
Unlocking the Complexity of Mitochondrial DNA: A Key to Understanding Neurodegenerative Disease Caused by Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Fundamentals of Human Mitochondrial Genetics
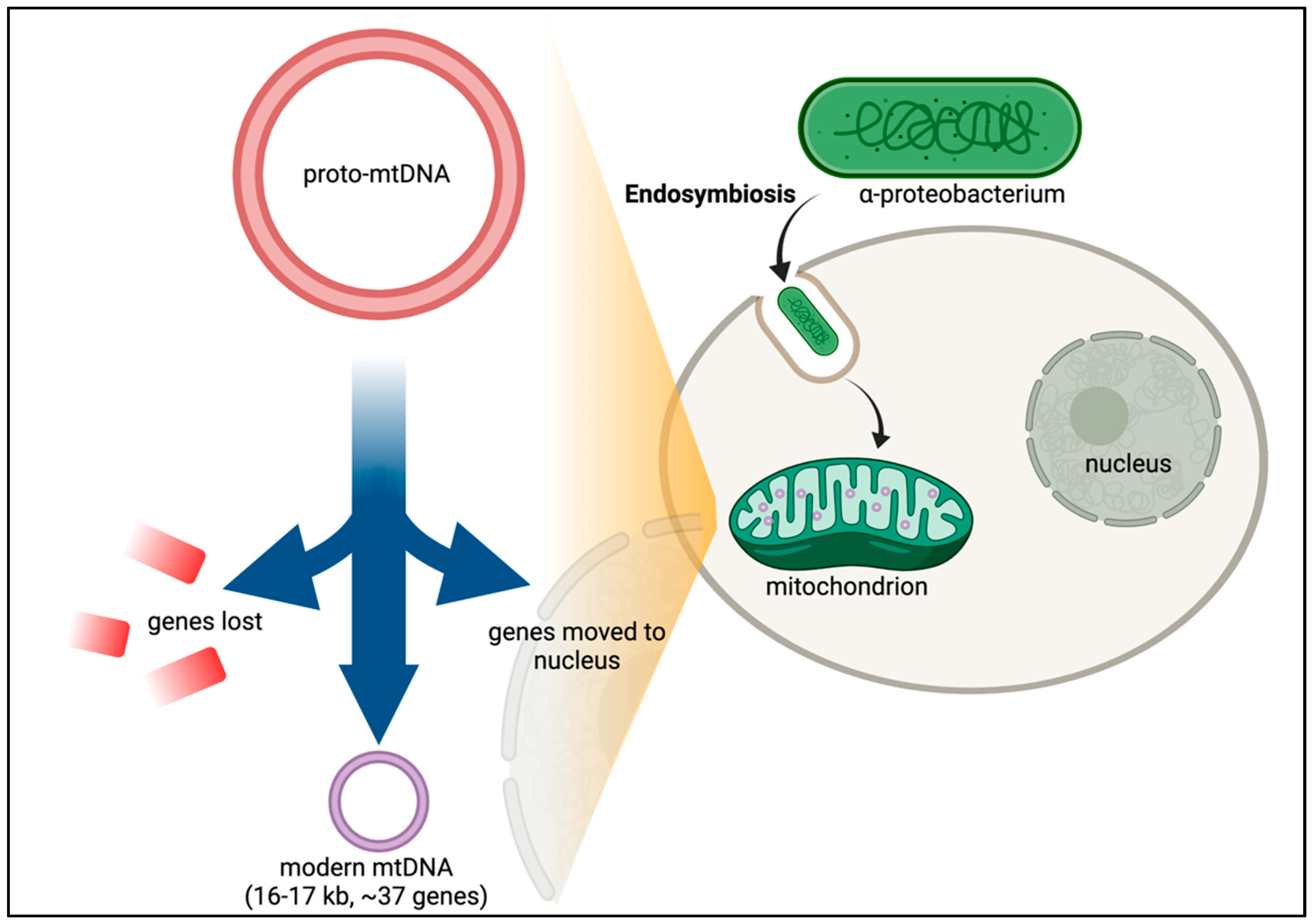
3. Origins of Mitochondria and mtDNA
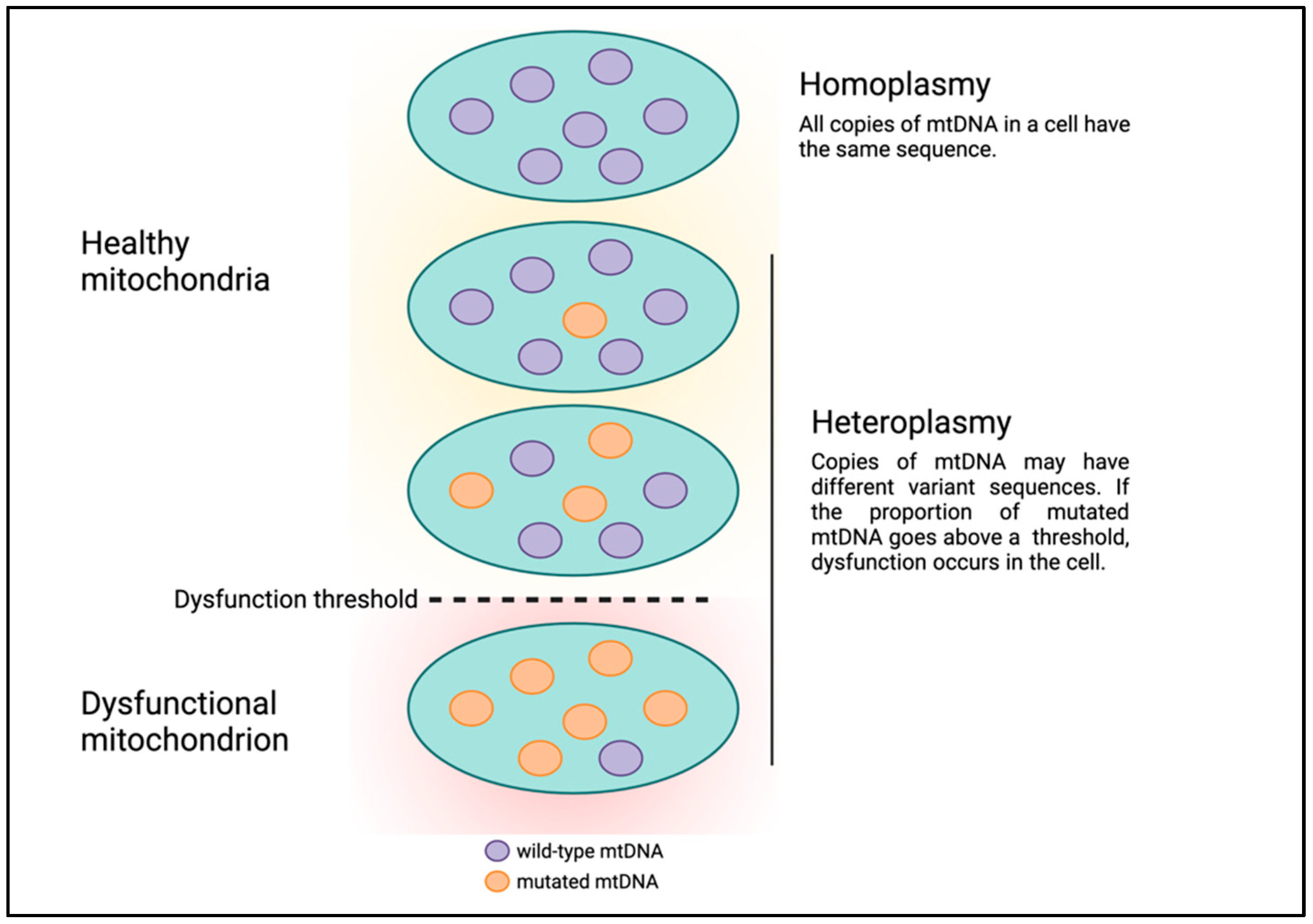
4. Mitochondrial DNA Variation
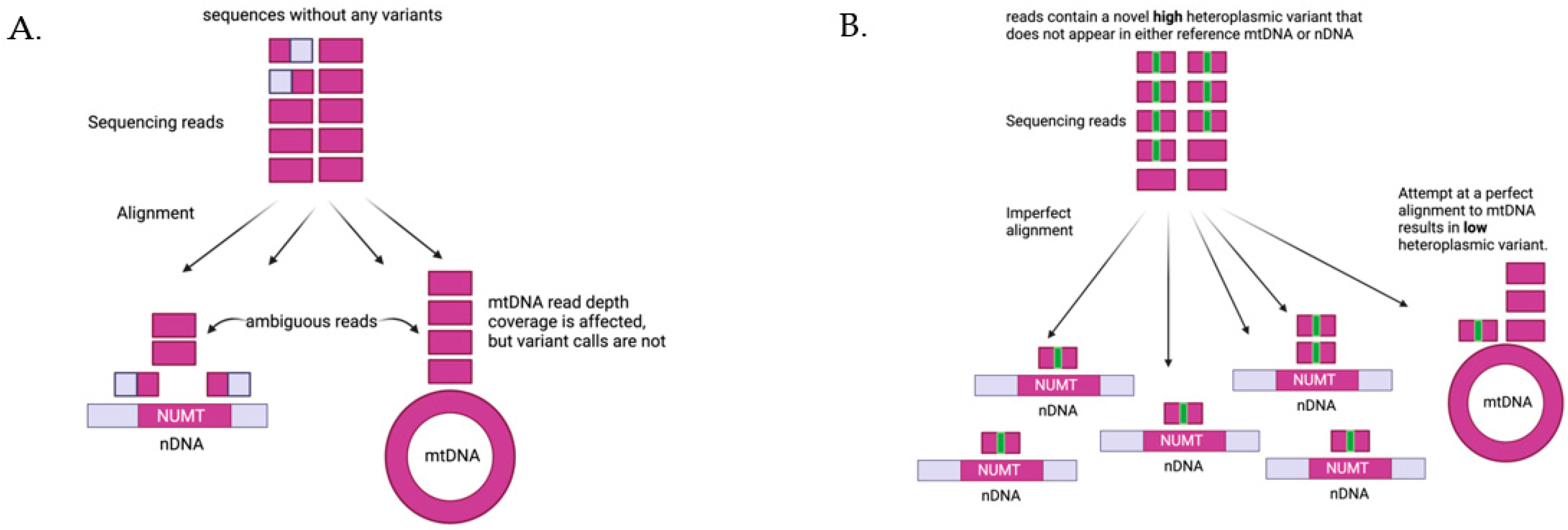
5. Quantifying Mitochondrial DNA Variation
6. Mitochondrial DNA Copy Number
7. Mitochondrial RNA
8. Secondary Neurodegenerative Disorders
8.1. Traumatic Brain Injury
8.2. Ischaemic Stroke
9. Future Directions and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Modesti, L.; Danese, A.; Vitto, V.A.M.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 1380. [Google Scholar] [CrossRef] [PubMed]
- Chalkia, D.; Singh, L.N.; Leipzig, J.; Lvova, M.; Derbeneva, O.; Lakatos, A.; Hadley, D.; Hakonarson, H.; Wallace, D.C. Association Between Mitochondrial DNA Haplogroup Variation and Autism Spectrum Disorders. JAMA Psychiatry 2017, 74, 1161–1168. [Google Scholar] [CrossRef]
- Singh, L.N.; Crowston, J.G.; Sanchez, M.I.G.L.; Van Bergen, N.J.; Kearns, L.S.; Hewitt, A.W.; Yazar, S.; Mackey, D.A.; Wallace, U.C.; Trounce, I.A. Mitochondrial DNA Variation and Disease Susceptibility in Primary Open-Angle Glaucoma. Investig. Opthalmol. Vis. Sci. 2018, 59, 4598–4602. [Google Scholar] [CrossRef] [PubMed]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial DNA Variation in Human Radiation and Disease. Cell 2015, 163, 33–38. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. The art of mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Macdonald, J.A.; Bothun, A.M.; Annis, S.N.; Sheehan, H.; Ray, S.; Gao, Y.; Ivanov, A.R.; Khrapko, K.; Tilly, J.L.; Woods, D.C. A nanoscale, multi-parametric flow cytometry-based platform to study mitochondrial heterogeneity and mitochondrial DNA dynamics. Commun. Biol. 2019, 2, 258. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef]
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Luo, H. Dating Alphaproteobacteria evolution with eukaryotic fossils. Nat. Commun. 2021, 12, 3324. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wu, D.; Goremykin, V.; Xiao, J.; Xu, Y.; Garg, S.; Zhang, C.; Martin, W.F.; Zhu, R. Phylogenetic analyses with systematic taxon sampling show that mitochondria branch within Alphaproteobacteria. Nat. Ecol. Evol. 2020, 4, 1213–1219. [Google Scholar] [CrossRef]
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef]
- Johnston, I.G.; Williams, B.P. Evolutionary Inference across Eukaryotes Identifies Specific Pressures Favoring Mitochondrial Gene Retention. Cell Syst. 2016, 2, 101–111. [Google Scholar] [CrossRef]
- Govindaraju, D.R.; Innan, H.; Veitia, R.A. The Muller’s Ratchet and Aging. Trends Genet. 2020, 36, 395–402. [Google Scholar] [CrossRef]
