
Role of Senescence and Aging in SARS-CoV-2 Infection and COVID-19 Disease




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Features of Senescent Cells
Senescence-Associated Secretory Phenotype (SASP)
3. Senescence and Cancer
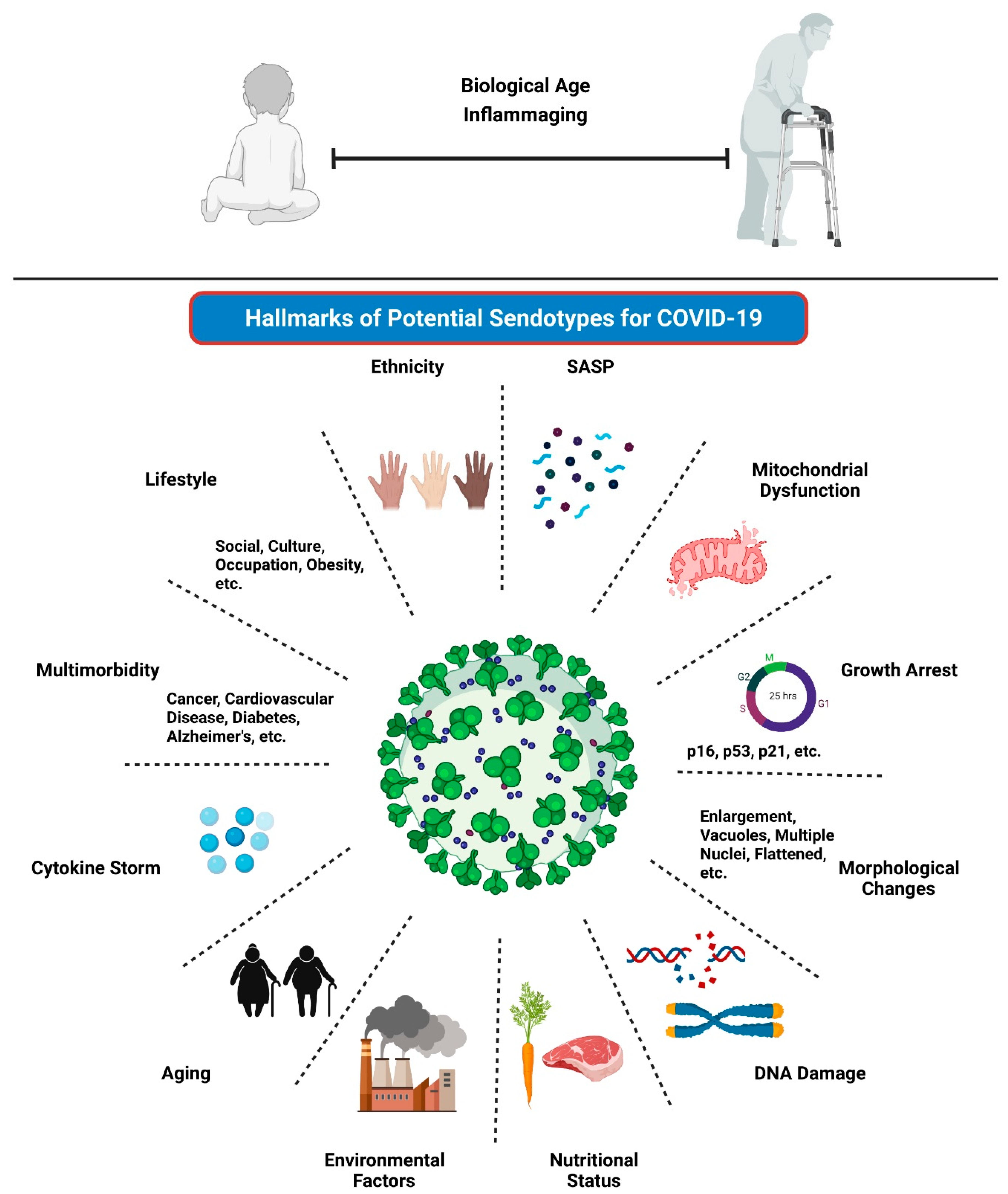
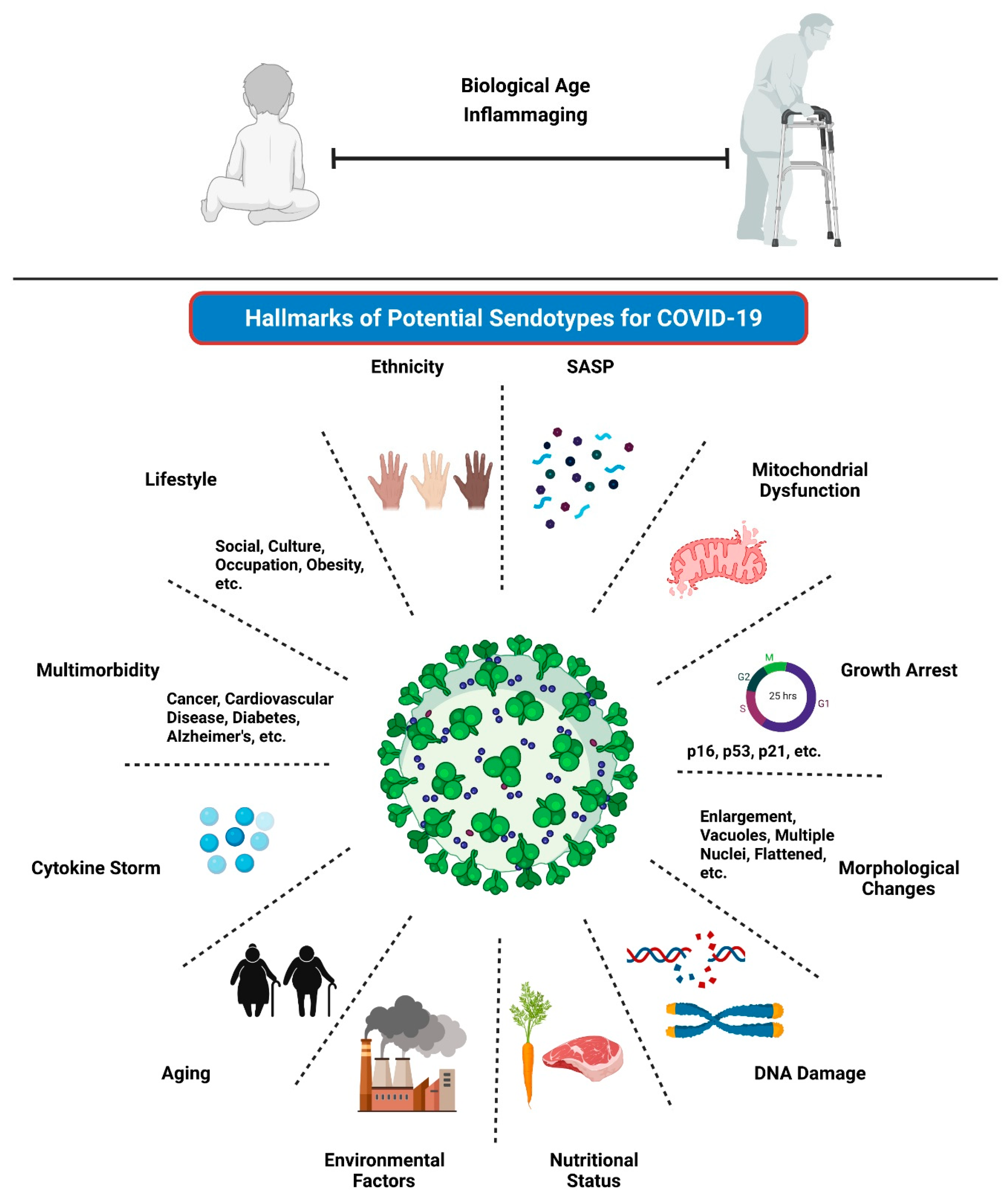
4. The Potential Implications and Clinical Translation of Senescence & Aging in COVID-19
4.1. Aging and COVID-19
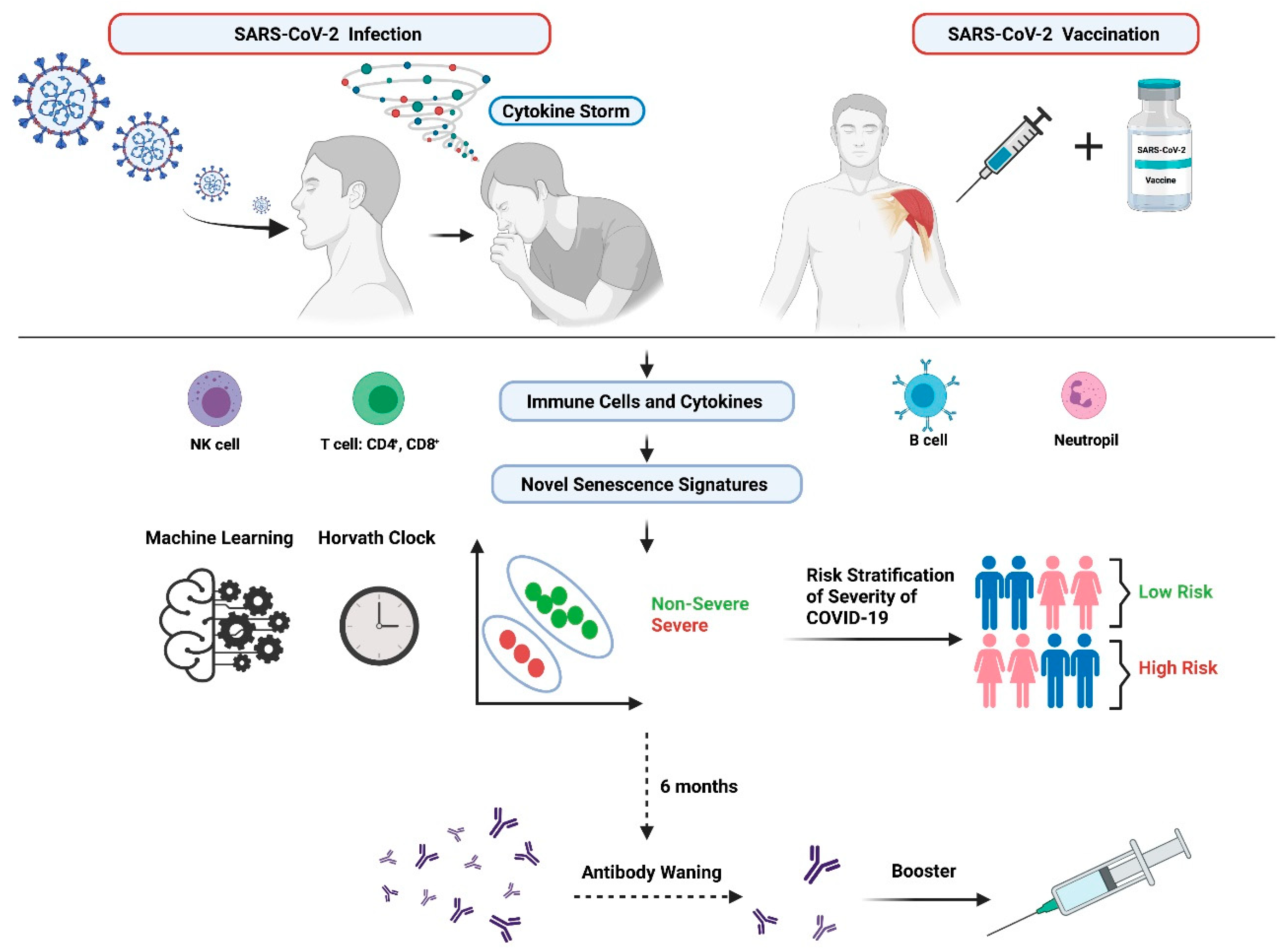
4.2. Senescence and COVID-19
4.3. Immune Dysregulation during COVID-19
4.3.1. Innate and Adaptive Immune Responses in the Pathogenesis of COVID-19
4.3.2. The Cytokine Storm and its Association with COVID-19
4.4. Immunosenescence and COVID-19 Severity and Mortality
4.5. Senescence Involvement in Multimorbidity and COVID-19
4.6. Senescence Involvement in Vaccination and the Association with COVID-19
4.6.1. Initial Vaccination Efficacies
4.6.2. Emerging Impact of Neutralization Responses to Vaccination
4.6.3. Future for Booster Vaccination Implementation
4.6.4. The Potential Impact from the Emergence of New Viral Variants
4.6.5. Potential Role of Senescence and Aging in Curtailing the COVID-19 Pandemic
5. Conclusions and Future Perspectives of Senescence as a Promising Biomarker and Target in COVID-19
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Carrel, A. On the permanent life of tissues outside of the organism. J. Exp. Med. 1912, 15, 516–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnet, F.M. Intrinsic Mutagenesis: A Genetic Approach to Ageing; Springer: Dordrecht, The Netherlands, 1974. [Google Scholar]
- Greider, C.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Szostak, J.W.; Blackburn, E.H. Cloning yeast telomeres on linear plasmid vectors. Cell 1982, 29, 245–255. [Google Scholar] [CrossRef]
- The Nobel Prize. The Nobel Prize in Physiology or Medicine. 2009. Available online: https://www.nobelprize.org/prizes/medicine/2009/summary/ (accessed on 17 November 2021).
