
Molecular Signaling to Preserve Mitochondrial Integrity against Ischemic Stress in the Heart: Rescue or Remove Mitochondria in Danger

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
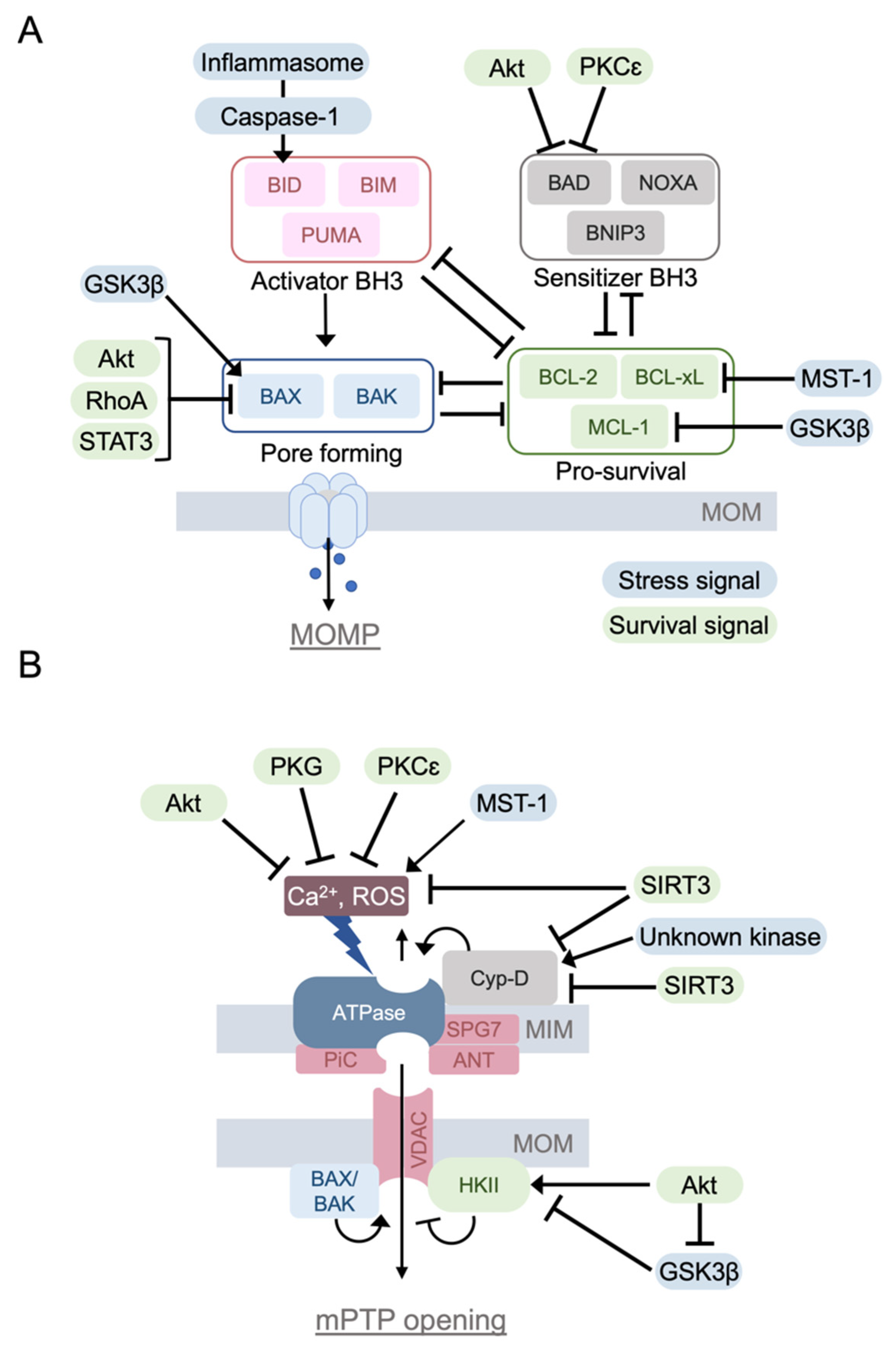
2. Mitochondrial Death Pathway
2.1. BCL-2 Family Protein and Apoptosis
2.2. Mitochondrial Permeability Transition Pore (mPTP)
2.3. Interaction between Cell Death Pathways
3. Signaling Pathways to Regulate the Mitochondrial Death Pathways
3.1. Protective Signaling to Inhibit the Mitochondrial Death Pathways
3.1.1. PKCε
3.1.2. Akt
3.1.3. RhoA
3.1.4. PKG
3.1.5. STAT3
3.1.6. SIRT3
3.2. Stress Signaling to Enhance the Mitochondrial Death Pathways
3.2.1. GSK-3β
3.2.2. Mst1
4. Mitochondrial Quality Control by Mitophagy
4.1. Conventional PINK1/Parkin Mediated Mitophagy
4.1.1. Positive Regulators of the PINK1/Parkin Pathway
BAG2, BAG3, and BAG6
SIRT3
PTENα (PTEN-L)
TBK1
AMPK1/ULK1
4.1.2. Negative Regulators of the PINK1/Parkin Pathway
Pro-Survival Members of BCL-2 Family Proteins
BAG5-HK-II Complex and BAG4
Mst1
4.2. Ubiquitination-Independent Mitophagy
4.2.1. FUNDC1
4.2.2. Cardiolipin
4.2.3. BCL2L13
4.3. Alternative (Non-Canonical) Autophagy
4.3.1. Rab9 Dependent Mitophagy
4.3.2. Endosome Pathway
4.3.3. Mitochondrial-Derived Vesicles (MDVs)
5. Mitochondrial Fission and Fusion
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ. Res. 2013, 113, 428–438. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: From endosymbionts to organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Colucci, W.S.; Arany, Z.; Bachschmid, M.M.; Ballinger, S.W.; Boudina, S.; Bruce, J.E.; Busija, D.W.; Dikalov, S.; Dorn, G.W., II; et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System: Path to a Cure in Heart Failure-A Report from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation 2019, 140, 1205–1216. [Google Scholar] [CrossRef]
- Hockenbery, D.; Nunez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef]
- Strasser, A.; Harris, A.W.; Bath, M.L.; Cory, S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 1990, 348, 331–333. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; Jockel, J.; Wei, M.C.; Korsmeyer, S.J. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998, 17, 3878–3885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chittenden, T.; Harrington, E.A.; O’Connor, R.; Flemington, C.; Lutz, R.J.; Evan, G.I.; Guild, B.C. Induction of apoptosis by the Bcl-2 homologue Bak. Nature 1995, 374, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Brocheriou, V.; Hagege, A.A.; Oubenaissa, A.; Lambert, M.; Mallet, V.O.; Duriez, M.; Wassef, M.; Kahn, A.; Menasche, P.; Gilgenkrantz, H. Cardiac functional improvement by a human Bcl-2 transgene in a mouse model of ischemia/reperfusion injury. J. Gene Med. 2000, 2, 326–333. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, H.; Xu, A.; Ross, T.; Bowler, E.; Hu, Y.; Lesnefsky, E.J. Inhibition of Bcl-2 sensitizes mitochondrial permeability transition pore (MPTP) opening in ischemia-damaged mitochondria. PLoS ONE 2015, 10, e0118834. [Google Scholar]
- Hochhauser, E.; Kivity, S.; Offen, D.; Maulik, N.; Otani, H.; Barhum, Y.; Pannet, H.; Shneyvays, V.; Shainberg, A.; Goldshtaub, V.; et al. Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2351–H2359. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Ycaza, J.E.; Gustafsson, A.B. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 2007, 405, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Toth, A.; Jeffers, J.R.; Nickson, P.; Min, J.Y.; Morgan, J.P.; Zambetti, G.P.; Erhardt, P. Targeted deletion of Puma attenuates cardiomyocyte death and improves cardiac function during ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H52–H60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yussman, M.G.; Toyokawa, T.; Odley, A.; Lynch, R.A.; Wu, G.; Colbert, M.C.; Aronow, B.J.; Lorenz, J.N.; Dorn, G.W., 2nd. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat. Med. 2002, 8, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.B.; Crofts, A.R. Calcium Ion Accumulation and Volume Changes of Isolated Liver Mitochondria. Calcium Ion-Induced Swelling. Biochem. J. 1965, 95, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crompton, M.; Costi, A.; Hayat, L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem. J. 1987, 245, 915–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Bauer, M.K.; Schubert, A.; Rocks, O.; Grimm, S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J. Cell Biol. 1999, 147, 1493–1502. [Google Scholar] [CrossRef]
- Gutierrez-Aguilar, M.; Douglas, D.L.; Gibson, A.K.; Domeier, T.L.; Molkentin, J.D.; Baines, C.P. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell Cardiol. 2014, 72, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef] [Green Version]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Forte, M. Commentary: SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Front. Physiol. 2015, 6, 320. [Google Scholar] [CrossRef] [Green Version]
- Tanveer, A.; Virji, S.; Andreeva, L.; Totty, N.F.; Hsuan, J.J.; Ward, J.M.; Crompton, M. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur. J. Biochem. 1996, 238, 166–172. [Google Scholar] [CrossRef]
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988, 255, 357–360. [Google Scholar] [PubMed]
