
Myofibre Hyper-Contractility in Horses Expressing the Myosin Heavy Chain Myopathy Mutation, MYH1E321G


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Solutions
2.3. Muscle Preparation and Fibre Permeabilisation
2.4. Mant-ATP Chase Experiments
2.5. Single Muscle Fibre Contractility
2.6. Fibre Typing
2.7. Statistics
3. Results
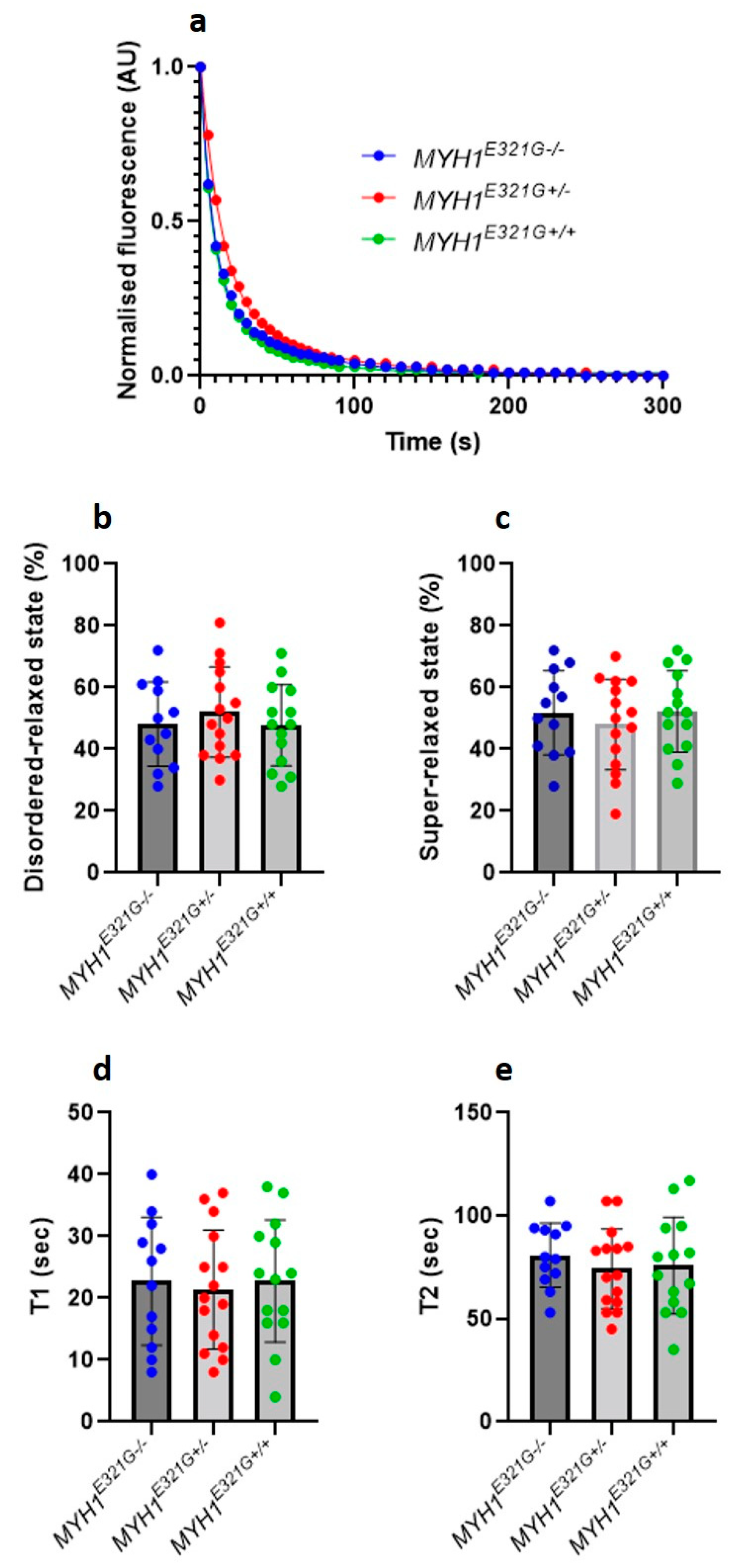
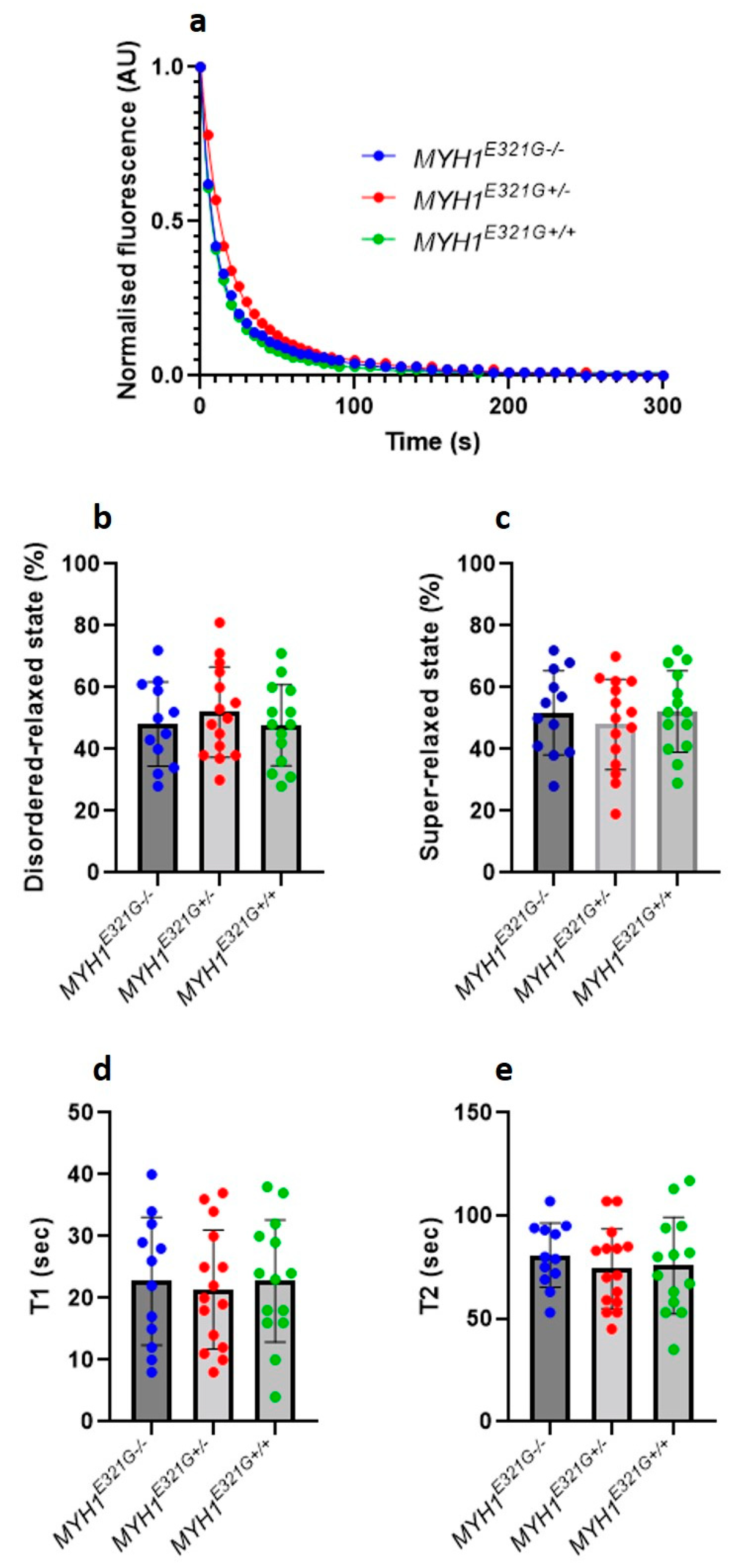
3.1. The Proportion of Myosin Heads in the Super-Relaxed State Is Preserved in the Presence of MYH1E321G
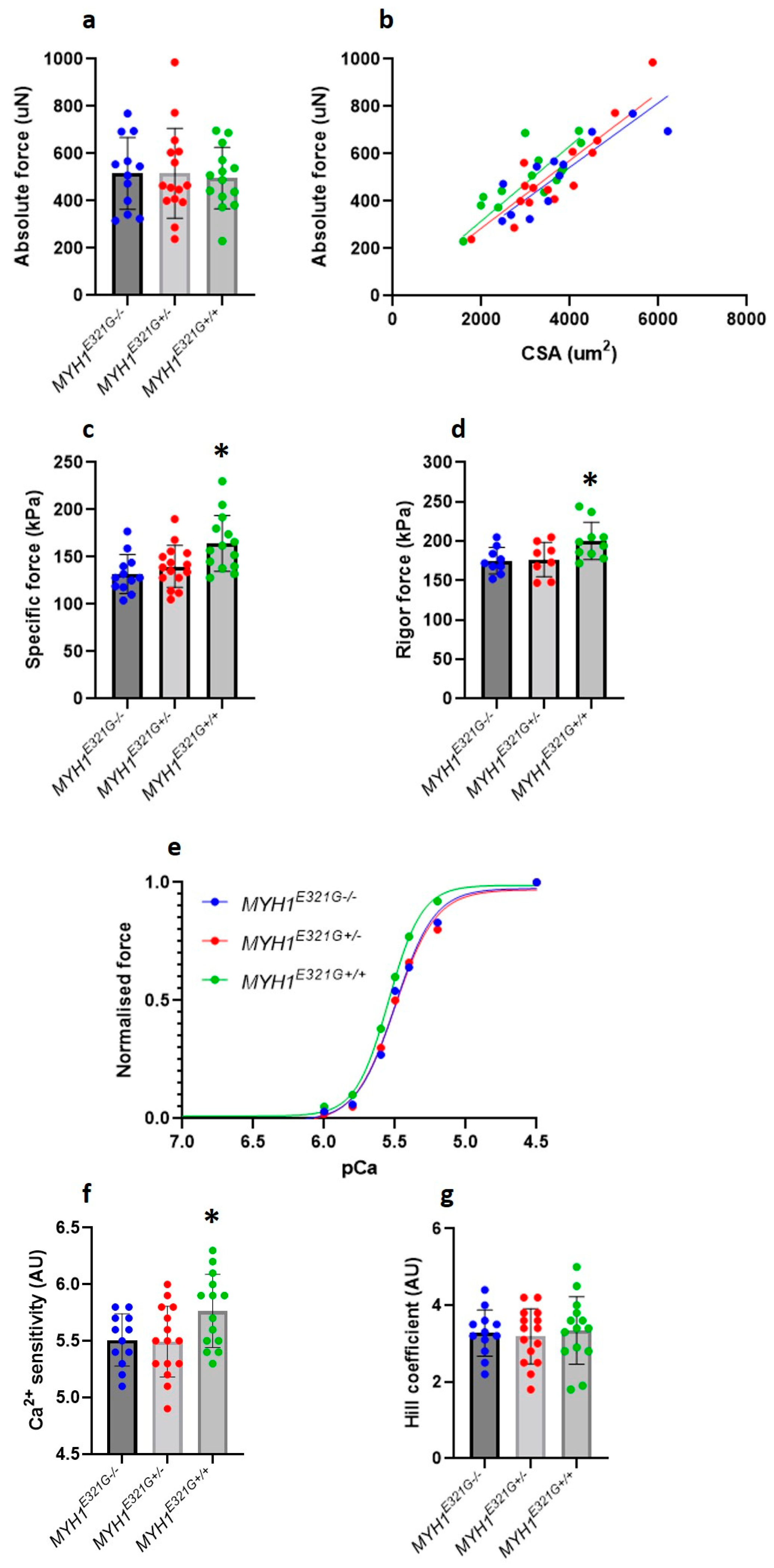
3.2. MYH1E321G Is Associated with an Increased Muscle Fibre Force Generating Capacity and Ca2+ Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oldfors, A. Hereditary myosin myopathies. Neuromuscul. Disord. 2007, 17, 355–367. [Google Scholar] [CrossRef]
- Tajsharghi, H.; Oldfors, A. Myosinopathies: Pathology and mechanisms. Acta Neuropathol. 2012, 125, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Rayment, I.; Holden, H.M.; Sellers, J.R.; Fananapazir, L.; Epstein, N.D. Structural interpretation of the mutations in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 1995, 92, 3864–3868. [Google Scholar] [CrossRef] [Green Version]
- Rayment, I.; Holden, H.M.; Whittaker, M.; Yohn, C.B.; Lorenz, M.; Holmes, K.C.; Milligan, R.A. Structure of the actin-myosin complex and its implications for muscle contraction. Science 1993, 261, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Rayment, I.; Rypniewski, W.R.; Schmidt-Base, K.; Smith, R.; Tomchick, D.R.; Benning, M.M.; Winkelmann, D.A.; Wesenberg, G.; Holden, H.M. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science 1993, 261, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Rayment, I.; Smith, C.; Yount, R.G. The active site of myosin. Annu. Rev. Physiol. 