 3 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 6 | 1.7 |
| APS2 * | 1 | 0 | 0.0 | NA | NA | 0 | 0.0 | 1 | 100.0 | 1 | 100.0 | 0 | 0.0 | 1 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 3 | 0.9 |
| APS3 (Graves) | 9 | 0 | 0.0 | 0 | 0.0 | NA | NA | NA | NA | 1 | 11.1 | 0 | 0.0 | 9 | 100.0 | 1 | 11.1 | 1 | 11.1 | 0 | 0.0 | 1 | 11.1 | 1 | 11.1 | 14 | 4.1 |
| APS3 (Hashimoto) | 43 | 2 | 4.6 | 1 | 2.3 | NA | NA | NA | NA | 6 | 13.9 | 9 | 20.9 | 28 | 65.1 | 13 | 30.2 | 7 | 16.3 | 6 | 13.9 | 7 | 16.3 | 15 | 34.9 | 88 | 25.7 |
| APS3A (DM1) | 8 | 1 | 12.5 | 1 | 12.5 | 1 | 12.5 | 6 | 75.0 | NA | NA | 1 | 12.5 | 6 | 75.0 | 3 | 37.5 | 0 | 0.0 | 1 | 12.5 | 0 | 0.0 | 0 | 0.0 | 19 | 5.6 |
| APS 3B (gNETs) | 11 | 3 | 27.3 | 0 | 0.0 | 0 | 0.0 | 9 | 81.8 | 1 | 9.1 | NA | NA | NA | NA | 1 | 9.1 | 1 | 9.1 | NA | NA | 4 | 36.36 | 3 | 27.3 | 22 | 6.4 |
| APS 3B (CAG) | 38 | 0 | 0.0 | 1 | 2.6 | 9 | 23.7 | 28 | 73.7 | 6 | 15.8 | NA | NA | NA | NA | 10 | 26.3 | 5 | 13.2 | NA | NA | 5 | 13.2 | 9 | 23.7 | 73 | 21.3 |
| APS3C (skin) | 15 | 0 | 0.0 | 0 | 0.0 | 1 | 6.7 | 13 | 86.7 | 3 | 20.0 | 1 | 6.7 | 10 | 66.7 | NA | NA | 3 | 20.0 | 4 | 26.7 | 2 | 13.3 | 2 | 13.3 | 35 | 10.2 |
| APS3D (collagen) | 9 | 0 | 0.0 | 0 | 0.0 | 1 | 11.1 | 7 | 77.8 | 0 | 0.0 | 1 | 11.1 | 5 | 55.6 | 3 | 33.3 | NA | NA | 3 | 33.3 | 0 | 0.0 | 6 | 66.7 | 23 | 6.7 |
| DM2 | 9 | 0 | 0.0 | 0 | 0.0 | 1 | 11.1 | 7 | 77.8 | 0 | 0.0 | 4 | 44.4 | 5 | 55.6 | 2 | 22.2 | 0 | 0.0 | 0 | 0.0 | NA | NA | 2 | 22.2 | 21 | 6.1 |
| Autoinflammatory | 16 | 0 | 0.0 | 0 | 0.0 | 1 | 6.2 | 15 | 93.7 | 0 | 0.0 | 3 | 18.7 | 9 | 56.2 | 2 | 12.5 | 6 | 37.5 | 4 | 25.0 | 2 | 12.5 | NA | NA | 38 | 11.1 |
| APS (non-gastric) | 6 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 6 | 100.0 | 1 | 16.7 | NA | NA | NA | NA | 4 | 66.7 | 3 | 50.0 | NA | NA | 0 | 0.0 | 4 | 66.7 | 18 | * |
| TOTAL | 162 | 6 | 3 | 14 | 88 | 19 | 22 | 73 | 35 | 23 | 18 | * | 21 | 38 | 342 | ||||||||||||
| APS1 * | APS2 * | APS3 (Graves) | APS3 (Hashimoto) | APS3A (DM1) | APS3B (gNET) | APS3B (CAG) | APS3C (skin) | APS3D (collagen) | non-gastric ** | DM2 | Autoinflammatory | TOTAL | |||||||||||||||
| SPORADIC | N | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) |
| APS1 * | 0 | NA | NA | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| APS2 * | 1 | 0 | 0.0 | NA | NA | 0 | 0.0 | 1 | 100.0 | 0 | 0.0 | 1 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 2.9 |