- Hurst, S.; Gonnot, F.; Dia, M.; Crola Da Silva, C.; Gomez, L.; Sheu, S.S. Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion. Cell Death Dis. 2020, 11, 661. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, J.G.; Hoek, J.B. Hexokinase II: The integration of energy metabolism and control of apoptosis. Curr. Med. Chem. 2003, 10, 1535–1551. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, C.J.; Eerbeek, O.; Meijer, A.J. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H496–H499. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Smeele, K.M.; Southworth, R.; Wu, R.; Xie, C.; Nederlof, R.; Warley, A.; Nelson, J.K.; van Horssen, P.; van den Wijngaard, J.P.; Heikkinen, S.; et al. Disruption of hexokinase II-mitochondrial binding blocks ischemic preconditioning and causes rapid cardiac necrosis. Circ. Res. 2011, 108, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Smeele, K.M.; Wyatt, E.; Ichikawa, Y.; Eerbeek, O.; Sun, L.; Chawla, K.; Hollmann, M.W.; Nagpal, V.; Heikkinen, S.; et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ. Res. 2011, 108, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martinez-Caballero, S.; Osinska, H.; Cheng, E.H.; Robbins, J.; et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013, 2, e00772. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.S.; Konstantinidis, K.; Wei, A.C.; Chen, Y.; Reyna, D.E.; Jha, S.; Yang, Y.; Calvert, J.W.; Lindsten, T.; Thompson, C.B.; et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 6566–6571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetomi, T.; Miyamoto, S.; Brown, J.H. Inflammation in nonischemic heart disease: Initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H877–H890. [Google Scholar] [CrossRef]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2017, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Oddi Erdle, C.; Abouzaki, N.A.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Heart Fail. 2017, 10, e004373. [Google Scholar] [CrossRef]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II d δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [Green Version]
- Willeford, A.; Suetomi, T.; Nickle, A.; Hoffman, H.M.; Miyamoto, S.; Heller Brown, J. CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 2018, 3, e97054. [Google Scholar] [CrossRef] [Green Version]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Shen, J.; Li, Y.; Wu, J.; Luo, X.; Yu, Y.; Zhang, Y.; Gu, L.; Zhang, X.; Jiang, C.; et al. Pyroptosis inhibition improves the symptom of acute myocardial infarction. Cell Death Dis. 2021, 12, 852. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Thi Nguyen, D.; Hattori, T.; Manh Le, T.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P.; Song, C.X.; Zheng, Y.T.; Wang, G.W.; Zhang, J.; Wang, O.L.; Guo, Y.; Bolli, R.; Cardwell, E.M.; Ping, P. Protein kinase Cε interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.S.; Cohen, M.V.; Mochly-Rosen, D.; Downey, J.M. Protein kinase C-ε is responsible for the protection of preconditioning in rabbit cardiomyocytes. J. Mol. Cell Cardiol. 1999, 31, 1937–1948. [Google Scholar] [CrossRef] [PubMed]
- Cross, H.R.; Murphy, E.; Bolli, R.; Ping, P.; Steenbergen, C. Expression of activated PKC epsilon (PKC ε) protects the ischemic heart, without attenuating ischemic H+ production. J. Mol. Cell Cardiol. 2002, 34, 361–367. [Google Scholar] [CrossRef]
- Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Sun, L.; Mochly-Rosen, D. Mitochondrial import of PKCε is mediated by HSP90: A role in cardioprotection from ischaemia and reperfusion injury. Cardiovasc. Res. 2010, 88, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [Green Version]
- Budas, G.R.; Disatnik, M.H.; Chen, C.H.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCε) knockout mice. J. Mol. Cell Cardiol. 2010, 48, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Ohnuma, Y.; Miura, T.; Miki, T.; Tanno, M.; Kuno, A.; Tsuchida, A.; Shimamoto, K. Opening of mitochondrial KATP channel occurs downstream of PKC-ε activation in the mechanism of preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H440–H447. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.Y.; Zhu, X.; Shao, Z.; Yao, Z. Preconditioning blocks cardiocyte apoptosis: Role of KATP channels and PKC-ε. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1380–H1386. [Google Scholar] [CrossRef]
- Garlid, K.D.; Halestrap, A.P. The mitochondrial KATP channel--fact or fiction? J. Mol. Cell Cardiol. 2012, 52, 578–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotto, C.; Maulon, L.; Filippa, N.; Baier, G.; Auberger, P. Protein kinase C θ and ε promote T-cell survival by a rsk-dependent phosphorylation and inactivation of BAD. J. Biol. Chem. 2000, 275, 37246–37250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, K.; Begley, R.; Ikeno, F.; Mochly-Rosen, D. Cardioprotection by ε-protein kinase C activation from ischemia: Continuous delivery and antiarrhythmic effect of an ε-protein kinase C-activating peptide. Circulation 2005, 111, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujio, Y.; Nguyen, T.; Wencker, D.; Kitsis, R.N.; Walsh, K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 2000, 101, 660–667. [Google Scholar] [CrossRef]
- Matsui, T.; Tao, J.; del Monte, F.; Lee, K.H.; Li, L.; Picard, M.; Force, T.L.; Franke, T.F.; Hajjar, R.J.; Rosenzweig, A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 2001, 104, 330–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [Green Version]
- Sussman, M.A.; Volkers, M.; Fischer, K.; Bailey, B.; Cottage, C.T.; Din, S.; Gude, N.; Avitabile, D.; Alvarez, R.; Sundararaman, B.; et al. Myocardial AKT: The omnipresent nexus. Physiol. Rev. 2011, 91, 1023–1070. [Google Scholar] [CrossRef]