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K. How long does telomerase extend telomeres? Regulation of telomerase release and telomere length homeostasis. Curr. Genet. 2018, 64, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Aksenova, A.Y.; Mirkin, S.M. At the Beginning of the End and in the Middle of the Beginning: Structure and Maintenance of Telomeric DNA Repeats and Interstitial Telomeric Sequences. Genes 2019, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, N.; Rachakonda, S.; Kumar, R. Telomeres and Telomere Length: A General Overview. Cancers 2020, 12, 558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence: Figure 1. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. JAMA 2018, 320, 1319–1320. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Solimando, A.G.; Susca, N.; Borrelli, P.; Prete, M.; Lauletta, G.; Pappagallo, F.; Buono, R.; Inglese, G.; Forina, B.M.; Bochicchio, D.; et al. Short-Term Variations in Neutrophil-to-Lymphocyte and Urea-to-Creatinine Ratios Anticipate Intensive Care Unit Admission of COVID-19 Patients in the Emergency Department. Front. Med. 2020, 7, 625176. [Google Scholar] [CrossRef]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Wengerodt, D.; Schmeer, C.; Witte, O.W.; Kretz, A. Amitosenescence and Pseudomitosenescence: Putative New Players in the Aging Process. Cells 2019, 8, 1546. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Muciño-Hernández, G.; Gerónimo-Olvera, C.; Maciel-Barón, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregón, S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2017, 217, 65–77. [Google Scholar] [CrossRef]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nat. Cell Biol. 2004, 432, 307–315. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Depinho, R.A. The mighty mouse: Genetically engineered mouse models in cancer drug development. Nat. Rev. Drug Discov. 2006, 5, 741–754. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [Green Version]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [Green Version]
- Biran, A.; Perelmutter, M.; Gal, H.; Burton, D.G.; Ovadya, Y.; Vadai, E.; Geiger, T.; Krizhanovsky, V. Senescent cells communicate via intercellular protein transfer. Genes Dev. 2015, 29, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biran, A.; Zada, L.; Karam, P.A.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasugi, M.; Okada, R.; Takahashi, A.; Chen, D.V.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, X.D.; Lapi, E.; Sullivan, A.; Jia, W.; He, Y.-W.; Ratnayaka, I.; Zhong, S.; Goldin, R.; Goemans, C.G.; et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc. Natl. Acad. Sci. USA 2012, 109, 13325–13330. [Google Scholar] [CrossRef] [Green Version]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55. [Google Scholar] [CrossRef]
- Zhang, H.; Alsaleh, G.; Feltham, J.; Sun, Y.; Napolitano, G.; Riffelmacher, T.; Charles, P.; Frau, L.; Hublitz, P.; Yu, Z.; et al. Polyamines Control eIF5A Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol. Cell 2019, 76, 110–125.e9. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, Y.; García-Cañaveras, J.C.; Guo, L.; Kan, M.; Yu, S.; Blair, I.A.; Rabinowitz, J.D.; Yang, X. Chaperone-mediated autophagy regulates the pluripotency of embryonic stem cells. Science 2020, 369, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nature 2020, 22, 1170–1179. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D. Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene 2007, 397, 84–93. [Google Scholar] [CrossRef] [Green Version]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.; Neretti, N.; Sedivy, J.M. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, T.; Ewels, P.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.; Kirschner, K.; Thuret, J.-Y.; Andrews, S.; Fraser, P.; Reik, W. Global Reorganization of the Nuclear Landscape in Senescent Cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef]
- Cheng, L.-Q.; Zhang, Z.-Q.; Chen, H.-Z.; Liu, D.-P. Epigenetic regulation in cell senescence. J. Mol. Med. 2017, 95, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Narita, M.; Nũnez, S.; Heard, E.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Funayama, R.; Saito, M.; Tanobe, H.; Ishikawa, F. Loss of linker histone H1 in cellular senescence. J. Cell Biol. 2006, 175, 869–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, T.S.; Cole, J.J.; Nelson, D.M.; Dikovskaya, D.; Faller, W.J.; Vizioli, M.G.; Hewitt, R.N.; Anannya, O.; McBryan, T.; Manoharan, I.; et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014, 28, 2712–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumendil, C.; Hari, P.; Olsen, K.C.F.; Acosta, J.C.; Bickmore, W.A. Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev. 2019, 33, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Swanson, E.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013, 203, 929–942. [Google Scholar] [CrossRef]
- Criscione, S.W.; Teo, Y.V.; Neretti, N. The Chromatin Landscape of Cellular Senescence. Trends Genet. 2016, 32, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.L.; McBryan, T.; Enders, G.H.; Johnson, F.B.; Zhang, R.; Adams, P.D. Senescent mouse cells fail to overtly regulate the HIRA histone chaperone and do not form robust Senescence Associated Heterochromatin Foci. Cell Div. 2010, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Suh, N. MicroRNA controls of cellular senescence. BMB Rep. 2018, 51, 493–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome signature of cellular senescence. Nucleic Acids Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef] [Green Version]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wang, X.; Wei, G.; Huang, Y.; Shi, Y.; Li, D.; Qiu, S.; Zhou, B.; Cao, J.; Chen, M.; et al. Single-Cell Transcriptome Analysis Reveals Six Subpopulations Reflecting Distinct Cellular Fates in Senescent Mouse Embryonic Fibroblasts. Front. Genet. 2020, 11, 867. [Google Scholar] [CrossRef] [PubMed]
- Philipot, D.; Guérit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.-M.; Piette, J.; Borzi, R.M.; et al. p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16, R58. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.M.; McKenna, M.M.; Walsh, C.P.; McKenna, D.J. miR-24 regulates CDKN1B/p27 expression in prostate cancer. Prostate 2016, 76, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Overhoff, M.G.; Garbe, J.C.; Koh, J.; Stampfer, M.R.; Beach, D.H.; Bishop, C.L. Cellular senescence mediated by p16INK4A-coupled miRNA pathways. Nucleic Acids Res. 2013, 42, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.B.; Anerillas, C.; Harris, S.C.; Munk, R.; Martindale, J.L.; Yang, X.; Mazan-Mamczarz, K.; Zhang, Y.; Heckenbach, I.J.; Scheibye-Knudsen, M.; et al. Reduction of lamin B receptor levels by miR-340-5p disrupts chromatin, promotes cell senescence and enhances senolysis. Nucleic Acids Res. 2021, 49, 7389–7405. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Bursuker, I.; Rhodes, J.M.; Goldman, R. Beta-galactosidase--an indicator of the maturational stage of mouse and human mononuclear phagocytes. J. Cell Physiol. 1982, 112, 385–390. [Google Scholar] [CrossRef]
- Kopp, H.-G.; Hooper, A.T.; Shmelkov, S.V.; Rafii, S. Beta-galactosidase staining on bone marrow. The osteoclast pitfall. Histol. Histopathol. 2007, 22, 971–976. [Google Scholar] [PubMed]
- Correia-Melo, C.; Jurk, D.; Passos, J.F. Robust Multiparametric Assessment of Cellular Senescence. Cell Senescence 2012, 965, 409–419. [Google Scholar] [CrossRef]
- Roche, B.; Arcangioli, B.; Martienssen, R. Transcriptional reprogramming in cellular quiescence. RNA Biol. 2017, 14, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [Green Version]
- Matjusaitis, M.; Chin, G.; Sarnoski, E.A.; Stolzing, A. Biomarkers to identify and isolate senescent cells. Ageing Res. Rev. 2016, 29, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Prata, L.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Coppé, J.P.; Lam, E.W. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- ONS. Living Longer. 2018. Available online: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/ageing/articles/livinglongerhowourpopulationischangingandwhyitmatters/2018-08-13 (accessed on 30 October 2021).
