
COVID-19 and Rheumatoid Arthritis Crosstalk: Emerging Association, Therapeutic Options and Challenges

 ,
,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. COVID-19 and RA Association
2.1. Can SARS-CoV-2 Infection Trigger RA Development?
2.2. Can RA Increase the Risk of Acquiring COVID-19 Infection?
2.3. Can RA Worsen COVID-19 Outcomes?
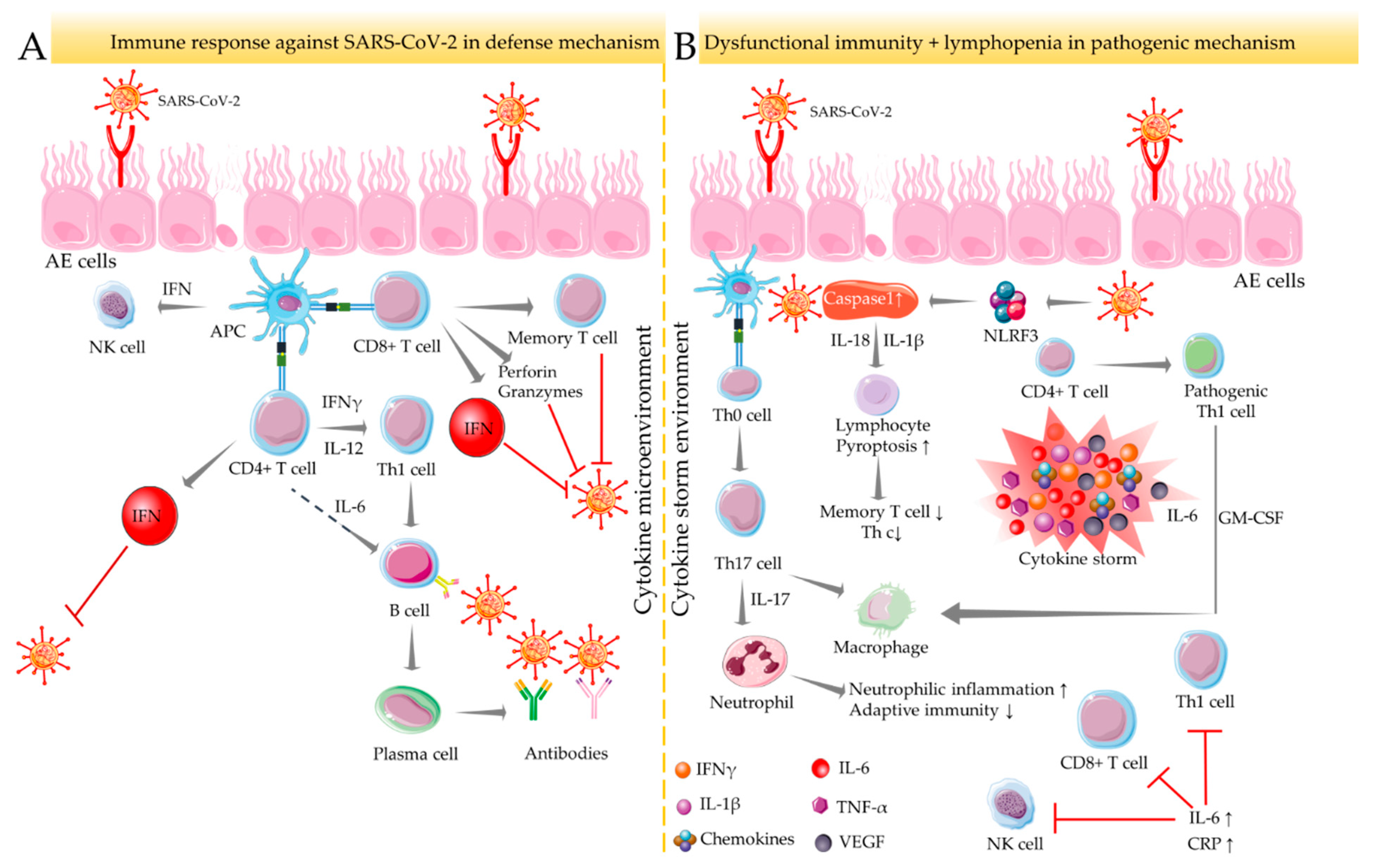
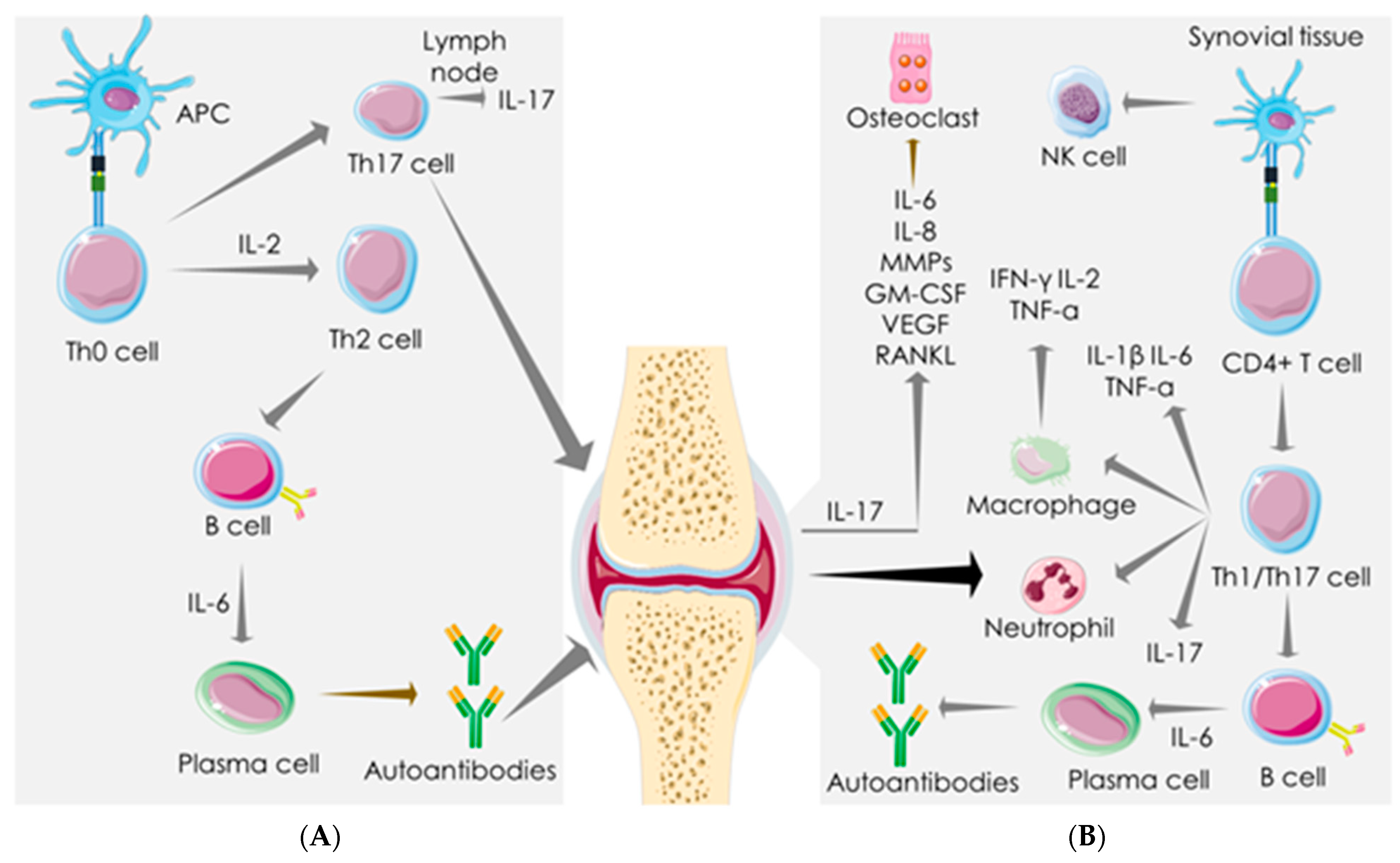
3. Immune-Inflammatory Activities in SARS-COV-2 Infection and RA
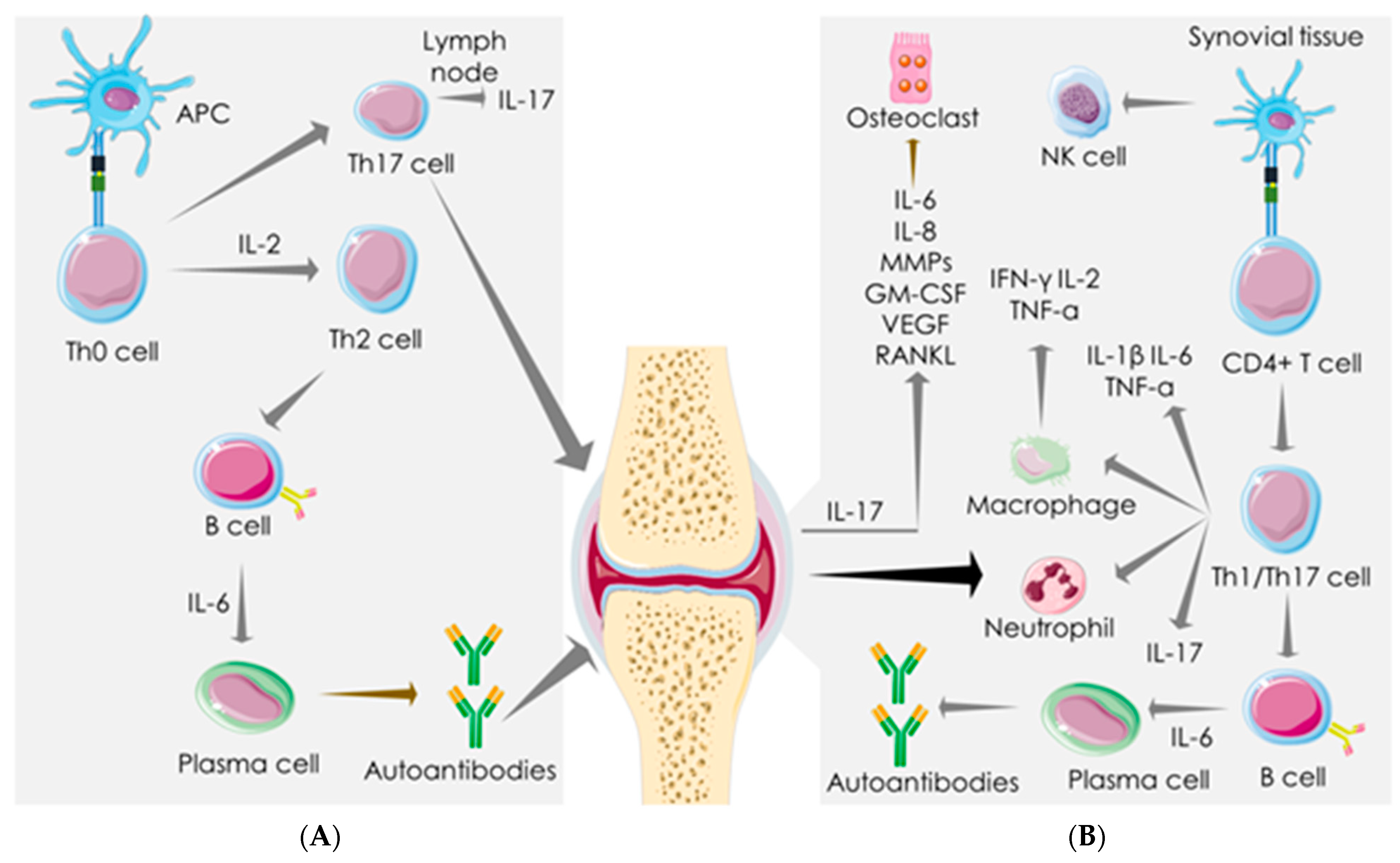
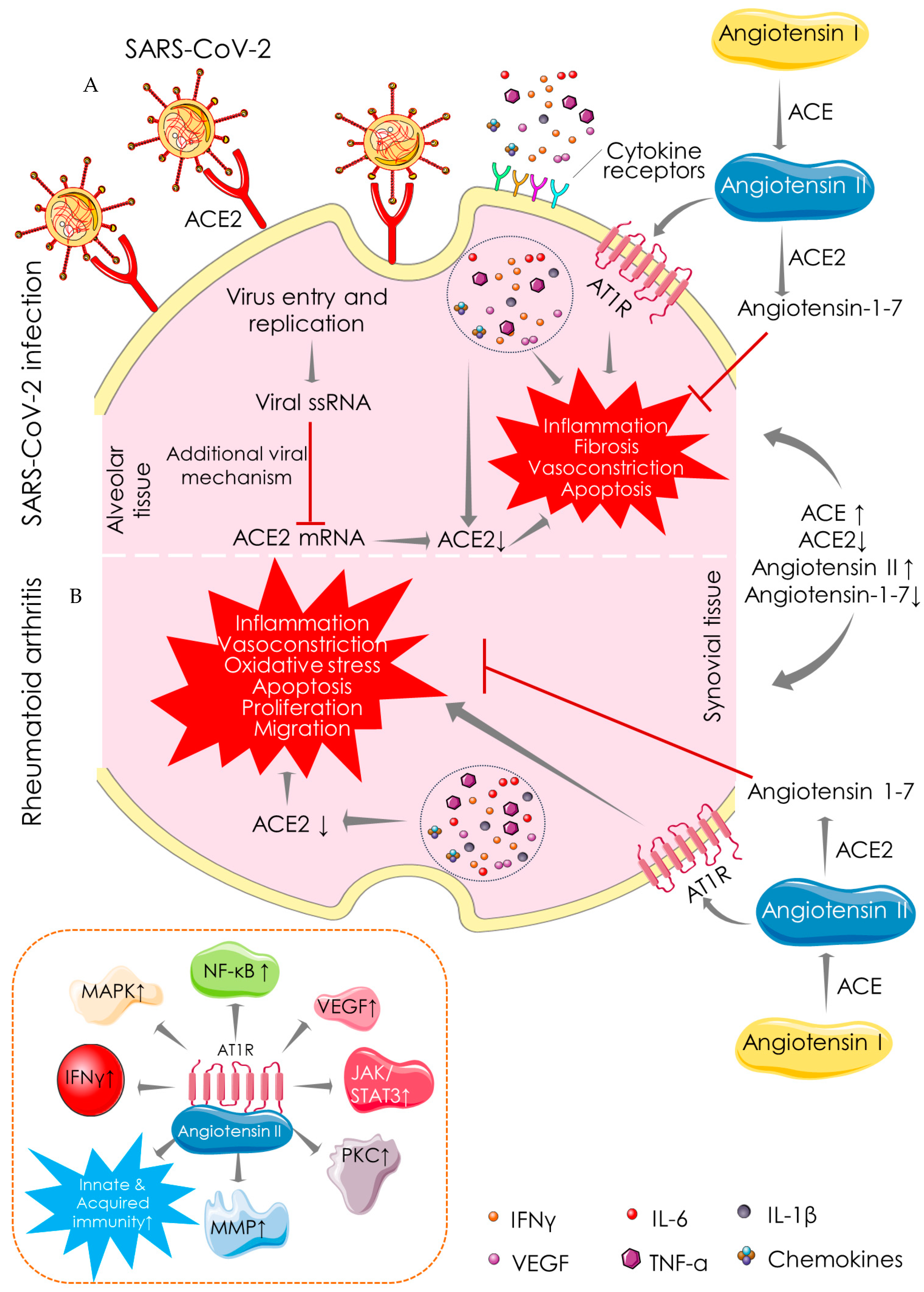
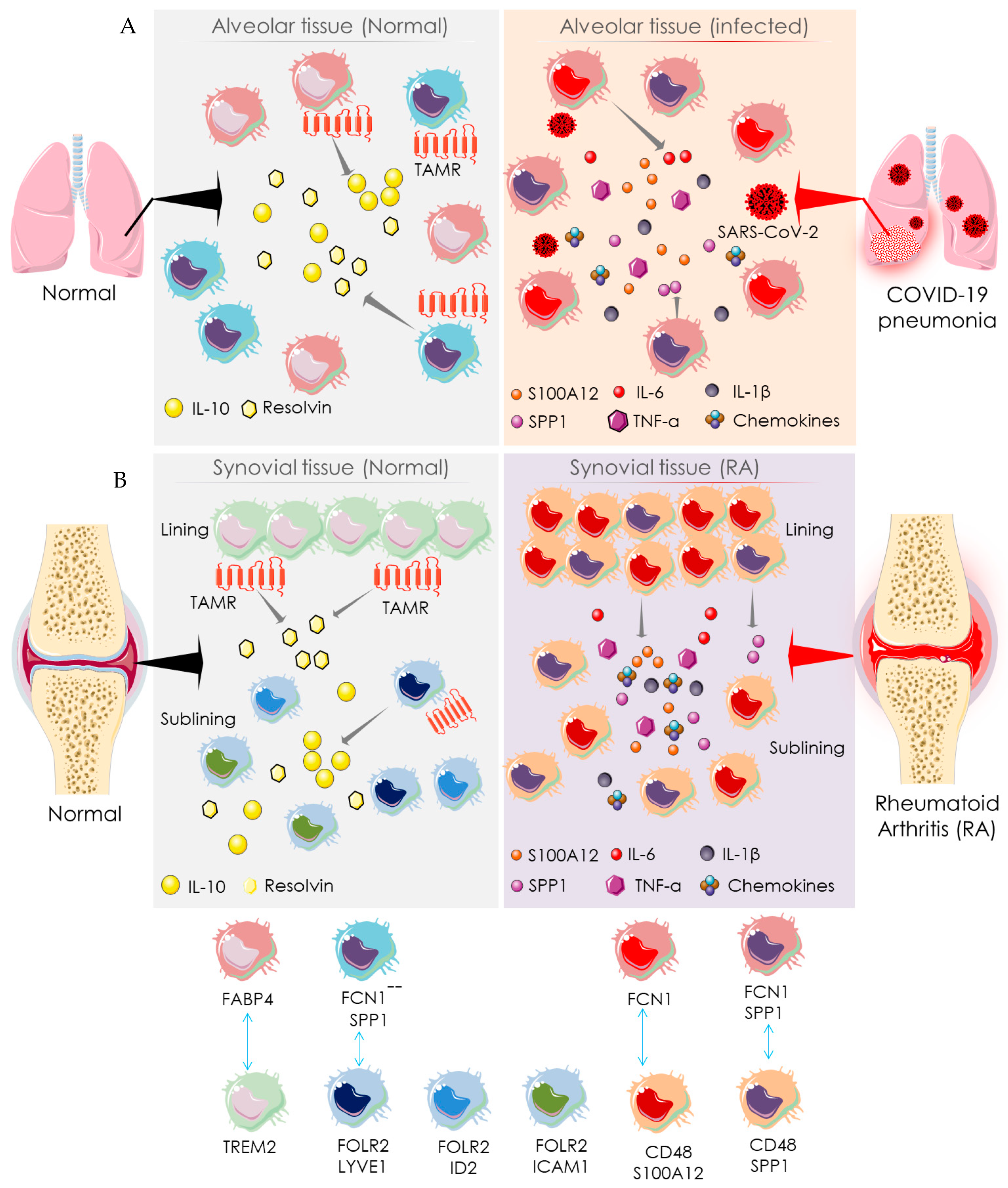
4. Mechanistic Similarity between SARS-COV-2 Infection and RA
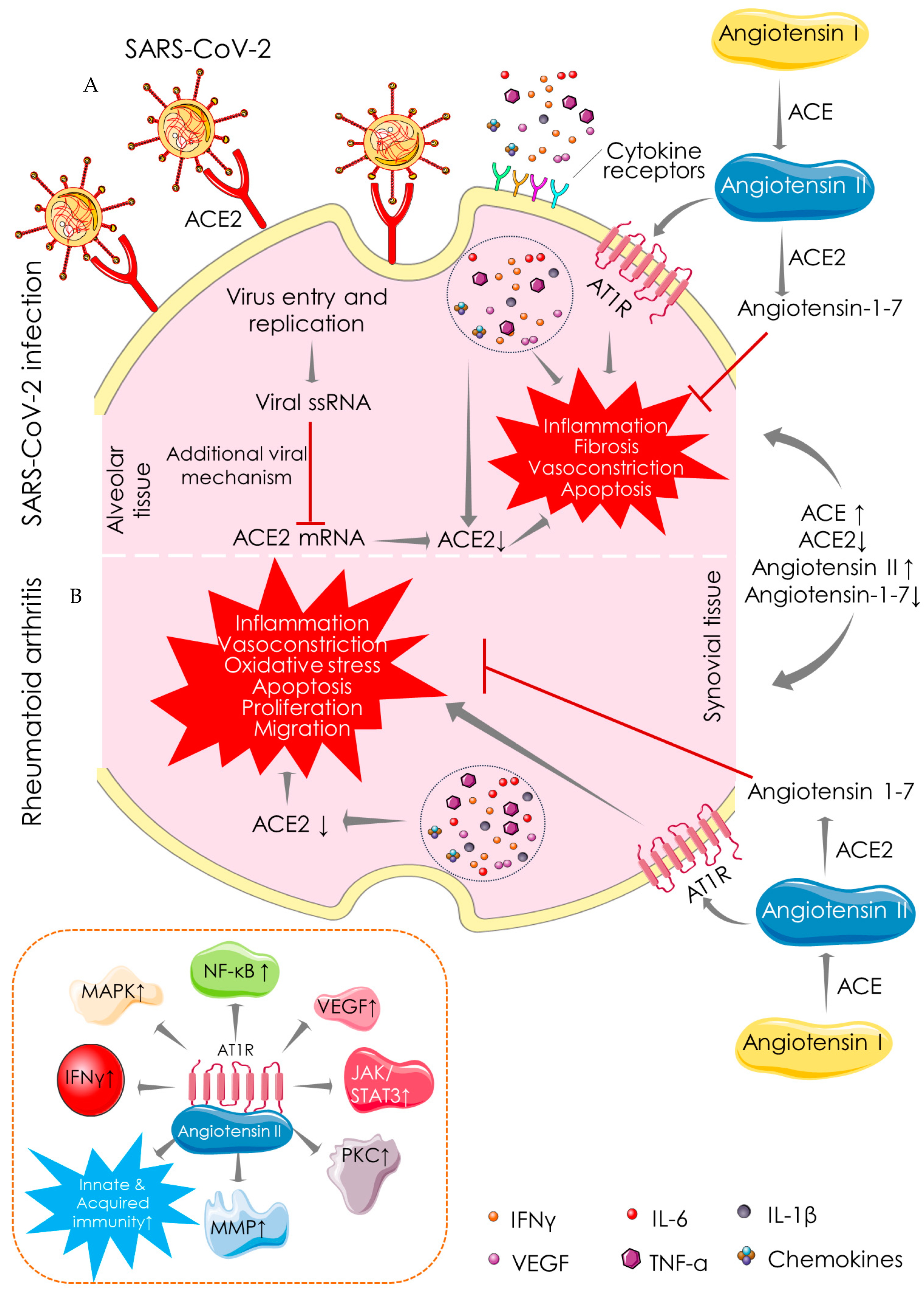
4.1. Angiotensin-Converting Enzyme (ACE)-Dependent Pathway
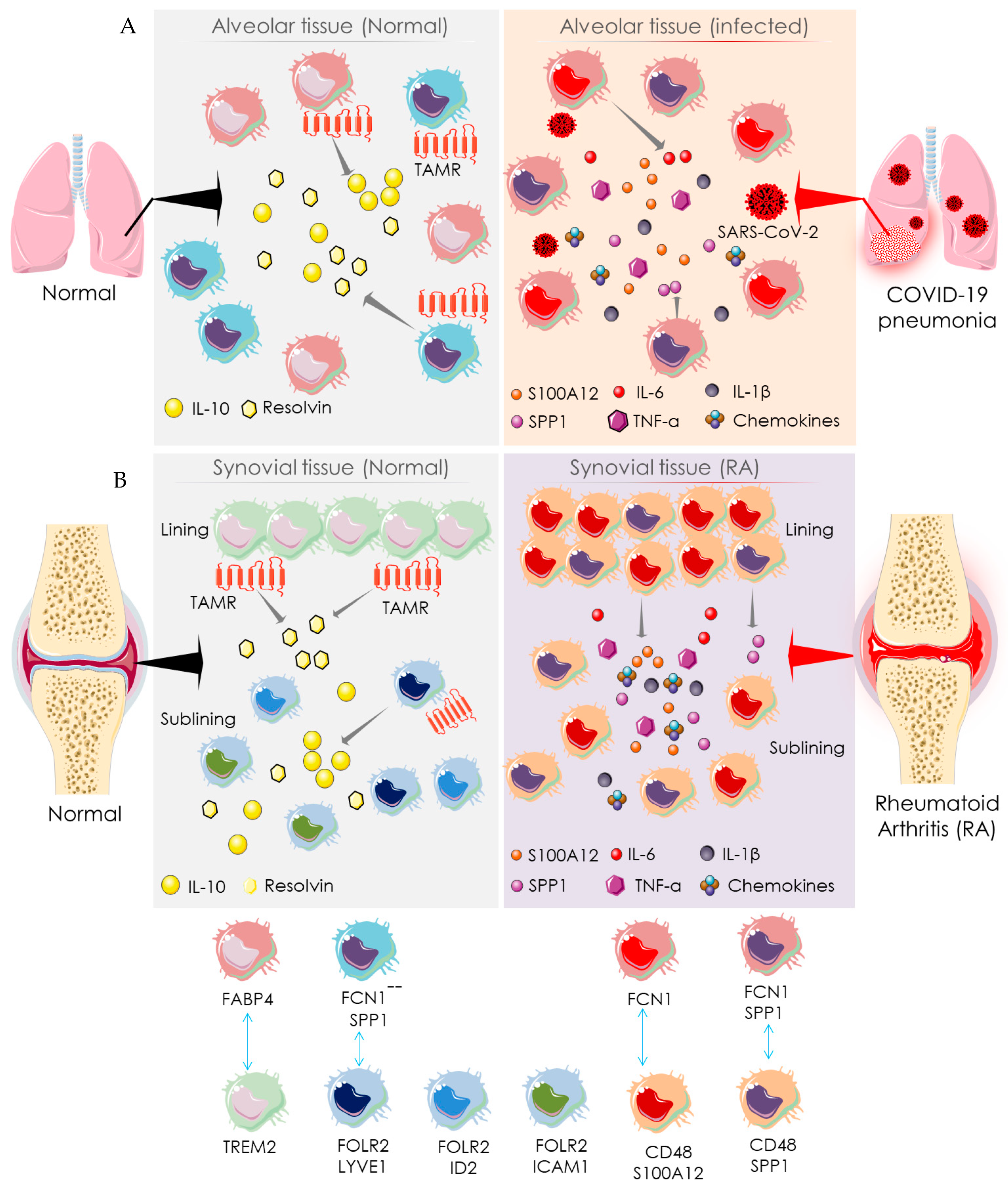
4.2. Macrophage-Mediated Pathway
5. Therapeutic Management and Challenges
5.1. Recommendation for Anti-Rheumatic Drugs in the COVID-19 Setting
5.2. Challenges with Anti-Rheumatic Agents in the COVID-19 Setting