- Miyamoto, S.; Rubio, M.; Sussman, M.A. Nuclear and mitochondrial signalling Akts in cardiomyocytes. Cardiovasc. Res. 2009, 82, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Tsujita, Y.; Muraski, J.; Shiraishi, I.; Kato, T.; Kajstura, J.; Anversa, P.; Sussman, M.A. Nuclear targeting of Akt antagonizes aspects of cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11946–11951. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- del Peso, L.; Gonzalez-Garcia, M.; Page, C.; Herrera, R.; Nunez, G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997, 278, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 2001, 20, 7779–7786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardai, S.J.; Hildeman, D.A.; Frankel, S.K.; Whitlock, B.B.; Frasch, S.C.; Borregaard, N.; Marrack, P.; Bratton, D.L.; Henson, P.M. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 2004, 279, 21085–21095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Howes, A.L.; Adams, J.W.; Dorn, G.W., 2nd; Brown, J.H. Ca2+ dysregulation induces mitochondrial depolarization and apoptosis: Role of Na+/Ca2+ exchanger and AKT. J. Biol. Chem. 2005, 280, 38505–38512. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Purcell, N.H.; Smith, J.M.; Gao, T.; Whittaker, R.; Huang, K.; Castillo, R.; Glembotski, C.C.; Sussman, M.A.; Newton, A.C.; et al. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ. Res. 2010, 107, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Means, C.K.; Miyamoto, S.; Chun, J.; Brown, J.H. S1P1 receptor localization confers selectivity for Gi-mediated cAMP and contractile responses. J. Biol. Chem. 2008, 283, 11954–11963. [Google Scholar] [CrossRef] [Green Version]
- Muraski, J.A.; Rota, M.; Misao, Y.; Fransioli, J.; Cottage, C.; Gude, N.; Esposito, G.; Delucchi, F.; Arcarese, M.; Alvarez, R.; et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat. Med. 2007, 13, 1467–1475. [Google Scholar] [CrossRef]
- Brown, J.H.; Del Re, D.P.; Sussman, M.A. The Rac and Rho hall of fame: A decade of hypertrophic signaling hits. Circ. Res. 2006, 98, 730–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S.; Del Re, D.P.; Xiang, S.Y.; Zhao, X.; Florholmen, G.; Brown, J.H. Revisited and revised: Is RhoA always a villain in cardiac pathophysiology? J. Cardiovasc. Transl. Res. 2010, 3, 330–343. [Google Scholar] [CrossRef] [Green Version]
- Kilian, L.S.; Voran, J.; Frank, D.; Rangrez, A.Y. RhoA: A dubious molecule in cardiac pathophysiology. J. Biomed. Sci. 2021, 28, 33. [Google Scholar] [CrossRef]
- Yung, B.S.; Brand, C.S.; Xiang, S.Y.; Gray, C.B.; Means, C.K.; Rosen, H.; Chun, J.; Purcell, N.H.; Brown, J.H.; Miyamoto, S. Selective coupling of the S1P3 receptor subtype to S1P-mediated RhoA activation and cardioprotection. J. Mol. Cell Cardiol. 2017, 103, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Ding, E.Y.; Yu, O.M.; Xiang, S.Y.; Tan-Sah, V.P.; Yung, B.S.; Hedgpeth, J.; Neubig, R.R.; Lau, L.F.; Brown, J.H.; et al. Induction of the matricellular protein CCN1 through RhoA and MRTF-A contributes to ischemic cardioprotection. J. Mol. Cell Cardiol. 2014, 75, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghajanian, A.; Wittchen, E.S.; Campbell, S.L.; Burridge, K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS ONE 2009, 4, e8045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Re, D.P.; Miyamoto, S.; Brown, J.H. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J. Biol. Chem. 2008, 283, 35622–35629. [Google Scholar] [CrossRef] [Green Version]
- Xiang, S.Y.; Ouyang, K.; Yung, B.S.; Miyamoto, S.; Smrcka, A.V.; Chen, J.; Heller Brown, J. PLCε, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal. 2013, 6, ra108. [Google Scholar] [CrossRef] [Green Version]
- Xiang, S.Y.; Vanhoutte, D.; Del Re, D.P.; Purcell, N.H.; Ling, H.; Banerjee, I.; Bossuyt, J.; Lang, R.A.; Zheng, Y.; Matkovich, S.J.; et al. RhoA protects the mouse heart against ischemia/reperfusion injury. J. Clin. Investig. 2011, 121, 3269–3276. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Hsu, C.P.; Kajimoto, K.; Shao, D.; Yang, Y.; Maejima, Y.; Zhai, P.; Yehia, G.; Yamada, C.; Zablocki, D.; et al. Activation of PKN mediates survival of cardiac myocytes in the heart during ischemia/reperfusion. Circ. Res. 2010, 107, 642–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; Sadoshima, J. Regulation of Myocardial Cell Growth and Death by the Hippo Pathway. Circ. J. 2016, 80, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [Green Version]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, V.P.; Minamisawa, S.; Tam, S.P.; Wu, T.H.; Dorn, G.W., 2nd; Ross, J., Jr.; Chien, K.R.; Brown, J.H. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J. Clin. Investig. 1999, 103, 1627–1634. [Google Scholar] [CrossRef] [Green Version]
- Lauriol, J.; Keith, K.; Jaffre, F.; Couvillon, A.; Saci, A.; Goonasekera, S.A.; McCarthy, J.R.; Kessinger, C.W.; Wang, J.; Ke, Q.; et al. RhoA signaling in cardiomyocytes protects against stress-induced heart failure but facilitates cardiac fibrosis. Sci. Signal. 2014, 7, ra100. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Sandner, P.; Krieg, T. cGMP at the centre of attention: Emerging strategies for activating the cardioprotective PKG pathway. Basic Res. Cardiol. 2018, 113, 24. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Lohmann, S.M.; Vaandrager, A.B.; Smolenski, A.; Walter, U.; De Jonge, H.R. Distinct and specific functions of cGMP-dependent protein kinases. Trends Biochem. Sci. 1997, 22, 307–312. [Google Scholar] [CrossRef]