- Crimmins, E.M. Lifespan and Healthspan: Past, Present, and Promise. Gerontologist 2015, 55, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lau, S.; Meijer, E.; Hu, P. Living Longer, With or Without Disability? A Global and Longitudinal Perspective. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2019, 75, 162–167. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Pilleron, S.; Sarfati, D.; Janssen-Heijnen, M.; Vignat, J.; Ferlay, J.; Bray, F.; Soerjomataram, I. Global cancer incidence in older adults, 2012 and 2035: A population-based study. Int. J. Cancer 2018, 144, 49–58. [Google Scholar] [CrossRef]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Campisi, J.; Andersen, J.K.; Kapahi, P.; Melov, S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011, 21, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Turnquist, C.; Beck, J.A.; Horikawa, I.; Obiorah, I.E.; Von Muhlinen, N.; Vojtesek, B.; Lane, D.P.; Grunseich, C.; Chahine, J.J.; Ames, H.M.; et al. Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro-Oncology 2019, 21, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.Y.; Kim, J.K.; Ham, S.W.; Oh, S.Y.; Kim, J.; Park, J.B.; Lee, J.Y.; Kim, S.C.; Kim, H. Irradiation induces glioblastoma cell senescence and senescence-associated secretory phenotype. Tumour Biol. 2016, 37, 5857–5867. [Google Scholar] [CrossRef]
- DeMaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- da Silva, P.; Ogrodnik, M.; Kucheryavenko, O.; Glibert, J.; Miwa, S.; Cameron, K.; Ishaq, A.; Saretzki, G.; Nagaraja-Grellscheid, S.; Nelson, G.; et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 2018, 18, e12848. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Kim, L.; Whitaker, M.; O’Halloran, A.; Cummings, C.; Holstein, R.; Prill, M.; Chai, S.J.; Kirley, P.D.; Alden, N.B.; et al. Hospitalization Rates and Characteristics of Patients Hospitalized with Laboratory-Confirmed Coronavirus Disease 2019—COVID-NET, 14 States, March 1–30, 2020. MMWR Morb. Mortal Wkly Rep. 2020, 69, 458–464. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Martínez, I.; García-Carpizo, V.; Guijarro, T.; García-Gomez, A.; Navarro, D.; Aranda, A.; Zambrano, A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence 2016, 7, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Brunetta, E.; Folci, M.; Bottazzi, B.; De Santis, M.; Gritti, G.; Protti, A.; Mapelli, S.N.; Bonovas, S.; Piovani, D.; Leone, R.; et al. Macrophage expression and prognostic significance of the long pentraxin PTX3 in COVID-19. Nat. Immunol. 2020, 22, 19–24. [Google Scholar] [CrossRef]
- Protti, A.; Meessen, J.; Bottazzi, B.; Garlanda, C.; Milani, A.; Bacci, M.; Mantovani, A.; Cecconi, M.; Latini, R.; Caironi, P. Circulating pentraxin 3 in severe COVID-19 or other pulmonary sepsis. Eur. J. Clin. Investig. 2021, 51, e13530. [Google Scholar] [CrossRef]
- Myhre, P.L.; Prebensen, C.; Strand, H.; Røysland, R.; Jonassen, C.M.; Rangberg, A.; Sørensen, V.; Søvik, S.; Røsjø, H.; Svensson, M.; et al. Growth Differentiation Factor 15 Provides Prognostic Information Superior to Established Cardiovascular and Inflammatory Biomarkers in Unselected Patients Hospitalized With COVID-19. Circulation 2020, 142, 2128–2137. [Google Scholar] [CrossRef]
- Shu, T.; Ning, W.; Wu, D.; Xu, J.; Han, Q.; Huang, M.; Zou, X.; Yang, Q.; Yuan, Y.; Bie, Y.; et al. Plasma Proteomics Identify Biomarkers and Pathogenesis of COVID-19. Immunity 2020, 53, 1108–1122.e5. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72. [Google Scholar] [CrossRef]
- Geyer, P.E.; Arend, F.M.; Doll, S.; Louiset, M.L.; Virreira Winter, S.; Müller-Reif, J.B.; Torun, F.M.; Weigand, M.; Eichhorn, P.; Bruegel, M.; et al. High-resolution serum proteome trajectories in COVID-19 reveal patient-specific seroconversion. EMBO Mol. Med. 2021, 13, e14167. [Google Scholar] [CrossRef]
- Wu, M.; Chen, Y.; Xia, H.; Wang, C.; Tan, C.Y.; Cai, X.; Liu, Y.; Ji, F.; Xiong, P.; Liu, R.; et al. Transcriptional and proteomic insights into the host response in fatal COVID-19 cases. Proc. Natl. Acad. Sci. USA 2020, 117, 28336–28343. [Google Scholar] [CrossRef]
- Sanchez-Vazquez, R.; Guío-Carrión, A.; Zapatero-Gaviria, A.; Martínez, P.; Blasco, M.A. Shorter telomere lengths in patients with severe COVID-19 disease. Aging 2021, 13, 1–15. [Google Scholar] [CrossRef]
- Carfì, A.; Bernabei, R.; Landi, F. For the Gemelli Against COVID-19 Post-Acute Care Study Group. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Mo, X.; Jian, W.; Su, Z.; Chen, M.; Peng, H.; Peng, P.; Lei, C.; Chen, R.; Zhong, N.; Li, S. Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur. Respir. J. 2020, 55, 2001217. [Google Scholar] [CrossRef]
- Wei, J.; Yang, H.; Lei, P.; Fan, B.; Qiu, Y.; Zeng, B.; Yu, P.; Lv, J.; Jian, Y.; Wan, C. Analysis of thin-section CT in patients with coronavirus disese (COVID-19) after hospital discharge. J. Xray Sci. Technol. 2020, 28, 383–389. [Google Scholar]
- Mandal, S.; Barnett, J.; Brill, S.E.; Brown, J.S.; Denneny, E.K.; Hare, S.S.; Heightman, M.; Hillman, T.E.; Jacob, J.; Jarvis, H.C.; et al. ‘Long-COVID’: A cross-sectional study of persisting symptoms, biomarker and imaging abnormalities following hospitalisation for COVID-19. Thorax 2021, 76, 396–398. [Google Scholar] [CrossRef]
- Halpin, S.J.; McIvor, C.; Whyatt, G.; Adams, A.; Harvey, O.; McLean, L.; Walshaw, C.; Kemp, S.; Corrado, J.; Singh, R.; et al. Postdischarge symptoms and rehabilitation needs in survivors of COVID-19 infection: A cross-sectional evaluation. J. Med. Virol. 2021, 93, 1013–1022. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 24 October 2021).
- Onder, G.; Rezza, G.; Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef]
- Gao, Z.; Xu, Y.; Sun, C.; Wang, X.; Guo, Y.; Qiu, S.; Ma, K. A systematic review of asymptomatic infections with COVID-19. J. Microbiol. Immunol. Infect. 2020, 54, 12–16. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Rothe, C.; Schunk, M.; Sothmann, P.; Bretzel, G.; Froeschl, G.; Wallrauch, C.; Zimmer, T.; Thiel, V.; Janke, C.; Guggemos, W.; et al. Transmission of 2019-nCoV Infection from an Asymptomatic Contact in Germany. N. Engl. J. Med. 2020, 382, 970–971. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, E.S.; Dufort, E.M.; Blog, D.S.; Hall, E.W.; Hoefer, D.; Backenson, B.P.; Muse, A.T.; Kirkwood, J.N.; St George, K.; Holtgrave, D.R.; et al. COVID-19 Testing, Epidemic Features, Hospital Outcomes, and Household Prevalence, New York State-March 2020. Clin. Infect. Dis. 2020, 71, 1953–1959. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Grasselli, G.; Pesenti, A.; Cecconi, M. Critical Care Utilization for the COVID-19 Outbreak in Lombardy, Italy: Early Experience and Forecast During an Emergency Response. JAMA 2020, 323, 1545–1546. [Google Scholar] [CrossRef] [Green Version]
- Halamka, J.; Cerrato, P.; Perlman, A. Redesigning COVID-19 Care with Network Medicine and Machine Learning. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 4, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Lalmuanawma, S.; Hussain, J.; Chhakchhuak, L. Applications of machine learning and artificial intelligence for Covid-19 (SARS-CoV-2) pandemic: A review. Chaos Solitons Fractals 2020, 139, 110059. [Google Scholar] [CrossRef] [PubMed]
- Minaee, S.; Kafieh, R.; Sonka, M.; Yazdani, S.; Soufi, G.J. Deep-COVID: Predicting COVID-19 from chest X-ray images using deep transfer learning. Med. Image Anal. 2020, 65, 101794. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Du, C.; Zhang, Y.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Garibaldi, B.T.; Fiksel, J.; Muschelli, J.; Robinson, M.L.; Rouhizadeh, M.; Perin, J.; Schumock, G.; Nagy, P.; Gray, J.H.; Malapati, H.; et al. Patient Trajectories Among Persons Hospitalized for COVID-19: A Cohort Study. Ann. Intern. Med. 2021, 174, 33–41. [Google Scholar] [CrossRef]
- Wang, L.; He, W.; Yu, X.; Hu, D.; Bao, M.; Liu, H.; Zhou, J.; Jiang, H. Coronavirus disease 2019 in elderly patients: Characteristics and prognostic factors based on 4-week follow-up. J. Infect. 2020, 80, 639–645. [Google Scholar] [CrossRef]
- Guan, W.J.; Liang, W.H.; Zhao, Y.; Liang, H.R.; Chen, Z.S.; Li, Y.M.; Liu, X.Q.; Chen, R.C.; Tang, C.L.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: A nationwide analysis. Eur. Respir. J. 2020, 55, 2000547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.J.; Dong, X.; Cao, Y.Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Munster, V.J.; Koopmans, M.; van Doremalen, N.; van Riel, D.; de Wit, E. A Novel Coronavirus Emerging in China—Key Questions for Impact Assessment. N. Engl. J. Med. 2020, 382, 692–694. [Google Scholar] [CrossRef]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 531–538. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Santesmasses, D.; Castro, J.P.; Zenin, A.A.; Shindyapina, A.V.; Gerashchenko, M.V.; Zhang, B.; Kerepesi, C.; Yim, S.H.; Fedichev, P.O.; Gladyshev, V.N. COVID-19 is an emergent disease of aging. Aging Cell 2020, 19. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC); National Center for Health Statistics (NCHS). COVID-19 Death Data and Resources, COVID-19 Mortality Overview. Available online: https://www.cdc.gov/nchs/covid19/mortality-overview.htm (accessed on 17 November 2021).