| APS3 (Graves) | 3 | 0 | 0.0 | 0 | 0.0 | NA | NA | NA | NA | 0 | 0.0 | 0 | 0.0 | 3 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 1 | 33.3 | 1 | 33.3 | 5 | 7.1 |
| APS3 (Hashimoto) | 8 | 0 | 0.0 | 1 | 12.5 | NA | NA | NA | NA | 2 | 25.0 | 4 | 50.0 | 4 | 50.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 25.0 | 1 | 12.5 | 14 | 20.0 |
| APS3A (DM1) | 2 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 100.0 | NA | NA | 0 | 0.0 | 2 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 1 | 50.0 | 1 | 50.0 | 6 | 8.6 |
| APS 3B (gNETs) | 11 | 0 | 0.0 | 1 | 9.1 | 0 | 0.0 | 4 | 36.4 | 0 | 0.0 | NA | NA | NA | NA | 0 | 0.0 | 0 | 0.0 | NA | NA | 1 | 9.1 | 1 | 9.1 | 7 | 10.0 |
| APS 3B (CAG) | 8 | 0 | 0.0 | 0 | 0.0 | 3 | 37.5 | 4 | 50.0 | 2 | 25.0 | NA | NA | NA | NA | 1 | 12.5 | 0 | 0.0 | NA | NA | 3 | 37.5 | 2 | 25.0 | 15 | 21.4 |
| APS3C (skin) | 1 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 1 | 100.0 | NA | NA | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 1 | 1.4 |
| APS3D (collagen) | 0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | NA | NA | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| DM2 | 4 | 0 | 0.0 | 0 | 0.0 | 1 | 25.00 | 2 | 50.0 | 1 | 25.0 | 1 | 25.0 | 3 | 75.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | NA | NA | 3 | 75.0 | 11 | 15.7 |
| Autoinflammatory | 3 | 0 | 0.0 | 0 | 0.0 | 1 | 33.3 | 1 | 33.3 | 1 | 33.3 | 1 | 33.3 | 2 | 66.7 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 3 | 100.0 | NA | NA | 9 | 12.9 |
| APS (non-gastric) | 0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | NA | NA | NA | NA | 0 | 0.0 | 0 | 0.0 | NA | NA | 0 | 0.0 | 0 | 0.0 | 0 | * |
| TOTAL | 41 | 0 | 2 | 5 | 14 | 6 | 7 | 15 | 1 | 0 | 0 | * | 11 | 9 | 70 | ||||||||||||
| APS1 * | APS2 * | APS3 (Graves) | APS3 (Hashimoto) | APS3A (DM1) | APS3B (gNET) | APS3B (CAG) | APS3C (skin) | APS3D (collagen) | non-gastric ** | DM2 | Autoinflammatory | TOTAL | |||||||||||||||
| TOTAL SERIES | N | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) |
| APS1 * | 3 | NA | NA | 0 | 0.0 | 0 | 0.0 | 2 | 66.7 | 1 | 33.3 | 3 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 6 | 1.5 |
| APS2 * | 2 | 0 | 0.0 | NA | NA | 0 | 0.0 | 2 | 100.0 | 1 | 50.0 | 1 | 50.0 | 1 | 50.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 5 | 1.2 |