- Borutaite, V.; Morkuniene, R.; Arandarcikaite, O.; Jekabsone, A.; Barauskaite, J.; Brown, G.C. Nitric oxide protects the heart from ischemia-induced apoptosis and mitochondrial damage via protein kinase G mediated blockage of permeability transition and cytochrome c release. J. Biomed. Sci. 2009, 16, 70. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.D.; Garlid, K.D.; West, I.C.; Lincoln, T.M.; Downey, J.M.; Cohen, M.V.; Critz, S.D. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ. Res. 2005, 97, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankenreiter, S.; Bednarczyk, P.; Kniess, A.; Bork, N.I.; Straubinger, J.; Koprowski, P.; Wrzosek, A.; Mohr, E.; Logan, A.; Murphy, M.P.; et al. cGMP-Elevating Compounds and Ischemic Conditioning Provide Cardioprotection Against Ischemia and Reperfusion Injury via Cardiomyocyte-Specific BK Channels. Circulation 2017, 136, 2337–2355. [Google Scholar] [CrossRef] [Green Version]
- Inserte, J.; Barba, I.; Poncelas-Nozal, M.; Hernando, V.; Agullo, L.; Ruiz-Meana, M.; Garcia-Dorado, D. cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion. J. Mol. Cell Cardiol. 2011, 50, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Garcia-Dorado, D. The cGMP/PKG pathway as a common mediator of cardioprotection: Translatability and mechanism. Br. J. Pharmacol. 2015, 172, 1996–2009. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Lee, S.R.; Cho, H.; Woo, J.S.; Kang, J.H.; Jeong, Y.M.; Cheng, X.W.; Kim, W.S.; Kim, W. Cardioprotective effects of PKG activation by soluble GC activator, BAY 60-2770, in ischemia-reperfusion-injured rat hearts. PLoS ONE 2017, 12, e0180207. [Google Scholar] [CrossRef] [PubMed]
- Ranek, M.J.; Oeing, C.; Sanchez-Hodge, R.; Kokkonen-Simon, K.M.; Dillard, D.; Aslam, M.I.; Rainer, P.P.; Mishra, S.; Dunkerly-Eyring, B.; Holewinski, R.J.; et al. CHIP phosphorylation by protein kinase G enhances protein quality control and attenuates cardiac ischemic injury. Nat. Commun. 2020, 11, 5237. [Google Scholar] [CrossRef]
- Das, A.; Smolenski, A.; Lohmann, S.M.; Kukreja, R.C. Cyclic GMP-dependent protein kinase Iα attenuates necrosis and apoptosis following ischemia/reoxygenation in adult cardiomyocyte. J. Biol. Chem. 2006, 281, 38644–38652. [Google Scholar] [CrossRef] [Green Version]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Zouein, F.A.; Altara, R.; Chen, Q.; Lesnefsky, E.J.; Kurdi, M.; Booz, G.W. Pivotal Importance of STAT3 in Protecting the Heart from Acute and Chronic Stress: New Advancement and Unresolved Issues. Front. Cardiovasc. Med. 2015, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Harhous, Z.; Booz, G.W.; Ovize, M.; Bidaux, G.; Kurdi, M. An Update on the Multifaceted Roles of STAT3 in the Heart. Front. Cardiovasc. Med. 2019, 6, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G.; Musiolik, J.; Gedik, N.; Skyschally, A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ. Res. 2011, 109, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Fuglesteg, B.N.; Suleman, N.; Tiron, C.; Kanhema, T.; Lacerda, L.; Andreasen, T.V.; Sack, M.N.; Jonassen, A.K.; Mjos, O.D.; Opie, L.H.; et al. Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res. Cardiol. 2008, 103, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G.; Boengler, K.; Schulz, R. Inhibition of mitochondrial permeability transition pore opening: The Holy Grail of cardioprotection. Basic Res. Cardiol. 2010, 105, 151–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 10, eaag2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanek, K.; Chen, Q.; Derecka, M.; Salloum, F.N.; Zhang, Q.; Szelag, M.; Cichy, J.; Kukreja, R.C.; Dulak, J.; Lesnefsky, E.J.; et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 2011, 286, 29610–29620. [Google Scholar] [CrossRef] [Green Version]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Fuchs, M.; Kaminski, K.; Schaefer, A.; Schieffer, B.; Hillmer, A.; Schmiedl, A.; Ding, Z.; Podewski, E.; et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004, 95, 187–195. [Google Scholar] [CrossRef]
- Enomoto, D.; Obana, M.; Miyawaki, A.; Maeda, M.; Nakayama, H.; Fujio, Y. Cardiac-specific ablation of the STAT3 gene in the subacute phase of myocardial infarction exacerbated cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H471–H480. [Google Scholar] [CrossRef] [Green Version]
- Parodi-Rullan, R.M.; Chapa-Dubocq, X.R.; Javadov, S. Acetylation of Mitochondrial Proteins in the Heart: The Role of SIRT3. Front. Physiol. 2018, 9, 1094. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, S.; Sadoshima, J. The role of sirtuins in cardiac disease. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1375–H1389. [Google Scholar] [CrossRef] [Green Version]
- Koentges, C.; Bode, C.; Bugger, H. SIRT3 in Cardiac Physiology and Disease. Front. Cardiovasc. Med. 2016, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Parodi-Rullan, R.M.; Chapa-Dubocq, X.; Rullan, P.J.; Jang, S.; Javadov, S. High Sensitivity of SIRT3 Deficient Hearts to Ischemia-Reperfusion Is Associated with Mitochondrial Abnormalities. Front. Pharmacol. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1602–H1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koentges, C.; Pfeil, K.; Meyer-Steenbuck, M.; Lother, A.; Hoffmann, M.M.; Odening, K.E.; Hein, L.; Bode, C.; Bugger, H. Preserved recovery of cardiac function following ischemia-reperfusion in mice lacking SIRT3. Can. J. Physiol. Pharmacol. 2016, 94, 72–80. [Google Scholar] [CrossRef]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Ohori, K.; Miura, T.; Tanno, M.; Miki, T.; Sato, T.; Ishikawa, S.; Horio, Y.; Shimamoto, K. Ser9 phosphorylation of mitochondrial GSK-3β is a primary mechanism of cardiomyocyte protection by erythropoietin against oxidant-induced apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2079–H2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef] [Green Version]