- Kontopantelis, E.; A Mamas, M.; Deanfield, J.; Asaria, M.; Doran, T. Excess mortality in England and Wales during the first wave of the COVID-19 pandemic. J. Epidemiol. Community Health 2020, 75, 213–223. [Google Scholar] [CrossRef]
- Veronese, N.; Koyanagi, A.; Stangherlin, V.; Mantoan, P.; Chiavalin, M.; Tudor, F.; Pozzobon, G.; Tessarin, M.; Pilotto, A. Mortality attributable to COVID-19 in nursing home residents: A retrospective study. Aging Clin. Exp. Res. 2021, 33, 1745–1751. [Google Scholar] [CrossRef]
- The New York Times. Nearly One-Third of U.S. Coronavirus Deaths Are Linked to Nursing Homes. Available online: https://www.nytimes.com/interactive/2020/us/coronavirus-nursing-homes.html (accessed on 17 November 2021).
- Comas-Herrera, A.; Zalakaín, J.; Lemmon, E.; Henderson, D.; Litwin, C.; Hsu, A.T.; Schmidt, A.E.; Arling, G.; Kruse, F.; Fernández, J.-L. Mortality Associated with COVID-19 in Care Homes: International Evidence; International Long-Term Care Policy Network: London, UK, 2021. [Google Scholar]
- Panagiotou, O.A.; Kosar, C.M.; White, E.M.; Bantis, L.E.; Yang, X.; Santostefano, C.M.; Feifer, R.A.; Blackman, C.; Rudolph, J.L.; Gravenstein, S.; et al. Risk Factors Associated with All-Cause 30-Day Mortality in Nursing Home Residents With COVID-19. JAMA Intern. Med. 2021, 181, 439. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [Green Version]
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age- and Tissue-Specific Expression of Senescence Biomarkers in Mice. Front. Genet. 2018, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, C.S.L.; Waaijer, M.E.C.; Slee-Valentijn, M.S.; Stijnen, T.; Westendorp, R.; Maier, A.B. Cellular senescence and chronological age in various human tissues: A systematic review and meta-analysis. Aging Cell 2019, 19, e13083. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, C.S.; Luesken, S.W.; Waaijer, M.E.; Maier, A.B. Senescence in tissue samples of humans with age-related diseases: A systematic review. Ageing Res. Rev. 2021, 68, 101334. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tyshkovskiy, A.; Muñoz-Espín, D.; Tian, X.; Serrano, M.; de Magalhaes, J.P.; Nevo, E.; Gladyshev, V.N.; Seluanov, A.; Gorbunova, V. Naked mole rats can undergo developmental, oncogene-induced and DNA damage-induced cellular senescence. Proc. Natl. Acad. Sci. USA 2018, 115, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez-Ribes, M.; Gualda, E.G.; Doherty, G.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Newton, A.H.; Cardani, A.; Braciale, T.J. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin. Immunopathol. 2016, 38, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Kirkland, J.L.; Schwartz, S. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging 2017, 9, 2411–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.S.; Kang, K.W.; Seu, Y.B.; Baek, S.H.; Kim, J.R. Interferon-gamma induces cellular senescence through p53-dependent DNA damage signaling in human endothelial cells. Mech. Ageing Dev. 2009, 130, 179–188. [Google Scholar] [CrossRef]
- Keilich, S.R.; Bartley, J.M.; Haynes, L. Diminished immune responses with aging predispose older adults to common and uncommon influenza complications. Cell. Immunol. 2019, 345, 103992. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, L.-M.; Wan, L.; Xiang, T.-X.; Le, A.; Liu, J.-M.; Peiris, M.; Poon, L.L.M.; Zhang, W. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect. Dis. 2020, 20, 656–657. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.; Tsang, O.T.; Leung, W.S.; Tam, A.R.; Wu, T.C.; Lung, D.C.; Yip, C.C.; Cai, J.P.; Chan, J.M.; Chik, T.S.; et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: An observational cohort study. Lancet Infect. Dis. 2020, 20, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Nehme, J.; Borghesan, M.; Mackedenski, S.; Bird, T.G.; DeMaria, M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell 2020, 19. [Google Scholar] [CrossRef]
- Yang, M. Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV infection. SSRN 2020. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. The meteorology of cytokine storms, and the clinical usefulness of this knowledge. Semin. Immunopathol. 2017, 39, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotta, F.; Rocco, D.; Vitiello, F.; De Palma, R.; Guerra, G.; De Luca, A.; Navani, N.; Bianco, A. Immune Checkpoint Blockade for Advanced NSCLC: A New Landscape for Elderly Patients. Int. J. Mol. Sci. 2019, 20, 2258. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Yazdanpanah, F.; Hamblin, M.R.; Rezaei, N. The immune system and COVID-19: Friend or foe? Life Sci. 2020, 256, 117900. [Google Scholar] [CrossRef]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Pekosz, A.; Park, H.S.; Ursin, R.L.; Shapiro, J.R.; Benner, S.E.; Littlefield, K.; Kumar, S.; Naik, H.M.; Betenbaugh, M.J.; et al. Sex, age, and hospitalization drive antibody responses in a COVID-19 convalescent plasma donor population. J. Clin. Investig. 2020, 130, 6141–6150. [Google Scholar] [CrossRef]
- Iyer, A.S.; Jones, F.K.; Nodoushani, A.; Kelly, M.; Becker, M.; Slater, D.; Mills, R.; Teng, E.; Kamruzzaman, M.; Garcia-Beltran, W.F.; et al. Persistence and decay of human antibody responses to the receptor binding domain of SARS-CoV-2 spike protein in COVID-19 patients. Sci. Immunol. 2020, 5, eabe0367. [Google Scholar] [CrossRef]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 2021, 371, eabf4063. [Google Scholar] [CrossRef]
- Altmann, D.M.; Boyton, R.J. SARS-CoV-2 T cell immunity: Specificity, function, durability, and role in protection. Sci. Immunol. 2020, 5, eabd6160. [Google Scholar] [CrossRef]
- Kilic, G.; Bulut, O.; Jaeger, M.; Ter Horst, R.; Koeken, V.A.C.M.; Moorlag, S.J.C.F.; Mourits, V.P.; de Bree, C.; Domínguez-Andrés, J.; Joosten, L.A.B.; et al. The Immunological Factors Predisposing to Severe Covid-19 Are Already Present in Healthy Elderly and Men. Front. Immunol. 2021, 12, 720090. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168. [Google Scholar] [CrossRef]
- Prokop, J.W.; Hartog, N.L.; Chesla, D.; Faber, W.; Love, C.P.; Karam, R.; Abualkheir, N.; Feldmann, B.; Teng, L.; McBride, T.; et al. High-Density Blood Transcriptomics Reveals Precision Immune Signatures of SARS-CoV-2 Infection in Hospitalized Individuals. Front. Immunol. 2021, 12, 694243. [Google Scholar] [CrossRef]
- Lowery, S.A.; Sariol, A.; Perlman, S. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe 2021, 29, 1052–1062. [Google Scholar] [CrossRef]
- Franzo, G. SARS-CoV-2 and other human coronavirus show genome patterns previously associated to reduced viral recognition and altered immune response. Sci. Rep. 2021, 11, 10696. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.R.S.; Alrubayyi, A.; Pring, E.; Bart, V.M.T.; Jones, R.; Coveney, C.; Lu, F.; Tellier, M.; Maleki-Toyserkani, S.; Richter, F.C.; et al. Innate immunology in COVID-19-a living review. Part II: Dysregulated inflammation drives immunopathology. Oxf. Open Immunol. 2020, 1, iqaa005. [Google Scholar] [CrossRef] [PubMed]