| APS3 (Graves) | 12 | 0 | 0.0 | 0 | 0.0 | NA | NA | NA | NA | 1 | 8.3 | 0 | 0.0 | 12 | 100.0 | 1 | 8.3 | 1 | 8.3 | 0 | 0.0 | 2 | 16.7 | 2 | 16.7 | 19 | 4.6 |
| APS3 (Hashimoto) | 51 | 2 | 3.9 | 2 | 3.9 | NA | NA | NA | NA | 8 | 15.7 | 13 | 25.5 | 32 | 62.7 | 13 | 25.5 | 7 | 13.7 | 6 | 11.8 | 9 | 17.6 | 16 | 31.4 | 102 | 24.8 |
| APS3A (DM1) | 10 | 1 | 10.0 | 1 | 10.0 | 1 | 10.0 | 8 | 80.0 | NA | NA | 1 | 10.0 | 8 | 80.0 | 3 | 30.0 | 0 | 0.0 | 1 | 10.0 | 1 | 10.0 | 1 | 10.0 | 25 | 6.1 |
| APS 3B (gNETs) | 22 | 3 | 13.6 | 1 | 4.5 | 0 | 0.0 | 13 | 59.1 | 1 | 4.5 | NA | NA | NA | NA | 1 | 4.5 | 1 | 4.5 | NA | NA | 5 | 22.7 | 4 | 18.2 | 29 | 7.0 |
| APS 3B (CAG) | 46 | 0 | 0.0 | 1 | 2.2 | 12 | 26.1 | 32 | 69.6 | 8 | 17.4 | NA | NA | NA | NA | 11 | 23.9 | 5 | 10.9 | NA | NA | 8 | 17.4 | 11 | 23.9 | 88 | 21.4 |
| APS3C (skin) | 16 | 0 | 0.0 | 0 | 0.0 | 1 | 6.2 | 13 | 81.2 | 3 | 18.7 | 1 | 6.2 | 11 | 68.7 | NA | NA | 3 | 18.5 | 4 | 25.0 | 2 | 12.5 | 2 | 12.5 | 36 | 8.7 |
| APS3D (collagen) | 9 | 0 | 0.0 | 0 | 0.0 | 1 | 11.11 | 7 | 77.8 | 0 | 0.0 | 1 | 11.1 | 5 | 55.6 | 3 | 33.3 | NA | NA | 3 | 33.3 | 0 | 0.0 | 6 | 66.7 | 23 | 5.6 |
| DM2 | 13 | 0 | 0.0 | 0 | 0.0 | 2 | 15.4 | 9 | 69.2 | 1 | 7.7 | 5 | 38.5 | 8 | 61.5 | 2 | 15.4 | 0 | 0.0 | 0 | 0.0 | NA | NA | 5 | 38.5 | 32 | 7.8 |
| Autoinflammatory | 19 | 0 | 0.0 | 0 | 0.0 | 2 | 10.5 | 16 | 84.2 | 1 | 5.3 | 4 | 21.0 | 11 | 57.9 | 2 | 10.5 | 6 | 31.6 | 4 | 21.0 | 5 | 26.3 | NA | NA | 47 | 11.4 |
| APS (non-gastric) | 6 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 6 | 100.0 | 1 | 16.7 | NA | NA | NA | NA | 4 | 66.7 | 3 | 50.0 | NA | NA | 0 | 0.0 | 4 | 66.7 | 18 | * |
| TOTAL | 203 | 6 | 5 | 19 | 102 | 25 | 29 | 88 | 36 | 23 | 18 | * | 32 | 47 | 412 | ||||||||||||
| Total Alleles | Autoimmune Polyendocrine Syndrome (APS) | NON-APS | TOTAL | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| APS1 * | APS2 * | APS3 (Graves) | APS3 (Hashimoto) | APS3A (DM1) | APS3B (gNET) | APS3B (CAG) | APS3C (skin) | APS3D (collagen) | Non-gastric | DM2 | Autoinfl. | |||||||||||||||||||||||||||||||
| Patients | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) | N | P | (%) |