- Tria, F.D.K.; Brueckner, J.; Skejo, J.; Xavier, J.C.; Kapust, N.; Knopp, M.; Wimmer, J.L.E.; Nagies, F.S.P.; Zimorski, V.; Gould, S.B.; et al. Gene Duplications Trace Mitochondria to the Onset of Eukaryote Complexity. Genome Biol. Evol. 2021, 13, evab055. [Google Scholar] [CrossRef]
- Nei, M.; Suzuki, Y.; Nozawa, M. The Neutral Theory of Molecular Evolution in the Genomic Era. Annu. Rev. Genom. Hum. Genet. 2010, 11, 265–289. [Google Scholar] [CrossRef]
- Wallace, D.C. Why Do We Still Have a Maternally Inherited Mitochondrial DNA? Insights from Evolutionary Medicine. Annu. Rev. Biochem. 2007, 76, 781–821. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.-G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The Maintenance of Mitochondrial DNA Integrity—Critical Analysis and Update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.R.; Singh, K.K. Mitochondrial determinants of cancer health disparities. Semin. Cancer Biol. 2017, 47, 125–146. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Li, F.; Chen, D.; Wang, G.; Truong, C.K.; Enns, G.M.; Graham, B.; Milone, M.; Landsverk, M.L.; Wang, J.; et al. Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genet. Med. 2013, 15, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Weerts, M.J.A.; Timmermans, E.C.; Vossen, R.H.A.M.; Van Strijp, D.; Van den Hout–van Vroonhoven, M.C.G.N.; Van Ijcken, W.F.J.; Van Der Zaag, P.J.; Anvar, S.Y.; Sleijfer, S.; Martens, J.W.M. Sensitive detection of mitochondrial DNA variants for analysis of mitochondrial DNA-enriched extracts from frozen tumor tissue. Sci. Rep. 2018, 8, 2261. [Google Scholar] [CrossRef] [PubMed]
- Sondheimer, N.; Glatz, C.E.; Tirone, J.E.; Deardorff, M.A.; Krieger, A.M.; Hakonarson, H. Neutral mitochondrial heteroplasmy and the influence of aging. Hum. Mol. Genet. 2011, 20, 1653–1659. [Google Scholar] [CrossRef]
- Clark, M.J.; Chen, R.; Lam, H.Y.K.; Karczewski, K.J.; Chen, R.; Euskirchen, G.; Butte, A.J.; Snyder, M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011, 29, 908–914. [Google Scholar] [CrossRef]
- Falk, M.; Pierce, E.; Consugar, M.; Xie, M.H.; Guadalupe, M.; Hardy, O.; Rappaport, E.F.; Wallace, U.C.; Leproust, E.; Gai, X. Mitochondrial Disease Genetic Diagnostics: Optimized whole-exome analysis for all MitoCarta nuclear genes and the mitochondrial genome. Discov. Med. 2012, 14, 389–399. [Google Scholar]
- Srinivasainagendra, V.; Sandel, M.W.; Singh, B.; Sundaresan, A.; Mooga, V.P.; Bajpai, P.; Tiwari, H.K.; Singh, K.K. Migration of mitochondrial DNA in the nuclear genome of colorectal adenocarcinoma. Genome Med. 2017, 9, 31. [Google Scholar] [CrossRef]
- Dayama, G.; Emery, S.B.; Kidd, J.M.; Mills, R.E. The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res. 2014, 42, 12640–12649. [Google Scholar] [CrossRef] [PubMed]