- Masso-Silva, J.A.; Moshensky, A.; Lam, M.T.Y.; Odish, M.; Patel, A.; Xu, L.; Hansen, E.; Trescott, S.; Nguyen, C.; Kim, R.; et al. Increased peripheral blood neutrophil activation phenotypes and NETosis in critically ill COVID-19 patients: A case series and review of the literature. Clin. Infect. Dis. 2021, ciab437. [Google Scholar] [CrossRef]
- Youn, Y.J.; Lee, Y.B.; Kim, S.H.; Jin, H.K.; Bae, J.S.; Hong, C.W. Nucleocapsid and Spike Proteins of SARS-CoV-2 Drive Neutrophil Extracellular Trap Formation. Immune Netw. 2021, 21, e16. [Google Scholar] [CrossRef]
- Woo, M.S.; Haag, F.; Nierhaus, A.; Jarczak, D.; Roedl, K.; Mayer, C.; Brehm, T.T.; van der Meirschen, M.; Hennigs, A.; Christopeit, M.; et al. Multi-dimensional and longitudinal systems profiling reveals predictive pattern of severe COVID-19. iScience 2021, 24, 102752. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Miyara, M.; Costantino, C.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef]
- Harb, H.; Benamar, M.; Lai, P.S.; Contini, P.; Griffith, J.W.; Crestani, E.; Schmitz-Abe, K.; Chen, Q.; Fong, J.; Marri, L.; et al. Notch4 signaling limits regulatory T-cell-mediated tissue repair and promotes severe lung inflammation in viral infections. Immunity 2021, 54, 1186–1199. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Kidd, C.K.; Dan, J.M.; Ramirez, S.I.; Yu, E.D.; Mateus, J.; da Silva Antunes, R.; Moore, E.; Rubiro, P.; et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep. Med. 2021, 2, 100204. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Methot, N.; Yu, E.D.; Zhang, Y.; Dan, J.M.; Goodwin, B.; Rubiro, P.; Sutherland, A.; Wang, E.; et al. Impact of SARS-CoV-2 variants on the total CD4. Cell Rep. Med. 2021, 2, 100355. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Wegman, A.D.; Ripich, K.; Friberg, H.; Currier, J.R.; Thomas, S.J.; Endy, T.P.; Waickman, A.T. Persistent COVID-19 Symptoms Minimally Impact the Development of SARS-CoV-2-Specific T Cell Immunity. Viruses 2021, 13, 916. [Google Scholar] [CrossRef]
- Osuchowski, M.F.; Winkler, M.S.; Skirecki, T.; Cajander, S.; Shankar-Hari, M.; Lachmann, G.; Monneret, G.; Venet, F.; Bauer, M.; Brunkhorst, F.M.; et al. The COVID-19 puzzle: Deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir. Med. 2021, 9, 622–642. [Google Scholar] [CrossRef]
- Rodriguez-Smith, J.J.; Verweyen, E.L.; Clay, G.M.; Esteban, Y.M.; de Loizaga, S.R.; Baker, E.J.; Do, T.; Dhakal, S.; Lang, S.M.; Grom, A.A.; et al. Inflammatory biomarkers in COVID-19-associated multisystem inflammatory syndrome in children, Kawasaki disease, and macrophage activation syndrome: A cohort study. Lancet Rheumatol. 2021, 3, e574–e584. [Google Scholar] [CrossRef]
- Hammoudeh, S.M.; Hammoudeh, A.M.; Bhamidimarri, P.M.; Al Safar, H.; Mahboub, B.; Künstner, A.; Busch, H.; Halwani, R.; Hamid, Q.; Rahmani, M.; et al. Systems Immunology Analysis Reveals the Contribution of Pulmonary and Extrapulmonary Tissues to the Immunopathogenesis of Severe COVID-19 Patients. Front. Immunol. 2021, 12, 595150. [Google Scholar] [CrossRef] [PubMed]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, Immunity, and COVID-19: How Age Influences the Host Immune Response to Coronavirus Infections? Front. Physiol. 2020, 11, 571416. [Google Scholar] [CrossRef] [PubMed]
- Nikolich-Žugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [Green Version]
- Barbé-Tuana, F.; Funchal, G.; Schmitz, C.R.R.; Maurmann, R.; Bauer, M.E. The interplay between immunosenescence and age-related diseases. Semin. Immunopathol. 2020, 42, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Qun, S.; Wang, Y.; Chen, J.; Huang, X.; Guo, H.; Lu, Z.; Wang, J.; Zheng, C.; Ma, Y.; Zhu, Y.; et al. Neutrophil-to-Lymphocyte Ratios Are Closely Associated with the Severity and Course of Non-mild COVID-19. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Ulhaq, Z.S.; Soraya, G.V. Interleukin-6 as a potential biomarker of COVID-19 progression. Med. Mal. Infect. 2020, 50, 382–383. [Google Scholar] [CrossRef]
- Nikolich-Zugich, J.; Knox, K.S.; Rios, C.T.; Natt, B.; Bhattacharya, D.; Fain, M.J. SARS-CoV-2 and COVID-19 in older adults: What we may expect regarding pathogenesis, immune responses, and outcomes. Geroscience 2020, 42, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavel, A.B.; Glickman, J.W.; Michels, J.R.; Kim-Schulze, S.; Miller, R.L.; Guttman-Yassky, E. Th2/Th1 Cytokine Imbalance Is Associated with Higher COVID-19 Risk Mortality. Front. Genet. 2021, 12, 706902. [Google Scholar] [CrossRef]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.C.; Masoli, J.; Delgado, J.; Tignanelli, C.; Kuchel, G.; Melzer, D.; Beckman, K.B.; Levine, M. COVID-19 severity is predicted by earlier evidence of accelerated aging. medRxiv 2020. [Google Scholar] [CrossRef]
- Berthelot, J.-M.; Lioté, F. COVID-19 as a STING disorder with delayed over-secretion of interferon-beta. EBioMedicine 2020, 56, 102801. [Google Scholar] [CrossRef]
- Hamann, L.; Szwed, M.; Mossakowska, M.; Chudek, J.; Puzianowska-Kuznicka, M. First evidence for STING SNP R293Q being protective regarding obesity-associated cardiovascular disease in age-advanced subjects – a cohort study. Immun. Ageing 2020, 17, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kelley, W.J.; Zemans, R.L.; Goldstein, D.R. Cellular senescence: Friend or foe to respiratory viral infections? Eur. Respir. J. 2020, 56, 2002708. [Google Scholar] [CrossRef] [PubMed]
- Mair, F.S.; Foster, H.M.; I Nicholl, B. Multimorbidity and the COVID-19 pandemic – An urgent call to action. J. Comorbidity 2020, 10. [Google Scholar] [CrossRef]
- Helfand, B.K.I.; Webb, M.; Gartaganis, S.L.; Fuller, L.; Kwon, C.-S.; Inouye, S.K. The Exclusion of Older Persons from Vaccine and Treatment Trials for Coronavirus Disease 2019—Missing the Target. JAMA Intern. Med. 2020, 180, 1546. [Google Scholar] [CrossRef]
- Fernández-Niño, J.A.; Guerra-Gómez, J.A.; Idrovo, A.J. Multimorbidity patterns among COVID-19 deaths: Proposal for the construction of etiological models. Rev. Panam. Salud Públ. 2020, 44. [Google Scholar] [CrossRef] [PubMed]
- Lisco, G.; De Tullio, A.; Stragapede, A.; Solimando, A.; Albanese, F.; Capobianco, M.; Giagulli, V.; Guastamacchia, E.; De Pergola, G.; Vacca, A.; et al. COVID-19 and the Endocrine System: A Comprehensive Review on the Theme. J. Clin. Med. 2021, 10, 2920. [Google Scholar] [CrossRef] [PubMed]
- Medicines and Healthcare Products Regulatory Agency (MHRA). Vaccine BNT162b2: Conditions of Authorisation under Regulation 174. 2020. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1007802/Conditions_of_authorisation_for_Pfizer_BioNTech_vaccine_29Jul21-CLEAN.pdf (accessed on 17 November 2021).