- Linseman, D.A.; Butts, B.D.; Precht, T.A.; Phelps, R.A.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Florez-McClure, M.L.; Heidenreich, K.A. Glycogen synthase kinase-3β phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004, 24, 9993–10002. [Google Scholar] [CrossRef]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Paillard, M.; Thibault, H.; Derumeaux, G.; Ovize, M. Inhibition of GSK3β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation 2008, 117, 2761–2768. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Imahashi, K.; Steenbergen, C.; Murphy, E. Phosphorylation of glycogen synthase kinase-3β during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective. Circ. Res. 2002, 90, 377–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, M.; Miura, T.; Miki, T.; Tanno, M.; Yano, T.; Naitoh, K.; Ohori, K.; Hotta, H.; Terashima, Y.; Shimamoto, K. Modulation of the mitochondrial permeability transition pore complex in GSK-3β-mediated myocardial protection. J. Mol. Cell Cardiol. 2007, 43, 564–570. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 Family as Therapeutic Target for Myocardial Diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential Roles of GSK-3β During Myocardial Ischemia and Ischemia/Reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P. Hippo Signaling in the Heart- Non-Canonical Pathways Impact Growth, Survival and Function. Circ. J. 2016, 80, 1504–1510. [Google Scholar] [CrossRef] [Green Version]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Maejima, Y.; Jain, M.R.; Liu, T.; Li, H.; Hsu, C.P.; Sadoshima, J. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol. Cell 2014, 54, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Zhai, P.; Del Re, D.P.; Maejima, Y.; Sadoshima, J. Mst1-mediated phosphorylation of Bcl-xL is required for myocardial reperfusion injury. JCI Insight 2016, 1, e86217. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yang, G.; Zablocki, D.; Liu, J.; Hong, C.; Kim, S.J.; Soler, S.; Odashima, M.; Thaisz, J.; Yehia, G.; et al. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J. Clin. Investig. 2003, 111, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Lavandero, S.; Chiong, M.; Rothermel, B.A.; Hill, J.A. Autophagy in cardiovascular biology. J. Clin. Investig. 2015, 125, 55–64. [Google Scholar] [CrossRef]
- Delbridge, L.M.D.; Mellor, K.M.; Taylor, D.J.; Gottlieb, R.A. Myocardial stress and autophagy: Mechanisms and potential therapies. Nat. Rev. Cardiol. 2017, 14, 412–425. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Guberman, M.; Kirshenbaum, L.A. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc. Med. 2018, 28, 246–260. [Google Scholar] [CrossRef]
- Gustafsson, A.B.; Dorn, G.W., 2nd. Evolving and Expanding the Roles of Mitophagy as a Homeostatic and Pathogenic Process. Physiol. Rev. 2019, 99, 853–892. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Piplani, H.; Sin, J.; Sawaged, S.; Hamid, S.M.; Taylor, D.J.; de Freitas Germano, J. At the heart of mitochondrial quality control: Many roads to the top. Cell Mol. Life Sci. 2021, 78, 3791–3801. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Liu, Y.; Ginet, V.; Shi, M.; Nah, J.; Zou, Z.; Zhou, A.; Posner, B.A.; Xiao, G.; Tanguy, M.; et al. Interaction between the autophagy protein Beclin 1 and Na+,K+-ATPase during starvation, exercise, and ischemia. JCI Insight 2020, 5, e133282. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, A.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W., 2nd. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddall, H.K.; Yellon, D.M.; Ong, S.B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PLoS ONE 2013, 8, e62400. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Quinsay, M.N.; Gustafsson, A.B. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun. Integr. Biol. 2013, 6, e24511. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Uno, M.; Koyano, F.; Go, E.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J. Biol. Chem. 2013, 288, 36372–36384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Koyano, F.; Kimura, M.; Kosako, H.; Saeki, Y.; Tanaka, K.; Matsuda, N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 2015, 209, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamano, K.; Wang, C.; Sarraf, S.A.; Munch, C.; Kikuchi, R.; Noda, N.N.; Hizukuri, Y.; Kanemaki, M.T.; Harper, W.; Tanaka, K.; et al. Endosomal Rab cycles regulate Parkin-mediated mitophagy. eLife 2018, 7, e31326. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sun, S.; Xu, H.; Li, C.; Ren, J.; Zhang, Y. TBC1D15/RAB7-regulated mitochondria-lysosome interaction confers cardioprotection against acute myocardial infarction-induced cardiac injury. Theranostics 2020, 10, 11244–11263. [Google Scholar] [CrossRef]
- Takayama, S.; Sato, T.; Krajewski, S.; Kochel, K.; Irie, S.; Millan, J.A.; Reed, J.C. Cloning and functional analysis of BAG-1: A novel Bcl-2-binding protein with anti-cell death activity. Cell 1995, 80, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Behl, C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol. Sci. 2016, 37, 672–688. [Google Scholar] [CrossRef]
- Kabbage, M.; Dickman, M.B. The BAG proteins: A ubiquitous family of chaperone regulators. Cell Mol. Life Sci. 2008, 65, 1390–1402. [Google Scholar] [CrossRef]
- Schanzer, A.; Rupp, S.; Graf, S.; Zengeler, D.; Jux, C.; Akinturk, H.; Gulatz, L.; Mazhari, N.; Acker, T.; Van Coster, R.; et al. Dysregulated autophagy in restrictive cardiomyopathy due to Pro209Leu mutation in BAG3. Mol. Genet. Metab. 2018, 123, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, K.; Zhang, X.; Tang, Y.; Xu, D. Advances in the role and mechanism of BAG3 in dilated cardiomyopathy. Heart Fail. Rev. 2021, 26, 183–194. [Google Scholar] [CrossRef]