- Hazkani-Covo, E.; Covo, S. Numt-Mediated Double-Strand Break Repair Mitigates Deletions during Primate Genome Evolution. PLoS Genet. 2008, 4, e1000237. [Google Scholar] [CrossRef]
- Mourier, T.; Hansen, A.J.; Willerslev, E.; Arctander, P. The Human Genome Project Reveals a Continuous Transfer of Large Mitochondrial Fragments to the Nucleus. Mol. Biol. Evol. 2001, 18, 1833–1837. [Google Scholar] [CrossRef]
- Calabrese, C.; Simone, D.; Diroma, M.A.; Santorsola, M.; Guttà, C.; Gasparre, G.; Picardi, E.; Pesole, G.; Attimonelli, M. MToolBox: A highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 2014, 30, 3115–3117. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, H.; Forer, L.; Fuchsberger, C.; Schöpf, B.; Kloss-Brandstätter, A.; Specht, G.; Kronenberg, F.; Schonherr, S. mtDNA-Server: Next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 2016, 44, W64–W69. [Google Scholar] [CrossRef]
- Ekblom, R.; Smeds, L.; Ellegren, H. Patterns of sequencing coverage bias revealed by ultra-deep sequencing of vertebrate mitochondria. BMC Genom. 2014, 15, 467. [Google Scholar] [CrossRef]
- Singh, L.N.; Ennis, B.; Loneragan, B.; Tsao, N.L.; Sanchez, M.I.G.L.; Li, J.; Acheampong, P.; Tran, O.; Trounce, I.A.; Zhu, Y.; et al. MitoScape: A big-data, machine-learning platform for obtaining mitochondrial DNA from next-generation sequencing data. PLoS Comput. Biol. 2021, 17, e1009594. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; Ludwig, L.S.; Muus, C.; Gohil, S.H.; Zhao, T.; Chiang, Z.; Pelka, K.; Verboon, J.M.; Luo, W.; Christian, E.; et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 2020, 39, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J.; Frith, M.C.; Tomii, K.; Horton, P. Mammalian NUMT insertion is non-random. Nucleic Acids Res. 2012, 40, 9073–9088. [Google Scholar] [CrossRef]
- Marom, S.; Blumberg, A.; Kundaje, A.; Mishmar, D. mtDNA Chromatin-like Organization Is Gradually Established during Mammalian Embryogenesis. iScience 2019, 12, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Choudhury, A.R.; Tiwari, H.K. Numtogenesis as a mechanism for development of cancer. Semin. Cancer Biol. 2017, 47, 101–109. [Google Scholar] [CrossRef]
- Picard, M. Blood mitochondrial DNA copy number: What are we counting? Mitochondrion 2021, 60, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sainz, A.G.; Shadel, G.S. Mitochondrial DNA: Cellular genotoxic stress sentinel. Trends Biochem. Sci. 2021, 46, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, H. Mitochondrial DNA Copy Number and Developmental Origins of Health and Disease (DOHaD). Int. J. Mol. Sci. 2021, 22, 6634. [Google Scholar] [CrossRef]