| Familial | 110 | 48 | 43.6 | 6 | 3 | 50.0 | 2 | 1 | 50.0 | 18 | 8 | 44.4 | 86 | 36 | 41.9 | 16 | 6 | 37.5 | 22 | 10 | 45.5 | 76 | 30 | 39.5 | 30 | 16 | 53.3 | 20 | 8 | 40.0 | 12 | 8 | 66.7 | 18 | 9 | 50.0 | 32 | 8 | 25.0 | 338 | 143 | 42.3 |
| Sporadic | 38 | 10 | 26.3 | 0 | 0 | NA | 2 | 1 | 50.0 | 6 | 3 | 50.0 | 14 | 5 | 35.7 | 4 | 2 | 50.0 | 22 | 3 | 13.6 | 16 | 7 | 43.8 | 2 | 0 | 0.0 | 0 | 0 | NA | 0 | 0 | NA | 6 | 0 | 0.0 | 6 | 0 | 0.0 | 78 | 21 | 26.9 |
| TOTAL | 148 | 58 | 39.2 | 6 | 3 | 50.0 | 4 | 2 | 50.0 | 24 | 11 | 45.8 | 100 | 41 | 41.0 | 20 | 8 | 40.0 | 44 | 13 | 29.5 | 92 | 37 | 40.2 | 32 | 16 | 50.0 | 20 | 8 | 40.0 | 12 | 8 | 66.7 | 24 | 9 | 37.5 | 38 | 8 | 21.1 | 416 | 164 | 39.4 |
| Total Variants | APS1 * | APS2 * | APS3 (Graves) | APS3 (Hashimoto) | APS3A (DM1) | APS3B (gNET) | APS3B (CAG) | APS3C (skin) | APS3D (collagen) | Non-gastric | DM2 | Autoinfl. | TOTAL | |||||||||||||||||||||||||||||
| Genes | N | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | ||||||||||||||
| SLC26A9 | 16 | 24.6 | 1 | 33.3 | 1 | 50.0 | 3 | 27.3 | 12 | 25.5 | 3 | 33.3 | 2 | 13.3 | 11 | 24.4 | 7 | 38.9 | 2 | 16.7 | 3 | 42.9 | 5 | 50.0 | 3 | 30.0 | 50 | 27.5 | ||||||||||||||
| SLC9A4 | 2 | 3.1 | 0 | 0.0 | 0 | 0.0 | 2 | 18.2 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 4.4 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 20.0 | 0 | 0.0 | 6 | 3.3 | ||||||||||||||
| SLC9A2 | 1 | 1.5 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 1 | 2.1 | 0 | 0.0 | 0 | 0.0 | 1 | 2.2 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 1.1 | ||||||||||||||
| PTH1R | 8 | 12.3 | 0 | 0.0 | 0 | 0.0 | 3 | 27.3 | 5 | 10.6 | 2 | 22.2 | 1 | 6.7 | 6 | 13.3 | 2 | 11.1 | 2 | 16.7 | 1 | 14.3 | 0 | 0.0 | 1 | 10.0 | 22 | 12.1 | ||||||||||||||
| PTH2R | 5 | 7.7 | 1 | 33.3 | 0 | 0.0 | 0 | 0.0 | 4 | 8.5 | 0 | 0.0 | 1 | 6.7 | 4 | 8.9 | 1 | 5.6 | 2 | 16.7 | 0 | 0.0 | 0 | 0.0 | 1 | 10.0 | 14 | 7.7 | ||||||||||||||
| SLC4A2 | 6 | 9.2 | 1 | 33.3 | 0 | 0.0 | 1 | 9.1 | 5 | 10.6 | 0 | 0.0 | 2 | 13.3 | 4 | 8.9 | 0 | 0.0 | 1 | 8.3 | 0 | 0.0 | 1 | 10.0 | 1 | 10.0 | 16 | 8.8 | ||||||||||||||
| SLC26A7 | 11 | 16.9 | 0 | 0.0 | 0 | 0.0 | 1 | 9.1 | 8 | 17.0 | 1 | 11.1 | 2 | 13.3 | 9 | 20.0 | 5 | 27.8 | 2 | 16.7 | 2 | 28.6 | 0 | 0.0 | 1 | 10.0 | 29 | 15.9 | ||||||||||||||
| KCNQ1 | 2 | 3.1 | 0 | 0.0 | 0 | 0.0 | 1 | 9.1 | 1 | 2.1 | 0 | 0.0 | 0 | 0.0 | 2 | 4.4 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 1 | 10.0 | 5 | 2.7 | ||||||||||||||