- Myers, V.D.; Tomar, D.; Madesh, M.; Wang, J.; Song, J.; Zhang, X.Q.; Gupta, M.K.; Tahrir, F.G.; Gordon, J.; McClung, J.M.; et al. Haplo-insufficiency of Bcl2-associated athanogene 3 in mice results in progressive left ventricular dysfunction, β-adrenergic insensitivity, and increased apoptosis. J. Cell Physiol. 2018, 233, 6319–6326. [Google Scholar] [CrossRef] [PubMed]
- Homma, S.; Iwasaki, M.; Shelton, G.D.; Engvall, E.; Reed, J.C.; Takayama, S. BAG3 deficiency results in fulminant myopathy and early lethality. Am. J. Pathol. 2006, 169, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Myers, V.D.; Knezevic, T.; Wang, J.; Gao, E.; Madesh, M.; Tahrir, F.G.; Gupta, M.K.; Gordon, J.; Rabinowitz, J.; et al. Bcl-2-associated athanogene 3 protects the heart from ischemia/reperfusion injury. JCI Insight 2016, 1, e90931. [Google Scholar] [CrossRef] [PubMed]
- Tahrir, F.G.; Knezevic, T.; Gupta, M.K.; Gordon, J.; Cheung, J.Y.; Feldman, A.M.; Khalili, K. Evidence for the Role of BAG3 in Mitochondrial Quality Control in Cardiomyocytes. J. Cell Physiol. 2017, 232, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Che, X.; Tang, B.; Wang, X.; Chen, D.; Yan, X.; Jiang, H.; Shen, L.; Xu, Q.; Wang, G.; Guo, J. The BAG2 protein stabilises PINK1 by decreasing its ubiquitination. Biochem. Biophys. Res. Commun. 2013, 441, 488–492. [Google Scholar] [CrossRef]
- Qu, D.; Hage, A.; Don-Carolis, K.; Huang, E.; Joselin, A.; Safarpour, F.; Marcogliese, P.C.; Rousseaux, M.W.; Hewitt, S.J.; Huang, T.; et al. BAG2 Gene-mediated Regulation of PINK1 Protein Is Critical for Mitochondrial Translocation of PARKIN and Neuronal Survival. J. Biol. Chem. 2015, 290, 30441–30452. [Google Scholar] [CrossRef] [Green Version]
- Ragimbeau, R.; El Kebriti, L.; Sebti, S.; Fourgous, E.; Boulahtouf, A.; Arena, G.; Espert, L.; Turtoi, A.; Gongora, C.; Houede, N.; et al. BAG6 promotes PINK1 signaling pathway and is essential for mitophagy. FASEB J. 2021, 35, e21361. [Google Scholar] [CrossRef]
- Yang, W.; Nagasawa, K.; Munch, C.; Xu, Y.; Satterstrom, K.; Jeong, S.; Hayes, S.D.; Jedrychowski, M.P.; Vyas, F.S.; Zaganjor, E.; et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016, 167, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Huang, G.; Gao, J.; Huang, C.; Sun, M.; Wu, J.; Bu, J.; Shen, W. Sirtuin 3 Deficiency Accelerates Hypertensive Cardiac Remodeling by Impairing Angiogenesis. J. Am. Heart Assoc. 2017, 6, e006114. [Google Scholar] [CrossRef]
- Huang, D.; Liu, M.; Jiang, Y. Mitochonic acid-5 attenuates TNF-α-mediated neuronal inflammation via activating Parkin-related mitophagy and augmenting the AMPK-Sirt3 pathways. J. Cell Physiol. 2019, 234, 22172–22182. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Ali-Hemmati, A.; Rahmati, M.; Charoudeh, H.N. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014, 19, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cho, Y.L.; Tang, Y.; Wang, J.; Park, J.E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef]
- Li, G.; Yang, J.; Yang, C.; Zhu, M.; Jin, Y.; McNutt, M.A.; Yin, Y. PTENα regulates mitophagy and maintains mitochondrial quality control. Autophagy 2018, 14, 1742–1760. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, P.; Li, C.; Wang, M.; Ren, J.; Zhang, Y.; Fu, G. TANK-binding kinase 1 alleviates myocardial ischemia/reperfusion injury through regulating apoptotic pathway. Biochem. Biophys. Res. Commun. 2020, 528, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, W.; Bi, Y.; Xu, H.; Abudureyimu, M.; Peng, H.; Zhang, Y.; Ren, J. NDP52 Protects against Myocardial Infarction-Provoked Cardiac Anomalies through Promoting Autophagosome-Lysosome Fusion via Recruiting TBK1 and RAB7. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.R., 3rd; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018. [Google Scholar] [CrossRef]
- Drake, J.C.; Wilson, R.J.; Laker, R.C.; Guan, Y.; Spaulding, H.R.; Nichenko, A.S.; Shen, W.; Shang, H.; Dorn, M.V.; Huang, K.; et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025932118. [Google Scholar] [CrossRef]
- Hung, C.M.; Lombardo, P.S.; Malik, N.; Brun, S.N.; Hellberg, K.; Van Nostrand, J.L.; Garcia, D.; Baumgart, J.; Diffenderfer, K.; Asara, J.M.; et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci. Adv. 2021, 7, eabg4544. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362 e346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Nah, J.; Oka, S.I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [Green Version]
- Hollville, E.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, Z.; Liu, R.; Liang, Q.; Peng, Z.; Yin, S.; Tang, J.; Gong, T.; Liu, Y. Bcl-2 Proteins Regulate Mitophagy in Lipopolysaccharide-Induced Acute Lung Injury via PINK1/Parkin Signaling Pathway. Oxid Med. Cell Longev. 2020, 2020, 6579696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Du, M.; Yin, A.; Mai, Z.; Wang, Y.; Zhao, M.; Wang, X.; Chen, T. Bcl-xL inhibits PINK1/Parkin-dependent mitophagy by preventing mitochondrial Parkin accumulation. Int. J. Biochem. Cell Biol. 2020, 122, 105720. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Brouwer, J.M.; Tan, I.K.; Sandow, J.J.; Huang, S.; Stafford, C.A.; Bankovacki, A.; Riffkin, C.D.; Wardak, A.Z.; Czabotar, P.E.; et al. Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. 2019, 38, e99916. [Google Scholar] [CrossRef]