- Fries, G.R.; Zamzow, M.J.; Andrews, T.; Pink, O.; Scaini, G.; Quevedo, J. Accelerated aging in bipolar disorder: A comprehensive review of molecular findings and their clinical implications. Neurosci. Biobehav. Rev. 2020, 112, 107–116. [Google Scholar] [CrossRef]
- Kelly, R.D.W.; Mahmud, A.; McKenzie, M.; Trounce, I.; St John, J.C.S. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 2012, 40, 10124–10138. [Google Scholar] [CrossRef]
- Lien, L.; Chiou, H.Y.; Yeh, H.; Chiu, S.; Jeng, J.-S.; Lin, H.; Hu, C.; Hsieh, F.; Wei, Y.-H. Significant Association Between Low Mitochondrial DNA Content in Peripheral Blood Leukocytes and Ischemic Stroke. J. Am. Heart Assoc. 2017, 6, e006157. [Google Scholar] [CrossRef]
- Knez, J.; Winckelmans, E.; Plusquin, M.; Thijs, L.; Cauwenberghs, N.; Gu, Y.; Staessen, J.A.; Nawrot, T.; Kuznetsova, T. Correlates of Peripheral Blood Mitochondrial DNA Content in a General Population. Am. J. Epidemiol. 2015, 183, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Giordano, C.; D’Amati, G. Pathogenic expression of homoplasmic mtDNA mutations needs a complex nuclear–mitochondrial interaction. Trends Genet. 2003, 19, 257–262. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The Human Mitochondrial Transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef]
- Maas, S.; Kawahara, Y.; Tamburro, K.M.; Nishikura, K. A-to-I RNA Editing and Human Disease. RNA Biol. 2006, 3, 1–9. [Google Scholar] [CrossRef]
- Khermesh, K.; D’Erchia, A.M.; Barak, M.; Annese, A.; Wachtel, C.; Levanon, E.Y.; Picardi, E.; Eisenberg, E. Reduced levels of protein recoding by A-to-I RNA editing in Alzheimer’s disease. RNA 2015, 22, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef]
- Moreira, S.; Valach, M.; Aoulad-Aissa, M.; Otto, C.; Burger, G. Novel modes of RNA editing in mitochondria. Nucleic Acids Res. 2016, 44, 4907–4919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Nakahira, K.; Choi, A.M.K.; Gu, Z. Heteroplasmy concordance between mitochondrial DNA and RNA. Sci. Rep. 2019, 9, 12942. [Google Scholar] [CrossRef]
- Bar-Yaacov, D.; Avital, G.; Levin, L.; Richards, A.L.; Hachen, N.; Jaramillo, B.R.; Nekrutenko, A.; Zarivach, R.; Mishmar, D. RNA–DNA differences in human mitochondria restore ancestral form of 16S ribosomal RNA. Genome Res. 2013, 23, 1789–1796. [Google Scholar] [CrossRef]
- Rackham, O.; Shearwood, A.-M.J.; Mercer, T.R.; Davies, S.M.; Mattick, J.S.; Filipovska, A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA 2011, 17, 2085–2093. [Google Scholar] [CrossRef]
- Rorbach, J.; Minczuk, M. The post-transcriptional life of mammalian mitochondrial RNA. Biochem. J. 2012, 444, 357–373. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Mayne, K.; White, J.A.; McMurran, C.E.; Rivera, F.J.; de la Fuente, A.G. Aging and Neurodegenerative Disease: Is the Adaptive Immune System a Friend or Foe? Front. Aging Neurosci. 2020, 12, 305. [Google Scholar] [CrossRef]