1996, 58, 671–702. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of Contraction in Striated Muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Finno, C.J.; Gianino, G.; Perumbakkam, S.; Williams, Z.J.; Bordbari, M.H.; Gardner, K.L.; Burns, E.; Peng, S.; Durward-Akhurst, S.A.; Valberg, S.J. A missense mutation in MYH1 is associated with susceptibility to immune-mediated myositis in Quarter Horses. Skelet Muscle 2018, 8, 7. [Google Scholar] [CrossRef]
- Gianino, G.M.; Valberg, S.J.; Perumbakkam, S.; Henry, M.L.; Gardner, K.; Penedo, C.; Finno, C.J. Prevalence of the E321G MYH1 variant for immune-mediated myositis and nonexertional rhabdomyolysis in performance subgroups of American Quarter Horses. J. Veter.-Intern. Med. 2018, 33, 897–901. [Google Scholar] [CrossRef] [Green Version]
- Valberg, S.J.; Henry, M.L.; Perumbakkam, S.; Gardner, K.L.; Finno, C.J. An E321G MYH1 mutation is strongly associated with nonexertional rhabdomyolysis in Quarter Horses. J. Veter.-Intern. Med. 2018, 32, 1718–1725. [Google Scholar] [CrossRef] [Green Version]
- Hunyadi, L.; Sundman, E.A.; Kass, P.H.; Williams, D.C.; Aleman, M. Clinical Implications and Hospital Outcome of Immune-Mediated Myositis in Horses. J. Vet. Intern. Med. 2017, 31, 170–175. [Google Scholar] [CrossRef]
- Durward-Akhurst, S.; Finno, C.; Barnes, N.; Shivers, J.; Guo, L.; Shelton, G.; Valberg, S. Major Histocompatibility Complex I and II Expression and Lymphocytic Subtypes in Muscle of Horses with Immune-Mediated Myositis. J. Vet.-Intern. Med. 2016, 30, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Polymyositis, dermatomyositis and inclusion-body myositis. N. Engl. J. Med. 1991, 325, 1487–1498. [Google Scholar] [CrossRef]