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class of Drugs | Drugs | Indications | Risk Manifestations | Recommendations for RA Patients Acquiring COVID-19 Infection | References |
|---|---|---|---|---|---|
| Glucocorticoids | Dexamethasone, hydrocortisone, methylprednisolone. | Immunosuppressive agents and reduction in inflammation (during late phases of infection), mortality, and length of hospitalization. | Increase the risk of acquiring infection; a moderate-to-high dose can yield poor outcomes. | Continue at the lowest possible dose; however, sudden withdrawal is not recommended. | [23,25,59,69,77,78,79] |
| NSAIDs | Naproxen, celecoxib, etoricoxib, ibuprofen, ketoprofen, aspirin, acetaminophen. | Suppress inflammation and reduce fever. | Impair humoral immune response, increase the risk of bacterial infection, and increase severity and mortality (non-selective COX inhibitors). | Continue unless the patient with severe systemic manifestations. | [62,65,66] |
| csDMARDs Antimalarials | Hydroxychloroquine, chloroquine. | Not clearly understood, believed to exhibit antiviral effect via preventing viral entry, transport, and post-invasion events. Hydroxychloroquine is more potent and less toxic. | Dangerous when overdosed, cardiovascular side effect (QT prolongation). Maculopathy, retinal alteration, G6PD deficiency, and hypersensitivity are other contraindications. Special attention is required for injections. | Temporary suspension for RA patients with suspected/confirmed SARS-CoV-2 infection, patients with chronic heart failure, and/or patients receiving QT prolonging agents, such as azithromycin. | [25,67,69,80,81,82,83,84] |
| Other csDMARDs | Methotrexate, leflunomide, sulfasalazine. | Immunosuppressive agents; suppress inflammation. | Increase the risk of poor outcomes. Combination therapy yields poorer outputs then monotherapy. | Suspension for RA patients with suspected/confirmed SARS-CoV-2 infection. | [25,59,69] |