- Medicines and Healthcare Products Regulatory Agency (MHRA). Vaccine ChAdOx1-S: Conditions of Authorisation under Regulation 174. 2020. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/963841/AZ_Conditions_for_Authorisation_final_23.02.21.pdf (accessed on 17 November 2021).
- Waaijer, M.E.C.; Parish, W.E.; Strongitharm, B.H.; Van Heemst, D.; Slagboom, P.E.; De Craen, A.J.M.; Sedivy, J.M.; Westendorp, R.G.J.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; Von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Lumley, S.F.; O’Donnell, D.; Stoesser, N.E.; Matthews, P.C.; Howarth, A.; Hatch, S.B.; Marsden, B.D.; Cox, S.; James, T.; Warren, F.; et al. Antibody Status and Incidence of SARS-CoV-2 Infection in Health Care Workers. N. Engl. J. Med. 2021, 384, 533–540. [Google Scholar] [CrossRef]
- Kim, J.H.; Marks, F.; Clemens, J.D. Looking beyond COVID-19 vaccine phase 3 trials. Nat. Med. 2021, 27, 205–211. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Robertson, C.; Stowe, J.; Tessier, E.; Simmons, R.; Cottrell, S.; Roberts, R.; O’Doherty, M.; et al. Effectiveness of the Pfizer-BioNTech and Oxford-AstraZeneca vaccines on covid-19 related symptoms, hospital admissions, and mortality in older adults in England: Test negative case-control study. BMJ 2021, 373, n1088. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Olson, S.M.; Self, W.H.; Talbot, H.K.; Lindsell, C.J.; Steingrub, J.S.; Shapiro, N.I.; Ginde, A.A.; Douin, D.J.; Prekker, M.E.; et al. Effectiveness of Pfizer-BioNTech and Moderna Vaccines Against COVID-19 Among Hospitalized Adults Aged ≥65 Years—United States, January-March 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 674–679. [Google Scholar] [CrossRef]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Kardani, K.; Bolhassani, A.; Shahbazi, S. Prime-boost vaccine strategy against viral infections: Mechanisms and benefits. Vaccine 2016, 34, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.F.; Zhao, F.; Huang, D.; Beutler, N.; Burns, A.; He, W.T.; Limbo, O.; Smith, C.; Song, G.; Woehl, J.; et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020, 369, 956–963. [Google Scholar] [CrossRef]
- McMahan, K.; Yu, J.; Mercado, N.B.; Loos, C.; Tostanoski, L.H.; Chandrashekar, A.; Liu, J.; Peter, L.; Atyeo, C.; Zhu, A.; et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 2021, 590, 630–634. [Google Scholar] [CrossRef]
- Israelow, B.; Mao, T.; Klein, J.; Song, E.; Menasche, B.; Omer, S.B.; Iwasaki, A. Adaptive immune determinants of viral clearance and protection in mouse models of SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Khoury, D.S.; Cromer, D.; Reynaldi, A.; Schlub, T.E.; Wheatley, A.K.; Juno, J.A.; Subbarao, K.; Kent, S.J.; Triccas, J.A.; Davenport, M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 1205–1211. [Google Scholar] [CrossRef]
- Bates, T.A.; Leier, H.C.; Lyski, Z.L.; Goodman, J.R.; Curlin, M.E.; Messer, W.B.; Tafesse, F.G. Age-Dependent Neutralization of SARS-CoV-2 and P.1 Variant by Vaccine Immune Serum Samples. JAMA 2021, 326, 868–869. [Google Scholar] [CrossRef]
- Wheatley, A.K.; Juno, J.A.; Wang, J.J.; Selva, K.J.; Reynaldi, A.; Tan, H.X.; Lee, W.S.; Wragg, K.M.; Kelly, H.G.; Esterbauer, R.; et al. Evolution of immune responses to SARS-CoV-2 in mild-moderate COVID-19. Nat. Commun 2021, 12, 1162. [Google Scholar] [CrossRef] [PubMed]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Haas, E.J.; Angulo, F.J.; McLaughlin, J.M.; Anis, E.; Singer, S.R.; Khan, F.; Brooks, N.; Smaja, M.; Mircus, G.; Pan, K.; et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: An observational study using national surveillance data. Lancet 2021, 397, 1819–1829. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Moreira, E.D.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Polack, F.P.; Zerbini, C.; et al. Six Month Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. medRxiv 2021. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; Macary, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Lanna, A.; O Gomes, D.C.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.; Lee, J.H.; Karin, M.; Akbar, A.N. A sestrin-dependent Erk–Jnk–p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017, 18, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Collier, D.A.; Ferreira, I.A.T.M.; Kotagiri, P.; Datir, R.P.; Lim, E.Y.; Touizer, E.; Meng, B.; Abdullahi, A.; Elmer, A.; Kingston, N.; et al. Age-related immune response heterogeneity to SARS-CoV-2 vaccine BNT162b2. Nature 2021, 596, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, M.; Penfold, R.S.; Merino, J.; Sudre, C.H.; Molteni, E.; Berry, S.; Canas, L.S.; Graham, M.S.; Klaser, K.; Modat, M.; et al. Risk factors and disease profile of post-vaccination SARS-CoV-2 infection in UK users of the COVID Symptom Study app: A prospective, community-based, nested, case-control study. Lancet Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Bar-On, Y.M.; Goldberg, Y.; Mandel, M.; Bodenheimer, O.; Freedman, L.; Kalkstein, N.; Mizrahi, B.; Alroy-Preis, S.; Ash, N.; Milo, R.; et al. BNT162b2 vaccine booster dose protection: A nationwide study from Israel. medRxiv 2021. [Google Scholar] [CrossRef]
- Patalon, T.; Gazit, S.; Pitzer, V.E.; Prunas, O.; Warren, J.L.; Weinberger, D.M. Short Term Reduction in the Odds of Testing Positive for SARS-CoV-2; a Comparison Between Two Doses and Three doses of the BNT162b2 Vaccine. medRxiv 2021. [Google Scholar] [CrossRef]
- Levine-Tiefenbrun, M.; Yelin, I.; Alapi, H.; Katz, R.; Herzel, E.; Kuint, J.; Chodick, G.; Gazit, S.; Patalon, T.; Kishony, R. Viral loads of Delta-variant SARS-CoV2 breakthrough infections following vaccination and booster with the BNT162b2 vaccine. medRxiv 2021. [Google Scholar] [CrossRef]
- Oberhardt, V.; Luxenburger, H.; Kemming, J.; Schulien, I.; Ciminski, K.; Giese, S.; Csernalabics, B.; Lang-Meli, J.; Janowska, I.; Staniek, J.; et al. Rapid and stable mobilization of CD8+ T cells by SARS-CoV-2 mRNA vaccine. Nature 2021, 597, 268–273. [Google Scholar] [CrossRef]
- Guaraldi, G.; Zona, S.; Silva, A.R.; Menozzi, M.; Dolci, G.; Milic, J.; Carli, F.; Mussini, C. The dynamic association between Frailty, CD4 and CD4/CD8 ratio in people aging with HIV. PLoS ONE 2019, 14, e0212283. [Google Scholar] [CrossRef]