- Evans, J.; Levesque, D.; Shelton, G.D. Canine inflammatory myopathies: A clinicopathologic review of 200 cases. J. Vet. Intern. Med. 2004, 18, 679–691. [Google Scholar] [CrossRef]
- Alamo, L.; Ware, J.S.; Pinto, A.; Gillilan, R.E.; Seidman, J.G.; Seidman, C.E.; Padron, R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife 2017, 6, e24634. [Google Scholar] [CrossRef]
- Stewart, M.A.; Franks-Skiba, K.; Chen, S.; Cooke, R. Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc. Natl. Acad. Sci. USA 2010, 107, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Frontera, W.R.; Larsson, L. Contractile studies of single human skeletal muscle fibers: A comparison of different muscles, permeabilization procedures, and storage techniques. Muscle Nerve 1997, 20, 948–952. [Google Scholar] [CrossRef]
- Toepfer, C.N.; Garfinkel, A.C.; Venturini, G.; Wakimoto, H.; Repetti, G.; Alamo, L.; Sharma, A.; Agarwal, R.; Ewoldt, J.F.; Cloonan, P.; et al. Myosin Sequestration Regulates Sarcomere Function, Cardiomyocyte Energetics, and Metabolism, Informing the Pathogenesis of Hypertrophic Cardiomyopathy. Circulation 2020, 141, 828–842. [Google Scholar] [CrossRef]
- Toepfer, C.N.; Wakimoto, H.; Garfinkel, A.C.; McDonough, B.; Liao, D.; Jiang, J.; Tai, A.C.; Gorham, J.M.; Lunde, I.G.; Lun, M.; et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 2019, 11, 476. [Google Scholar] [CrossRef]