- Walker, M.A.; Lareau, C.A.; Ludwig, L.S.; Karaa, A.; Sankaran, V.G.; Regev, A.; Mootha, V.K. Purifying Selection against Pathogenic Mitochondrial DNA in Human T Cells. N. Engl. J. Med. 2020, 383, 1556–1563. [Google Scholar] [CrossRef]
- Carelli, V.; Chan, D.C. Mitochondrial DNA: Impacting Central and Peripheral Nervous Systems. Neuron 2014, 84, 1126–1142. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Guardia-Laguarta, C.; Schon, E.A.; Przedborski, S. Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J. Clin. Investig. 2019, 129, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, F.; McFadyen, C.; Newcombe, V.; Synnot, A.; Donoghue, E.L.; Ripatti, S.; Steyerberg, E.W.; Gruen, R.L.; McAllister, T.W.; Rosand, J.; et al. Genetic Influences on Patient-Oriented Outcomes in Traumatic Brain Injury: A Living Systematic Review of Non-Apolipoprotein E Single-Nucleotide Polymorphisms. J. Neurotrauma 2021, 38, 1107–1123. [Google Scholar] [CrossRef] [PubMed]
- Nathoo, N.; Chetty, R.; Van Dellen, J.R.; Barnett, G.H. Genetic vulnerability following traumatic brain injury: The role of apolipoprotein E. Mol. Pathol. 2003, 56, 132–136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.; Lomnitski, L.; Michaelson, D.M.; Shohami, E. Motor and cognitive deficits in apolipoprotein E-deficient mice after closed head injury. Neuroscience 1997, 80, 1255–1262. [Google Scholar] [CrossRef]
- Lynch, J.R.; Tang, W.; Wang, H.; Vitek, M.P.; Bennett, E.R.; Sullivan, P.M.; Warner, D.S.; Laskowitz, D.T. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 2003, 278, 48529–48533. [Google Scholar] [CrossRef]
- Vitek, M.P.; Brown, C.M.; Colton, C.A. APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 2009, 30, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Huang, Y. Apolipoprotein E Sets the Stage: Response to Injury Triggers Neuropathology. Neuron 2012, 76, 871–885. [Google Scholar] [CrossRef]
- Chang, S.; Ma, T.R.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef]
- Chen, H.-K.; Liu, Z.; Meyer-Franke, A.; Brodbeck, J.; Miranda, R.D.; McGuire, J.G.; Pleiss, M.A.; Ji, Z.-S.; Balestra, M.E.; Walker, D.W.; et al. Small Molecule Structure Correctors Abolish Detrimental Effects of Apolipoprotein E4 in Cultured Neurons. J. Biol. Chem. 2012, 287, 5253–5266. [Google Scholar] [CrossRef]
- Nakamura, T.; Watanabe, A.; Fujino, T.; Hosono, T.; Michikawa, M. Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener. 2009, 4, 35. [Google Scholar] [CrossRef]
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Primers 2016, 2, 16084. [Google Scholar] [CrossRef] [PubMed]
- Sauerbeck, A.; Gao, J.; Readnower, R.; Liu, M.; Pauly, J.R.; Bing, G.; Sullivan, P.G. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp. Neurol. 2011, 227, 128–135. [Google Scholar] [CrossRef]