- Corbi, G.; Cacciatore, F.; Komici, K.; Rengo, G.; Vitale, D.F.; Furgi, G.; Pagano, G.; Bencivenga, L.; Davinelli, S.; Ferrara, N. Inter-relationships between Gender, Frailty and 10-Year Survival in Older Italian Adults: An observational longitudinal study. Sci. Rep. 2019, 9, 18416. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, Y.; Ling, Y.; Lu, G.; Liu, F.; Yi, Z.; Jia, X.; Wu, M.; Shi, B.; Xu, S.; et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 2020, 583, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Colonna-Romano, G.; Bulati, M.; Aquino, A.; Pellicanò, M.; Vitello, S.; Lio, D.; Candore, G.; Caruso, C. A double-negative (IgD-CD27-) B cell population is increased in the peripheral blood of elderly people. Mech. Ageing Dev. 2009, 130, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.; Appay, V.; Campisi, J.; Frasca, D.; Fülöp, T.; Sauce, D.; Larbi, A.; Weinberger, B.; Cossarizza, A. Aging of the immune system: Focus on inflammation and vaccination. Eur. J. Immunol. 2016, 46, 2286–2301. [Google Scholar] [CrossRef]
- Weinberger, B. Vaccines for the elderly: Current use and future challenges. Immun. Ageing 2018, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Perrotta, F.; Barra, G.; Malapelle, U.; Rocco, D.; De Palma, R. Prognostic Factors and Biomarkers of Responses to Immune Checkpoint Inhibitors in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 4931. [Google Scholar] [CrossRef] [Green Version]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Collier, D.A.; De Marco, A.; Ferreira, I.A.T.M.; Meng, B.; Datir, R.; Walls, A.C.; Kemp, S.S.A.; Bassi, J.; Pinto, D.; Fregni, C.S.; et al. SARS-CoV-2 B.1.1.7 sensitivity to mRNA vaccine-elicited, convalescent and monoclonal antibodies. medRxiv 2021. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef]
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021, 384, 1885–1898. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 21 November 2021).
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- CDC COVID-19 Vaccine Breakthrough Case Investigations Team. COVID-19 Vaccine Breakthrough Infections Reported to CDC—United States, 1 January–30 April 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 792–793. [Google Scholar]
- Gazit, S.; Shlezinger, R.; Perez, G.; Lotan, R.; Peretz, A.; Ben-Tov, A.; Cohen, D.; Muhsen, K.; Chodick, G.; Patalon, T. Comparing SARS-CoV-2 natural immunity to vaccine-induced immunity: Reinfections versus breakthrough infections. medRxiv 2021. [Google Scholar] [CrossRef]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, S.; Pilishvili, T.; Derado, G.; Soe, M.M.; Dollard, P.; Wu, H.; Li, Q.; Bagchi, S.; Dubendris, H.; Link-Gelles, R.; et al. Effectiveness of Pfizer-BioNTech and Moderna Vaccines in Preventing SARS-CoV-2 Infection Among Nursing Home Residents Before and During Widespread Circulation of the SARS-CoV-2 B.1.617.2 (Delta) Variant—National Healthcare Safety Network, March 1–August 1, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1163–1166. [Google Scholar] [PubMed]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000. [Google Scholar] [CrossRef]
- Netea, M.G.; Li, Y. Immune memory in individuals with COVID-19. Nature 2021, 23, 582–584. [Google Scholar] [CrossRef]
- You, M.; Chen, L.; Zhang, D.; Zhao, P.; Chen, Z.; Qin, E.-Q.; Gao, Y.; Davis, M.M.; Yang, P. Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nature 2021, 23, 620–630. [Google Scholar] [CrossRef]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.G.P.L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, F.; Derosa, G.; Maffioli, P.; Bertuccioli, A.; Togni, S.; Riva, A.; Allegrini, P.; Khan, A.; Khan, S.; Khan, B.A.; et al. Possible Therapeutic Effects of Adjuvant Quercetin Supplementation Against Early-Stage COVID-19 Infection: A Prospective, Randomized, Controlled, and Open-Label Study. Int. J. Gen. Med. 2021, 14, 2359–2366. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, F.; Iqtadar, S.; Khan, A.; Mumtaz, S.U.; Chaudhry, M.M.; Bertuccioli, A.; Derosa, G.; Maffioli, P.; Togni, S.; Riva, A.; et al. Potential Clinical Benefits of Quercetin in the Early Stage of COVID-19: Results of a Second, Pilot, Randomized, Controlled and Open-Label Clinical Trial. Int. J. Gen. Med. 2021, 14, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Verdoorn, B.P.; Evans, T.K.; Hanson, G.J.; Zhu, Y.; Prata, L.G.P.L.; Pignolo, R.J.; Atkinson, E.J.; Wissler-Gerdes, E.O.; Kuchel, G.A.; Mannick, J.B.; et al. Fisetin for COVID -19 in skilled nursing facilities: Senolytic trials in the COVID era. J. Am. Geriatr. Soc. 2021, 69, 3023–3033. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef]
- Bartleson, J.M.; Radenkovic, D.; Covarrubias, A.J.; Furman, D.; Winer, D.A.; Verdin, E. SARS-CoV-2, COVID-19 and the aging immune system. Nat. Aging 2021, 1, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Karami, H.; Derakhshani, A.; Ghasemigol, M.; Fereidouni, M.; Miri-Moghaddam, E.; Baradaran, B.; Tabrizi, N.J.; Najafi, S.; Solimando, A.G.; Marsh, L.M.; et al. Weighted Gene Co-Expression Network Analysis Combined with Machine Learning Validation to Identify Key Modules and Hub Genes Associated with SARS-CoV-2 Infection. J. Clin. Med. 2021, 10, 3567. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lynch, S.M.; Guo, G.; Gibson, D.S.; Bjourson, A.J.; Rai, T.S. Role of Senescence and Aging in SARS-CoV-2 Infection and COVID-19 Disease. Cells 2021, 10, 3367. https://doi.org/10.3390/cells10123367
Lynch SM, Guo G, Gibson DS, Bjourson AJ, Rai TS. Role of Senescence and Aging in SARS-CoV-2 Infection and COVID-19 Disease. Cells. 2021; 10(12):3367. https://doi.org/10.3390/cells10123367
Chicago/Turabian StyleLynch, Seodhna M., Guangran Guo, David S. Gibson, Anthony J. Bjourson, and Taranjit Singh Rai. 2021. "Role of Senescence and Aging in SARS-CoV-2 Infection and COVID-19 Disease" Cells 10, no. 12: 3367. https://doi.org/10.3390/cells10123367
APA StyleLynch, S. M., Guo, G., Gibson, D. S., Bjourson, A. J., & Rai, T. S. (2021). Role of Senescence and Aging in SARS-CoV-2 Infection and COVID-19 Disease. Cells, 10(12), 3367. https://doi.org/10.3390/cells10123367