| ATP4A | 14 | 21.5 | 0 | 0.0 | 1 | 50.0 | 0 | 0.0 | 11 | 23.4 | 3 | 33.3 | 7 | 46.7 | 6 | 13.3 | 3 | 16.7 | 3 | 25.0 | 1 | 14.3 | 2 | 20.0 | 2 | 20.0 | 38 | 20.9 | ||||||||||||||
| TOTAL | 65 | 3 | 4.6 | 2 | 3.1 | 11 | 16.9 | 47 | 72.3 | 9 | 13.8 | 15 | 23.1 | 45 | 69.2 | 18 | 27.7 | 12 | 18.5 | 7 | 10.8 | 10 | 15.4 | 10 | 15.4 | 182 | ||||||||||||||||
| Associated | Total | APS1 * | APS2 * | APS3 (Graves) | APS3 (Hashimoto) | APS3A (DM1) | APS3B (gNET) | APS3B (CAG) | APS3C (skin) | APS3D (collagen) | Non-gastric | DM2 | Autoinfl. | TOTAL | ||||||||||||||||||||||||||||
| Pathologies | N | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | P | (%) | ||||||||||||||
| APS1 * | 2 | 2.0 | NA | NA | 0 | 0.0 | 0 | 0.0 | 1 | 50.0 | 1 | 50.0 | 2 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 4 | 2.2 | ||||||||||||||
| APS2 * | 2 | 2.0 | 0 | 0.0 | NA | NA | 0 | 0.0 | 2 | 100.0 | 1 | 50.0 | 1 | 50.0 | 1 | 50.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 5 | 2.7 | ||||||||||||||
| (Graves) | 7 | 6.9 | 0 | 0.0 | 0 | 0.0 | NA | NA | NA | NA | 1 | 14.3 | 0 | 0.0 | 7 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 1 | 14.3 | 0 | 0.0 | 9 | 4.8 | ||||||||||||||
| (Hashimoto) | 27 | 26.7 | 1 | 3.7 | 2 | 7.4 | NA | NA | NA | NA | 4 | 14.8 | 7 | 25.9 | 15 | 55.6 | 8 | 29.6 | 3 | 11.1 | 5 | 18.5 | 4 | 14.8 | 5 | 18.5 | 49 | 26.3 | ||||||||||||||
| APS3A | 6 | 5.9 | 1 | 16.7 | 1 | 16.7 | 1 | 16.7 | 4 | 66.7 | NA | NA | 1 | 16.7 | 4 | 66.7 | 1 | 16.7 | 0 | 0.0 | 1 | 16.7 | 0 | 0.0 | 0 | 0.0 | 13 | 7.0 | ||||||||||||||
| (gNETs) | 10 | 9.9 | 2 | 20.0 | 1 | 10.0 | 0 | 0.0 | 7 | 70.0 | 1 | 10.0 | NA | NA | NA | NA | 1 | 10.0 | 0 | 0.0 | NA | NA | 3 | 30.0 | 1 | 10.0 | 16 | 8.6 | ||||||||||||||
| (CAG) | 23 | 22.8 | 0 | 0.0 | 1 | 4.3 | 7 | 30.4 | 15 | 65.2 | 4 | 17.4 | NA | NA | NA | NA | 4 | 17.4 | 2 | 8.7 | NA | NA | 3 | 13.0 | 1 | 4.3 | 37 | 19.9 | ||||||||||||||
| APS3C | 9 | 8.9 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 8 | 88.9 | 1 | 11.1 | 1 | 11.1 | 4 | 44.4 | NA | NA | 2 | 22.2 | 4 | 44.4 | 1 | 11.1 | 2 | 22.2 | 19 | 10.2 | ||||||||||||||
| APS3D | 4 | 4.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 3 | 75.0 | 0 | 0.0 | 0 | 0.0 | 2 | 50.0 | 2 | 50.0 | NA | NA | 2 | 50.0 | 0 | 0.0 | 3 | 75.0 | 10 | 5.4 | ||||||||||||||