- Cheng, G.; Kong, R.-H.; Zhang, L.-M.; Zhang, J.-N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef]
- Friedman, G.; Froom, P.; Sazbon, L.; Grinblatt, I.; Shochina, M.; Tsenter, J.; Babaey, S.; Yehuda, B.; Groswasser, Z. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology 1999, 52, 244–248. [Google Scholar] [CrossRef]
- Chamelian, L.; Reis, M.; Feinstein, A. Six-month recovery from mild to moderate Traumatic Brain Injury: The role of APOE-epsilon4 allele. Brain 2004, 127, 2621–2628. [Google Scholar] [CrossRef] [PubMed]
- Crawford, F.C.; Vanderploeg, R.D.; Freeman, M.J.; Singh, S.; Waisman, M.; Michaels, L.; Abdullah, L.; Warden, D.; Lipsky, R.; Salazar, A.; et al. APOE genotype influences acquisition and recall following traumatic brain injury. Neurology 2002, 58, 1115–1118. [Google Scholar] [CrossRef]
- Han, S.D.; Drake, A.I.; Cessante, L.M.; Jak, A.J.; Houston, W.S.; Delis, D.C.; Filoteo, J.V.; Bondi, M.W. Apolipoprotein E and traumatic brain injury in a military population: Evidence of a neuropsychological compensatory mechanism? J. Neurol. Neurosurg. Psychiatry 2007, 78, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.R.; Roberts, G.W.; Graham, D.I. Amyloid β-Protein, APOE Genotype and Head Injury. Ann. N. Y. Acad. Sci. 1996, 777, 271–275. [Google Scholar] [CrossRef]
- Teasdale, G.M.; Nicoll, J.A.; Murray, G.; Fiddes, M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet 1997, 350, 1069–1071. [Google Scholar] [CrossRef]
- Carrieri, G.; Bonafè, M.; De Luca, M.; Rose, G.; Varcasia, O.; Bruni, A.; Maletta, R.; Nacmias, B.; Sorbi, S.; Corsonello, F.; et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Qual. Life Res. 2001, 108, 194–198. [Google Scholar] [CrossRef]
- Bulstrode, H.; Nicoll, J.A.R.; Hudson, G.; Chinnery, P.F.; Di Pietro, V.; Belli, A. Mitochondrial DNA and traumatic brain injury. Ann. Neurol. 2014, 75, 186–195. [Google Scholar] [CrossRef]
- Hrelia, P.; Sita, G.; Ziche, M.; Ristori, E.; Marino, A.; Cordaro, M.; Molteni, R.; Spero, V.; Malaguti, M.; Morroni, F.; et al. Common Protective Strategies in Neurodegenerative Disease: Focusing on Risk Factors to Target the Cellular Redox System. Oxid. Med. Cell. Longev. 2020, 2020, 8363245. [Google Scholar] [CrossRef]
- Conley, Y.P.; Okonkwo, D.O.; Deslouches, S.; Alexander, S.; Puccio, A.M.; Beers, S.R.; Ren, D. Mitochondrial Polymorphisms Impact Outcomes after Severe Traumatic Brain Injury. J. Neurotrauma 2014, 31, 34–41. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Russo, E.; Napoli, E. Healthy mitochondria for stroke cells. Brain Circ. 2018, 4, 95–98. [Google Scholar] [CrossRef]