| bDMARDs Anti-TNF drugs | Adalimumab, infliximab, certolizumab pegol, etanercept, golimumab, secukinumab. | Suppress inflammation and reduce GM-CSF, VEGF, CRP, and blood coagulation. | Increase the risk of acquiring infection, hypersensitivity, and few cases of poor outcomes. | Suspension for RA patients with suspected/confirmed SARS-CoV-2 infection. | [25,29,69,71,84,85] |
| Anti-IL-1 drugs | Anakinra, canakinumab, rilonacept. | Suppress inflammation, prevent overpowering of innate immunity, improve oxygen saturation, reduce neutrophil counts, and inhibit Th17 cell induction. | Increase the risk of acquiring infection and hypersensitivity. | Suspension for RA patients with suspected/confirmed SARS-CoV-2 infection. | [25,69,84,86,87,88] |
| Anti-IL-6 drugs | Tocilizumab, sarilumab. | Suppress inflammation, prevent immune damage to target cells, improve oxygen saturation and reduce CRP, neutrophil counts, and fever. | Increase the risk of acquiring infection. Hypersensitivity, thrombocytopenia, leukopenia, aminotransferase elevation, and gastrointestinal perforations (rare) are other contraindications. | Initiation or continuation is recommended even in COVID-19-positive cases. | [68,84,89,90,91,92] |
| Anti-IL-17 drugs | Brodalumab, ixekizumab (LY2439821), secukinumab (AIN457). | Suppress inflammation; inhibit the production of IL-1, IL-8, and IL-6; exhibit immune-modulatory effect; and reduce neutrophil recruitment. | Increase the risk of acquiring infection. | Suspension for RA patients with suspected/confirmed SARS-CoV-2 infection. | [25,69,85,93] |
| Anti-IL-23 drugs | Guselkumab, risankizumab, tildrakizumab, ustekinumab. | Suppress inflammation, inhibit IL12/IL-23p40 or IL-23p19, and inhibit Th17 cell induction. | Increase the risk of acquiring infection and hypersensitivity. | Suspension for RA patients with suspected/confirmed SARS-CoV-2 infection. | [25,69,85,88] |