- Lindqvist, J.; Cheng, A.J.; Renaud, G.; Hardeman, E.C.; Ochala, J. Distinct Underlying Mechanisms of Limb and Respiratory Muscle Fiber Weaknesses in Nemaline Myopathy. J. Neuropathol. Exp. Neurol. 2013, 72, 472–481. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, J.; Levy, Y.; Pati-Alam, A.; Hardeman, E.C.; Gregorevic, P.; Ochala, J. Modulating myosin restores muscle function in a mouse model of nemaline myopathy. Ann. Neurol. 2016, 79, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Ochala, J.; Ravenscroft, G.; Laing, N.G.; Nowak, K.J. Nemaline myopathy-related skeletal muscle alpha-actin (ACTA1) mutation, Asp286Gly, prevents proper strong myosin binding and triggers muscle weakness. PLoS ONE 2012, 7, e45923. [Google Scholar] [CrossRef]
- Ochala, J.; Larsson, L. Effects of a preferential myosin loss on Ca2+ activation of force generation in single human skeletal muscle fibres. Exp. Physiol. 2008, 93, 486–495. [Google Scholar] [CrossRef]
- Ross, J.A.; Levy, Y.; Svensson, K.; Philp, A.; Schenk, S.; Ochala, J. SIRT1 regulates nuclear number and domain size in skeletal muscle fibers. J. Cell Physiol. 2018, 233, 7157–7163. [Google Scholar] [CrossRef] [Green Version]
- Levy, Y.; Ross, J.A.; Niglas, M.; Snetkov, V.A.; Lynham, S.; Liao, C.-Y.; Puckelwartz, M.J.; Hsu, Y.-M.; McNally, E.M.; Alsheimer, M.; et al. Prelamin A causes aberrant myonuclear arrangement and results in muscle fiber weakness. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Kraft, T.; Montag, J. Altered force generation and cell-to-cell contractile imbalance in hypertrophic cardiomyopathy. Pflug. Arch. Eur. J. Physiol. 2019, 471, 719–733. [Google Scholar] [CrossRef]