- Lin, K.-L.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Wang, P.-W.; Chuang, J.-H.; Lin, T.-K. Quality Matters? The Involvement of Mitochondrial Quality Control in Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 9, 641. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.; Cotman, C.; Lynch, G.; Blass, J. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement. 2017, 13, 178–182.e17. [Google Scholar] [CrossRef]
- LoBue, C.; Cullum, C.M. POINT/COUNTER-POINT—Beyond the headlines: The actual evidence that traumatic brain injury is a risk factor for later-in-life dementia. Arch. Clin. Neuropsychol. 2020, 35, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Testai, F.D.; Gorelick, P.B. Inherited Metabolic Disorders and Stroke Part 1: Fabry Disease and Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Strokelike Episodes. Arch. Neurol. 2010, 67, 19–24. [Google Scholar] [CrossRef]
- DiMauro, S. Mitochondrial diseases. Biochim. Biophys. Acta (BBA)—Bioenerg. 2004, 1658, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Tetsuka, S.; Ogawa, T.; Hashimoto, R.; Kato, H. Clinical features, pathogenesis, and management of stroke-like episodes due to MELAS. Metab. Brain Dis. 2021, 36, 2181–2193. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Elliott, H.R.; Syed, A.; Rothwell, P.M. Mitochondrial DNA haplogroups and risk of transient ischaemic attack and ischaemic stroke: A genetic association study. Lancet Neurol. 2010, 9, 498–503. [Google Scholar] [CrossRef]
- Umbria, M.; Ramos, A.; Aluja, M.P.; Santos, C. The role of control region mitochondrial DNA mutations in cardiovascular disease: Stroke and myocardial infarction. Sci. Rep. 2020, 10, 2766. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Kuo, C.-W.; Lin, T.-K.; Ho, C.-J.; Wang, P.-W.; Chuang, J.-H.; Liou, C.-W. Ischemic Stroke Risk Associated with Mitochondrial Haplogroup F in the Asian Population. Cells 2020, 9, 1885. [Google Scholar] [CrossRef]
- Trounce, I.A.; Kim, Y.L.; Jun, A.S.; Wallace, D.C. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1996; Volume 264, pp. 484–509. [Google Scholar]
- Wei, R.; Ni, Y.; Bazeley, P.; Grandhi, S.; Wang, J.; Li, S.T.; Hazen, S.L.; Tang, W.H.W.; LaFramboise, T. Mitochondrial DNA Content is Linked to Cardiovascular Disease Patient Phenotypes. J. Am. Heart Assoc. 2021, 10, e018776. [Google Scholar] [CrossRef]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. Mol. Mech. Mutagen. 2015, 776, 84–97. [Google Scholar] [CrossRef]
- Wang, J.; Xiong, S.; Xie, C.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 2005, 93, 953–962. [Google Scholar] [CrossRef]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA Damage Can Promote Atherosclerosis Independently of Reactive Oxygen Species Through Effects on Smooth Muscle Cells and Monocytes and Correlates with Higher-Risk Plaques in Humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef]