| DM II | 6 | 5.9 | 0 | 0.0 | 0 | 0.0 | 1 | 16.7 | 4 | 66.7 | 0 | 0.0 | 3 | 50.0 | 3 | 50.0 | 1 | 16.7 | 0 | 0.0 | 0 | 0.0 | NA | NA | 0 | 0.0 | 12 | 6.5 | ||||||||||||||
| Autoinfl. | 5 | 5.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 5 | 100.0 | 0 | 0.0 | 1 | 20.0 | 1 | 20.0 | 2 | 40.0 | 3 | 60.0 | 3 | 60.0 | 0 | 0.0 | NA | NA | 12 | 6.5 | ||||||||||||||
| non-gastric ** | 5 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 5 | 100.0 | 1 | 20.0 | NA | NA | NA | NA | 4 | 80.0 | 2 | 40.0 | NA | NA | 0 | 0.0 | 3 | 60.0 | 15 | 8.1 | |||||||||||||||
| TOTAL | 101 | 4 | 5 | 9 | 49 | 13 | 16 | 37 | 19 | 10 | 15 | 12 | 12 | 186 | ||||||||||||||||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvete, O.; Reyes, J.; Valdés-Socin, H.; Martin, P.; Marazuela, M.; Barroso, A.; Escalada, J.; Castells, A.; Torres-Ruiz, R.; Rodríguez-Perales, S.; et al. Alterations in SLC4A2, SLC26A7 and SLC26A9 Drive Acid–Base Imbalance in Gastric Neuroendocrine Tumors and Uncover a Novel Mechanism for a Co-Occurring Polyautoimmune Scenario. Cells 2021, 10, 3500. https://doi.org/10.3390/cells10123500
Calvete O, Reyes J, Valdés-Socin H, Martin P, Marazuela M, Barroso A, Escalada J, Castells A, Torres-Ruiz R, Rodríguez-Perales S, et al. Alterations in SLC4A2, SLC26A7 and SLC26A9 Drive Acid–Base Imbalance in Gastric Neuroendocrine Tumors and Uncover a Novel Mechanism for a Co-Occurring Polyautoimmune Scenario. Cells. 2021; 10(12):3500. https://doi.org/10.3390/cells10123500
Chicago/Turabian StyleCalvete, Oriol, José Reyes, Hernán Valdés-Socin, Paloma Martin, Mónica Marazuela, Alicia Barroso, Javier Escalada, Antoni Castells, Raúl Torres-Ruiz, Sandra Rodríguez-Perales, and et al. 2021. "Alterations in SLC4A2, SLC26A7 and SLC26A9 Drive Acid–Base Imbalance in Gastric Neuroendocrine Tumors and Uncover a Novel Mechanism for a Co-Occurring Polyautoimmune Scenario" Cells 10, no. 12: 3500. https://doi.org/10.3390/cells10123500
APA StyleCalvete, O., Reyes, J., Valdés-Socin, H., Martin, P., Marazuela, M., Barroso, A., Escalada, J., Castells, A., Torres-Ruiz, R., Rodríguez-Perales, S., Currás-Freixes, M., & Benítez, J. (2021). Alterations in SLC4A2, SLC26A7 and SLC26A9 Drive Acid–Base Imbalance in Gastric Neuroendocrine Tumors and Uncover a Novel Mechanism for a Co-Occurring Polyautoimmune Scenario. Cells, 10(12), 3500. https://doi.org/10.3390/cells10123500