- Lewis, S.S.; Valberg, S.J.; Nielsen, I.L. Suspected immune-mediated myositis in horses. J. Vet. Intern. Med. 2007, 21, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Montag, J.; Kowalski, K.; Makul, M.; Ernstberger, P.; Radocaj, A.; Beck, J.; Becker, E.; Tripathi, S.; Keyser, B.; Muhlfeld, C.; et al. Burst-Like Transcription of Mutant and Wildtype MYH7-Alleles as Possible Origin of Cell-to-Cell Contractile Imbalance in Hypertrophic Cardiomyopathy. Front. Physiol. 2018, 9, 359. [Google Scholar] [CrossRef]
- Williams, Z.J.; Velez-Irizarry, D.; Petersen, J.L.; Ochala, J.; Finno, C.J.; Valberg, S.J. Candidate gene expression and coding sequence variants in Warmblood horses with myofibrillar myopathy. Equine Vet. J. 2020, 53, 306–315. [Google Scholar] [CrossRef]
- Alsaif, H.S.; Alshehri, A.; Sulaiman, R.A.; Al-Hindi, H.; Guzmán-Vega, F.J.; Arold, S.T.; Alkuraya, F.S. MYH1 is a candidate gene for recurrent rhabdomyolysis in humans. Am. J. Med. Genet. Part A 2021, 185, 2131–2135. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochala, J.; Finno, C.J.; Valberg, S.J. Myofibre Hyper-Contractility in Horses Expressing the Myosin Heavy Chain Myopathy Mutation, MYH1E321G. Cells 2021, 10, 3428. https://doi.org/10.3390/cells10123428
Ochala J, Finno CJ, Valberg SJ. Myofibre Hyper-Contractility in Horses Expressing the Myosin Heavy Chain Myopathy Mutation, MYH1E321G. Cells. 2021; 10(12):3428. https://doi.org/10.3390/cells10123428
Chicago/Turabian StyleOchala, Julien, Carrie J. Finno, and Stephanie J. Valberg. 2021. "Myofibre Hyper-Contractility in Horses Expressing the Myosin Heavy Chain Myopathy Mutation, MYH1E321G" Cells 10, no. 12: 3428. https://doi.org/10.3390/cells10123428
APA StyleOchala, J., Finno, C. J., & Valberg, S. J. (2021). Myofibre Hyper-Contractility in Horses Expressing the Myosin Heavy Chain Myopathy Mutation, MYH1E321G. Cells, 10(12), 3428. https://doi.org/10.3390/cells10123428