- Johnson, B.N.; Berger, A.K.; Cortese, G.P.; Lavoie, M.J. The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc. Natl. Acad. Sci. USA 2012, 109, 6283–6288. [Google Scholar] [CrossRef] [Green Version]
- Kalia, S.K.; Lee, S.; Smith, P.D.; Liu, L.; Crocker, S.J.; Thorarinsdottir, T.E.; Glover, J.R.; Fon, E.A.; Park, D.S.; Lozano, A.M. BAG5 inhibits parkin and enhances dopaminergic neuron degeneration. Neuron 2004, 44, 931–945. [Google Scholar] [CrossRef] [Green Version]
- Tan, V.P.; Smith, J.M.; Tu, M.; Yu, J.D.; Ding, E.Y.; Miyamoto, S. Dissociation of mitochondrial HK-II elicits mitophagy and confers cardioprotection against ischemia. Cell Death Dis. 2019, 10, 730. [Google Scholar] [CrossRef]
- De Snoo, M.L.; Friesen, E.L.; Zhang, Y.T.; Earnshaw, R.; Dorval, G.; Kapadia, M.; O’Hara, D.M.; Agapova, V.; Chau, H.; Pellerito, O.; et al. Bcl-2-associated athanogene 5 (BAG5) regulates Parkin-dependent mitophagy and cell death. Cell Death Dis. 2019, 10, 907. [Google Scholar] [CrossRef] [Green Version]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Z.; Fan, Y.; Zhang, M.; Feng, X.; Lin, J.; Hu, J.; Cheng, Z.; Sun, C.; Liu, T.; et al. Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1905–1914. [Google Scholar] [CrossRef]
- Shang, X.; Lin, K.; Zhang, Y.; Li, M.; Xu, J.; Chen, K.; Zhu, P.; Yu, R. Mst1 deletion reduces septic cardiomyopathy via activating Parkin-related mitophagy. J. Cell Physiol. 2020, 235, 317–327. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, B.; Wang, L.; Wang, Z.; Jian, Z.; Deng, L. Mammalian STE20like kinase 1 regulates pancreatic cancer cell survival and migration through Mfn2mediated mitophagy. Mol. Med. Rep. 2020, 22, 398–404. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd. Mitochondrial pruning by Nix and BNip3: An essential function for cardiac-expressed death factors. J. Cardiovasc. Transl. Res. 2010, 3, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A.; Gustafsson, A.B. Autophagy as a protective response to Bnip3-mediated apoptotic signaling in the heart. Autophagy 2006, 2, 307–309. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Quinsay, M.N.; Huang, C.; Lee, Y.; Gustafsson, A.B. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2025–H2031. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-Terminus Inhibits Mitochondrial Damage and Cell Death without Blocking Autophagy. PLoS ONE 2015, 10, e0129667. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.V.; Suzuki, H.; Marinkovic, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Sprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef] [PubMed]
- da Silva Rosa, S.C.; Martens, M.D.; Field, J.T.; Nguyen, L.; Kereliuk, S.M.; Hai, Y.; Chapman, D.; Diehl-Jones, W.; Aliani, M.; West, A.R.; et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 2021, 17, 2257–2272. [Google Scholar] [CrossRef]
- Marinkovic, M.; Sprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhu, P.; Guo, J.; Hu, N.; Wang, S.; Li, D.; Hu, S.; Ren, J.; Cao, F.; Chen, Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol 2017, 13, 498–507. [Google Scholar] [CrossRef]
- Zhang, W.; Ren, H.; Xu, C.; Zhu, C.; Wu, H.; Liu, D.; Wang, J.; Liu, L.; Li, W.; Ma, Q.; et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. eLife 2016, 5, e21407. [Google Scholar] [CrossRef]
- Yu, W.; Xu, M.; Zhang, T.; Zhang, Q.; Zou, C. Mst1 promotes cardiac ischemia-reperfusion injury by inhibiting the ERK-CREB pathway and repressing FUNDC1-mediated mitophagy. J. Physiol. Sci. 2019, 69, 113–127. [Google Scholar] [CrossRef]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in Vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: Implications for pharmacological cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, T.; Holler, N.; Micheau, O.; Martinon, F.; Tinel, A.; Hofmann, K.; Tschopp, J. Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J. Biol. Chem. 2001, 276, 19548–19554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hamano, K.; Sadoshima, J. Molecular mechanisms and clinical implications of multiple forms of mitophagy in the heart. Cardiovasc. Res. 2020, cvaa340. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Kuo, W.W.; Ho, T.J.; Chiang, S.F.; Pai, P.Y.; Lin, J.Y.; Lin, D.Y.; Kuo, C.H.; Huang, C.Y. Rab9-dependent autophagy is required for the IGF-IIR triggering mitophagy to eliminate damaged mitochondria. J. Cell Physiol. 2018, 233, 7080–7091. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Shires, S.E.; Leon, L.J.; Cortez, M.Q.; Gustafsson, A.B. Isolation of Rab5-positive endosomes reveals a new mitochondrial degradation pathway utilized by BNIP3 and Parkin. Small GTPases 2020, 11, 69–76. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Najor, R.H.; Cortez, M.Q.; Shires, S.E.; Leon, L.J.; Gonzalez, E.R.; Boassa, D.; Phan, S.; Thor, A.; Jimenez, R.E.; et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat. Commun. 2017, 8, 14050. [Google Scholar] [CrossRef]
- McLelland, G.L.; Lee, S.A.; McBride, H.M.; Fon, E.A. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol. 2016, 214, 275–291. [Google Scholar] [CrossRef]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhao, H.; Wu, Y.; Zhu, Y.; Zhang, J.; Yang, G.; Yan, Q.; Li, J.; Li, T.; Liu, L. Mitochondrial-Derived Vesicles Protect Cardiomyocytes Against Hypoxic Damage. Front. Cell Dev. Biol. 2020, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Cadete, V.J.; Deschenes, S.; Cuillerier, A.; Brisebois, F.; Sugiura, A.; Vincent, A.; Turnbull, D.; Picard, M.; McBride, H.M.; Burelle, Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 2016, 594, 5343–5362. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd; Kitsis, R.N. The Mitochondrial Dynamism-Mitophagy-Cell Death Interactome: Multiple Roles Performed by Members of a Mitochondrial Molecular Ensemble. Circ. Res. 2015, 116, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Adaniya, S.M.; O-Uchi, J.; Cypress, M.W.; Kusakari, Y.; Jhun, B.S. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am. J. Physiol. Cell Physiol. 2019, 316, C583–C604. [Google Scholar] [CrossRef]