- Yue, P.; Jing, S.; Liu, L.; Ma, F.; Zhang, Y.; Wang, C.; Duan, H.; Zhou, K.; Hua, Y.; Wu, G.; et al. Association between mitochondrial DNA copy number and cardiovascular disease: Current evidence based on a systematic review and meta-analysis. PLoS ONE 2018, 13, e0206003. [Google Scholar] [CrossRef]
- Akbari, M.; Morevati, M.; Croteau, D.; Bohr, V.A. The role of DNA base excision repair in brain homeostasis and disease. DNA Repair 2015, 32, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Kirkwood, T.B.; Bohr, V.A. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 2019, 54, 100940. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186, 111207. [Google Scholar] [CrossRef] [PubMed]
- Sia, E.A.; Stein, A. Chapter 8—Human mitochondrial DNA repair. In The Human Mitochondrial Genome; Gasparre, G., Porcelli, A.M., Eds.; Academic Press: London, UK; San Diego, CA, USA, 2020; pp. 173–194. [Google Scholar]
- Schumacker, P.T.; Gillespie, M.N.; Nakahira, K.; Choi, A.M.K.; Crouser, E.D.; Piantadosi, C.A.; Bhattacharya, J. Mitochondria in lung biology and pathology: More than just a powerhouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L962–L974. [Google Scholar] [CrossRef]
- Weissman, L.; Jo, D.-G.; Sørensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef]
- Simon, R.; Meller, R.; Yang, T.; Pearson, A.; Wilson, G. Enhancing Base Excision Repair of Mitochondrial DNA to Reduce Ischemic Injury Following Reperfusion. Transl. Stroke Res. 2019, 10, 664–671. [Google Scholar] [CrossRef]
- SenGupta, T.; Palikaras, K.; Esbensen, Y.Q.; Konstantinidis, G.; Galindo, F.J.N.; Achanta, K.; Kassahun, H.; Stavgiannoudaki, I.; Bohr, V.A.; Akbari, M.; et al. Base excision repair causes age-dependent accumulation of single-stranded DNA breaks that contribute to Parkinson disease pathology. Cell Rep. 2021, 36, 109668. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, L.N.; Kao, S.-H.; Wallace, D.C. Unlocking the Complexity of Mitochondrial DNA: A Key to Understanding Neurodegenerative Disease Caused by Injury. Cells 2021, 10, 3460. https://doi.org/10.3390/cells10123460
Singh LN, Kao S-H, Wallace DC. Unlocking the Complexity of Mitochondrial DNA: A Key to Understanding Neurodegenerative Disease Caused by Injury. Cells. 2021; 10(12):3460. https://doi.org/10.3390/cells10123460
Chicago/Turabian StyleSingh, Larry N., Shih-Han Kao, and Douglas C. Wallace. 2021. "Unlocking the Complexity of Mitochondrial DNA: A Key to Understanding Neurodegenerative Disease Caused by Injury" Cells 10, no. 12: 3460. https://doi.org/10.3390/cells10123460
APA StyleSingh, L. N., Kao, S.-H., & Wallace, D. C. (2021). Unlocking the Complexity of Mitochondrial DNA: A Key to Understanding Neurodegenerative Disease Caused by Injury. Cells, 10(12), 3460. https://doi.org/10.3390/cells10123460