- Tong, M.; Zablocki, D.; Sadoshima, J. The role of Drp1 in mitophagy and cell death in the heart. J. Mol. Cell Cardiol. 2020, 142, 138–145. [Google Scholar] [CrossRef]
- Zaja, I.; Bai, X.; Liu, Y.; Kikuchi, C.; Dosenovic, S.; Yan, Y.; Canfield, S.G.; Bosnjak, Z.J. Cdk1, PKCδ and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem. Biophys. Res. Commun. 2014, 453, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic β-AR stimulation. Nat. Commun. 2016, 7, 13189. [Google Scholar] [CrossRef]
- Yu, J.; Maimaitili, Y.; Xie, P.; Wu, J.J.; Wang, J.; Yang, Y.N.; Ma, H.P.; Zheng, H. High glucose concentration abrogates sevoflurane post-conditioning cardioprotection by advancing mitochondrial fission but dynamin-related protein 1 inhibitor restores these effects. Acta Physiol. 2017, 220, 83–98. [Google Scholar] [CrossRef]
- Brand, C.S.; Tan, V.P.; Brown, J.H.; Miyamoto, S. RhoA regulates Drp1 mediated mitochondrial fission through ROCK to protect cardiomyocytes. Cell Signal. 2018, 50, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhun, B.S.; O-Uchi, J.; Adaniya, S.M.; Mancini, T.J.; Cao, J.L.; King, M.E.; Landi, A.K.; Ma, H.; Shin, M.; Yang, D.; et al. Protein kinase D activation induces mitochondrial fragmentation and dysfunction in cardiomyocytes. J. Physiol. 2018, 596, 827–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Din, S.; Mason, M.; Volkers, M.; Johnson, B.; Cottage, C.T.; Wang, Z.; Joyo, A.Y.; Quijada, P.; Erhardt, P.; Magnuson, N.S.; et al. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc. Natl. Acad. Sci. USA 2013, 110, 5969–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn II, G.W.; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zepeda, R.; Kuzmicic, J.; Parra, V.; Troncoso, R.; Pennanen, C.; Riquelme, J.A.; Pedrozo, Z.; Chiong, M.; Sanchez, G.; Lavandero, S. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. 2014, 63, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Finkel, T. Cardiac mitochondria: A surprise about size. J. Mol. Cell Cardiol. 2015, 82, 213–215. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Ngoh, G.A.; Dabkowski, E.R.; O’Connell, K.A.; Ribeiro, R.F., Jr.; Stanley, W.C.; Walsh, K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H167–H179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Dasgupta, A.; Chen, K.H.; Neuber-Hess, M.; Patel, J.; Hurst, T.E.; Mewburn, J.D.; Lima, P.D.A.; Alizadeh, E.; Martin, A.; et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020, 34, 1447–1464. [Google Scholar] [CrossRef] [Green Version]
- Bouche, L.; Kamel, R.; Tamareille, S.; Garcia, G.; Villedieu, C.; Pillot, B.; Gueguen, N.; Chehaitly, A.; Chao de la Barca, J.M.; Beaumont, J.; et al. DRP1 haploinsufficiency attenuates cardiac ischemia/reperfusion injuries. PLoS ONE 2021, 16, e0248554. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Combs, C.A.; Femnou, A.; Sun, J.; Murphy, E.; Subramaniam, S.; Balaban, R.S. Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep. 2017, 19, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Leopold, J.A. Antioxidants and coronary artery disease: From pathophysiology to preventive therapy. Coron. Artery Dis. 2015, 26, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forini, F.; Canale, P.; Nicolini, G.; Iervasi, G. Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics 2020, 12, 1122. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, G.; Matoba, T.; Ishikita, A.; Nagaoka, K.; Nakano, K.; Koga, J.I.; Tsutsui, H.; Egashira, K. Nanoparticle-Mediated Simultaneous Targeting of Mitochondrial Injury and Inflammation Attenuates Myocardial Ischemia-Reperfusion Injury. J. Am. Heart Assoc. 2021, 10, e019521. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, D.Z.; Zhang, C.X.; Cui, H.; Liu, M.; Zhang, B.L.; Mei, Q.B.; Lu, Z.F.; Zhou, S.Y. Mitochondria-targeted antioxidant delivery for precise treatment of myocardial ischemia-reperfusion injury through a multistage continuous targeted strategy. Nanomedicine 2019, 16, 236–249. [Google Scholar] [CrossRef]
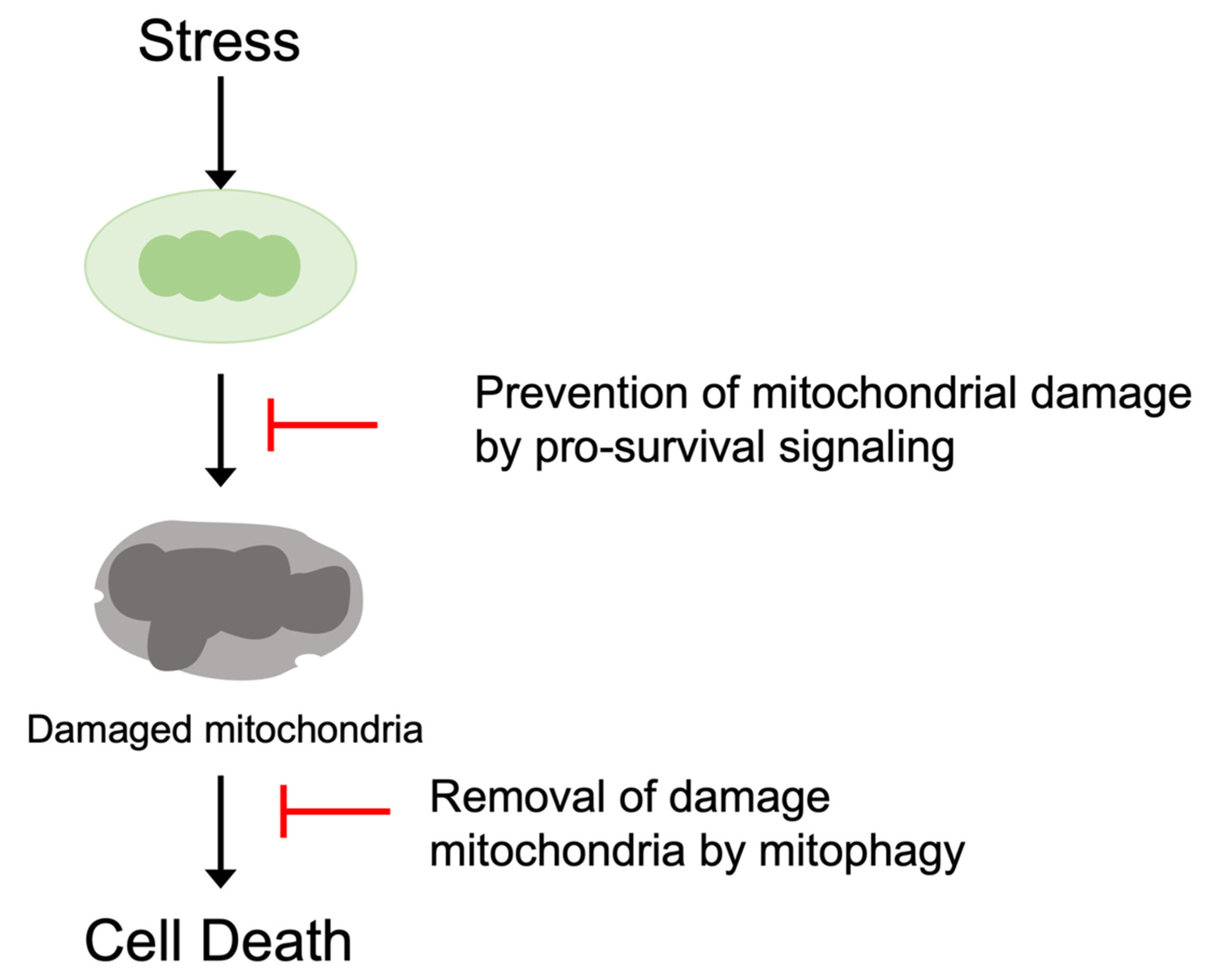
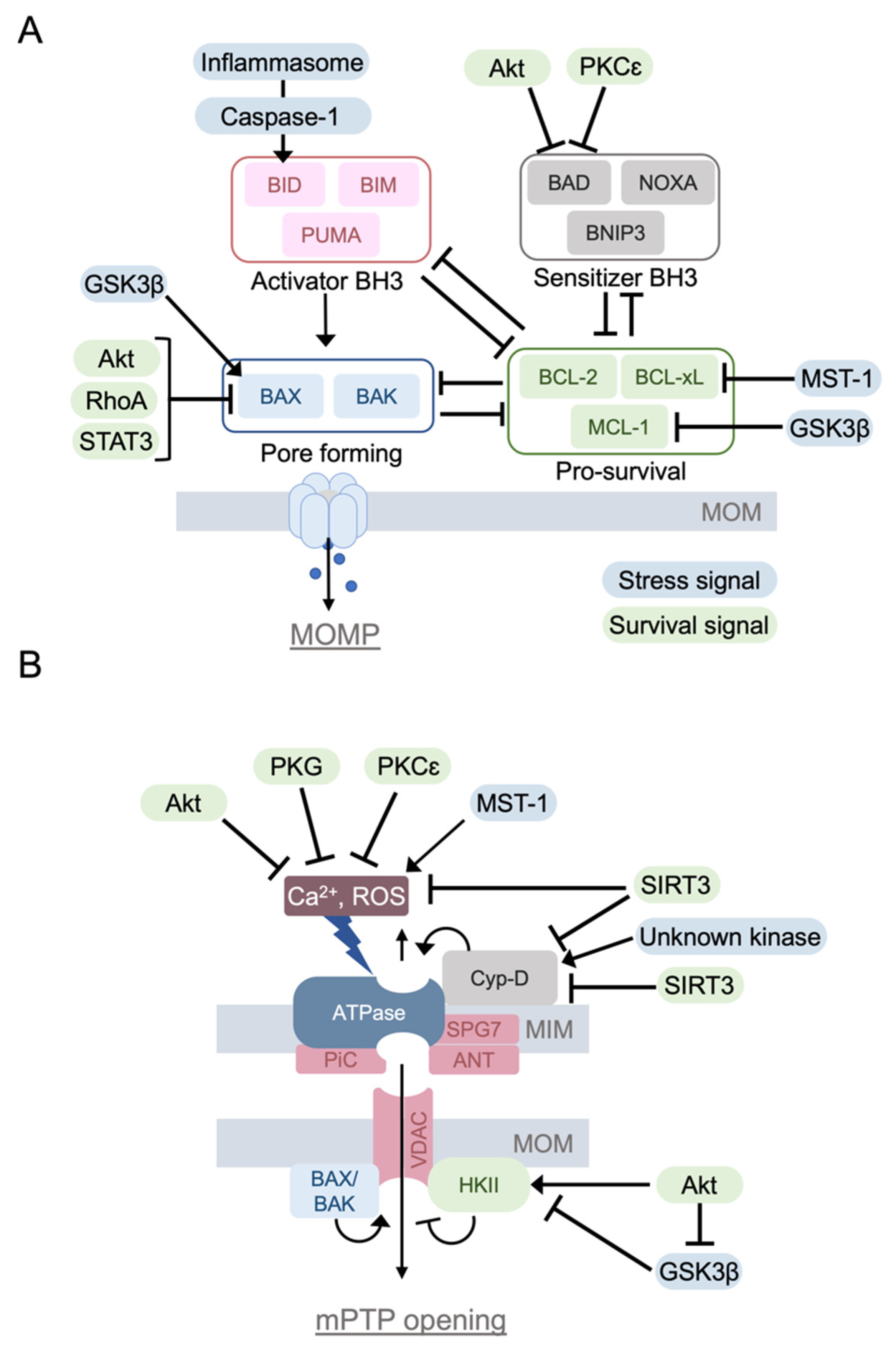
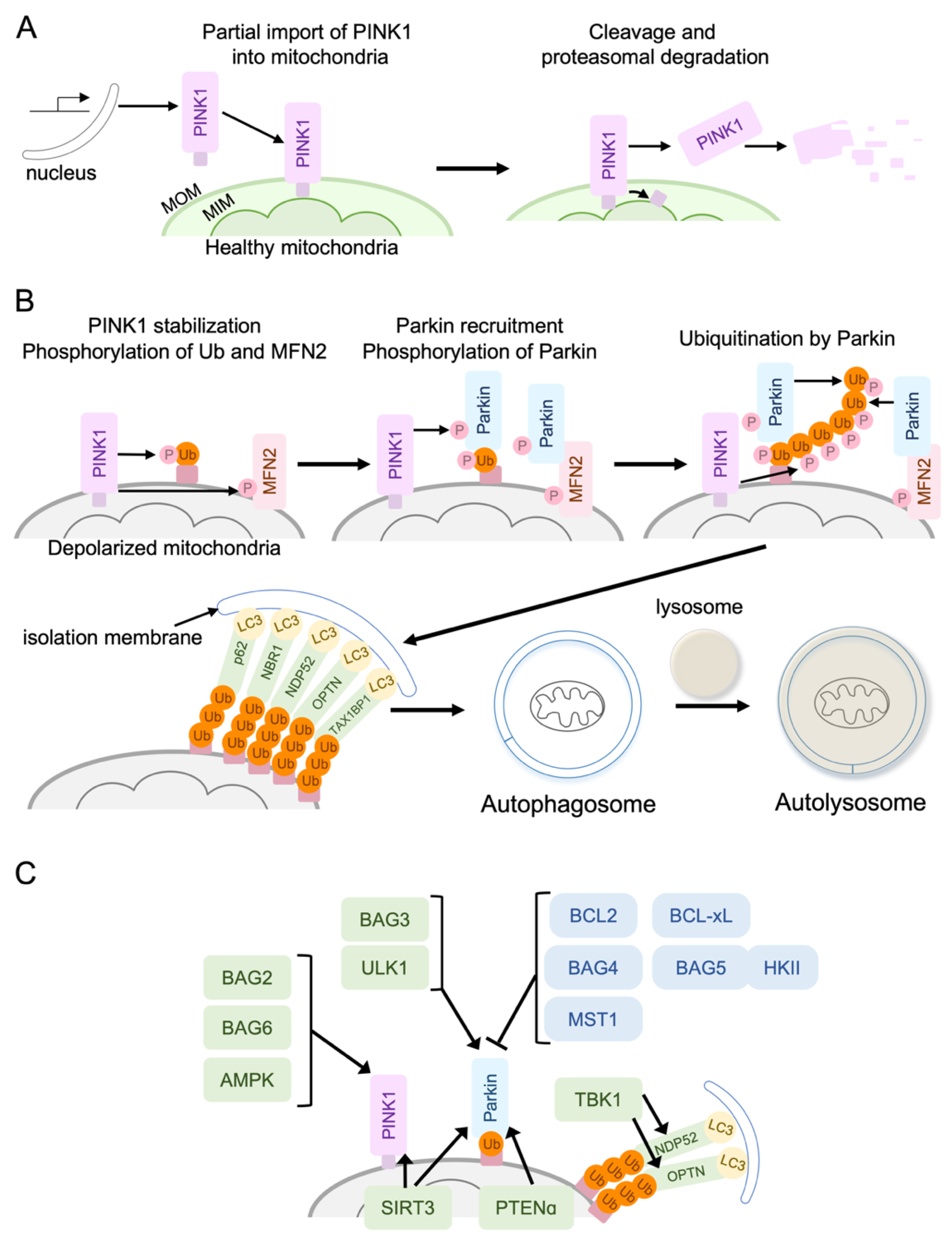
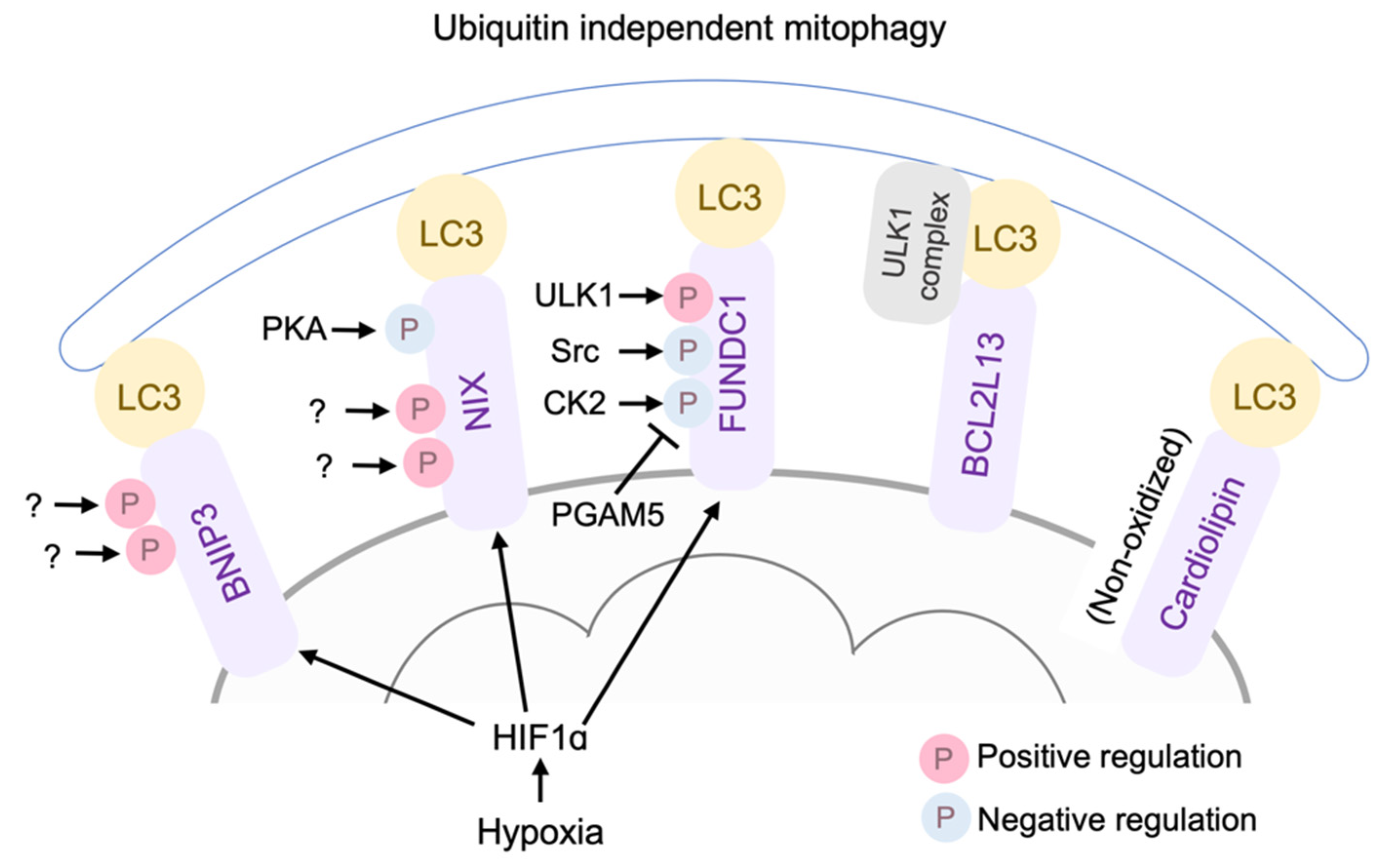
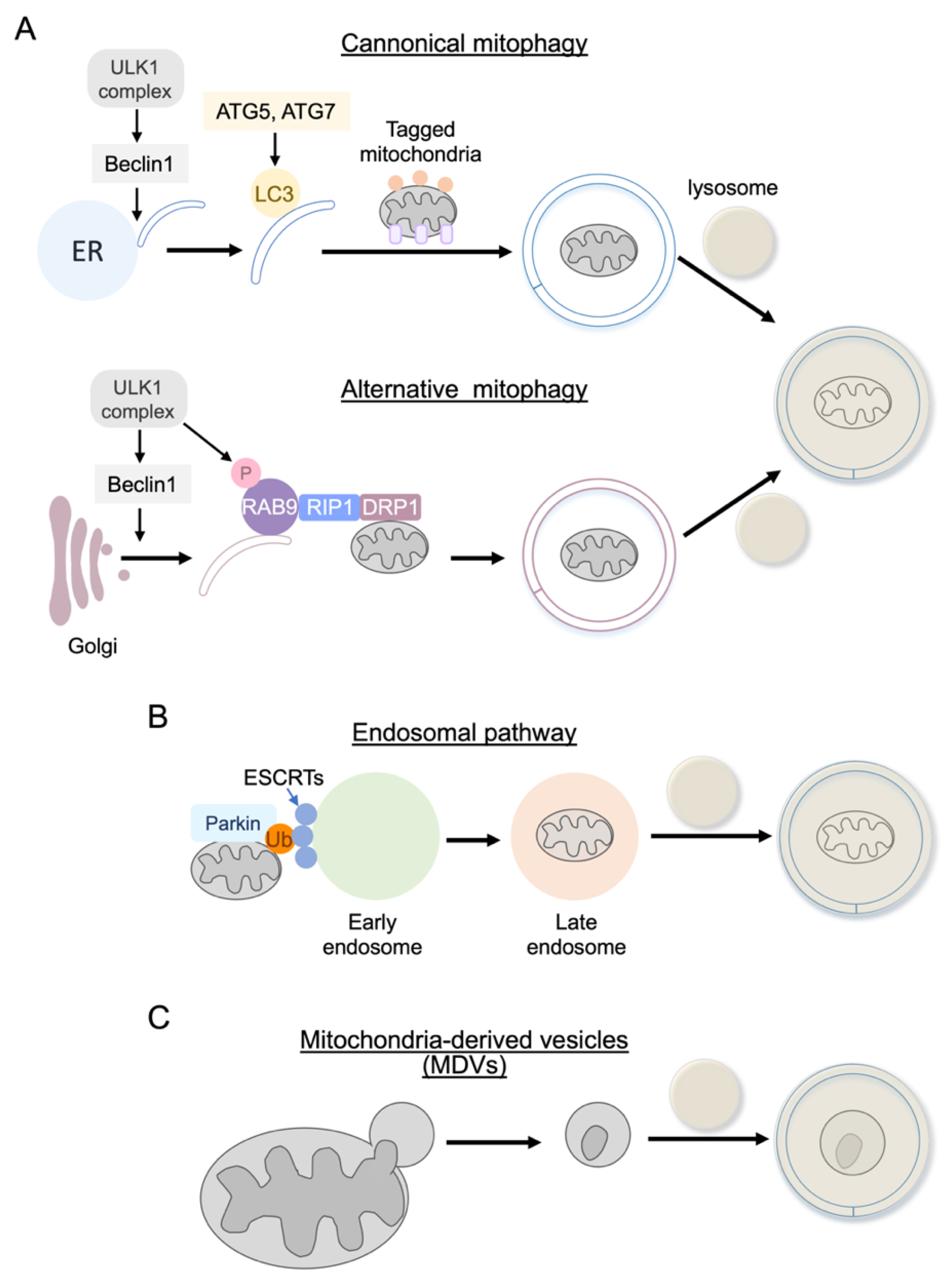
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.D.; Miyamoto, S. Molecular Signaling to Preserve Mitochondrial Integrity against Ischemic Stress in the Heart: Rescue or Remove Mitochondria in Danger. Cells 2021, 10, 3330. https://doi.org/10.3390/cells10123330
Yu JD, Miyamoto S. Molecular Signaling to Preserve Mitochondrial Integrity against Ischemic Stress in the Heart: Rescue or Remove Mitochondria in Danger. Cells. 2021; 10(12):3330. https://doi.org/10.3390/cells10123330
Chicago/Turabian StyleYu, Justin D., and Shigeki Miyamoto. 2021. "Molecular Signaling to Preserve Mitochondrial Integrity against Ischemic Stress in the Heart: Rescue or Remove Mitochondria in Danger" Cells 10, no. 12: 3330. https://doi.org/10.3390/cells10123330
APA StyleYu, J. D., & Miyamoto, S. (2021). Molecular Signaling to Preserve Mitochondrial Integrity against Ischemic Stress in the Heart: Rescue or Remove Mitochondria in Danger. Cells, 10(12), 3330. https://doi.org/10.3390/cells10123330