| tsDMARDs JAK inhibitors | Baricitinib, ruxolitinib, tofacitinib, upadacitinib. | Decrease virus infectivity, inhibit type-I/II cytokine receptors, reduce inflammation, and decrease neutrophil counts. | Impair IFN-mediated anti-viral response, increase the risk of secondary infection, venous thromboembolism, and hypersensitivity. | Suspension for RA patients with suspected/confirmed SARS-CoV-2 infection | [25,69,74,75,84,94] |
5.3. Monitoring RA Patients in the COVID-19 Setting
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| ACE2 | Angiotensin-converting enzyme 2 |
| ACPA | Anti-citrullinated peptide antibody |
| APC | Antigen presenting cell |
| bDMARDs | Biological disease-modifying antirheumatic drugs, |
| BMD | Bone marrow density |
| COVID-19 | Coronavirus disease 2019 |
| CRP | C reactive protein |
| csDMARDs | Conventional synthetic disease-modifying antirheumatic drugs |
| FABP | Fatty-acid-binding protein |
| FCN | Ficolin |
| FOLR | Folate receptor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| IFN | Interferon |
| IL | Interleukin |
| JAK | Janus tyrosine kinase |
| LYVE | Lymphaticvessel hyaluronan receptor |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSAID | Non-steroidal anti-inflammatory drug |
| RA | Rheumatoid arthritis |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| Th cells | T helper cells |
| TNF | Tumor necrosis factor |
| tsDMARDs | Targeted synthetic disease-modifying anti-rheumatic drugs |
| VEGF | Vascular endothelial growth factor |
References
- Kalra, R.S.; Tomar, D.; Meena, A.S.; Kandimalla, R. SARS-CoV-2, ACE2, and Hydroxychloroquine: Cardiovascular Complications, Therapeutics, and Clinical Readouts in the Current Settings. Pathogens 2020, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Manger, B.; Simon, D.; Caporali, R. COVID-19 revisiting inflammatory pathways of arthritis. Nat. Rev. Rheumatol. 2020, 16, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Kalra, R.S.; Dhanjal, J.K.; Meena, A.S.; Kalel, V.C.; Dahiya, S.; Singh, B.; Dewanjee, S.; Kandimalla, R. COVID-19, Neuropathology, and Aging: SARS-CoV-2 Neurological Infection, Mechanism, and Associated Complications. Front. Aging Neurosci. 2021, 13, 662786. [Google Scholar] [CrossRef]
- Mukarram, M.S.; Ishaq Ghauri, M.; Sethar, S.; Afsar, N.; Riaz, A.; Ishaq, K. COVID-19: An Emerging Culprit of Inflammatory Arthritis. Case Rep. Rheumatol. 2021, 2021, 6610340. [Google Scholar] [CrossRef]
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; Puvvada, N.; Kandimalla, R.; Reddy, P.H. Emerging COVID-19 Neurological Manifestations: Present Outlook and Potential Neurological Challenges in COVID-19 Pandemic. Mol. Neurobiol. 2021, 58, 4694–4715. [Google Scholar] [CrossRef]
- Joo, Y.B.; Lim, Y.H.; Kim, K.J.; Park, K.S.; Park, Y.J. Respiratory viral infections and the risk of rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 199. [Google Scholar] [CrossRef] [Green Version]
- Favalli, E.G.; Ingegnoli, F.; De Lucia, O.; Cincinelli, G.; Cimaz, R.; Caporali, R. COVID-19 infection and rheumatoid arthritis: Faraway, so close! Autoimmun. Rev. 2020, 19, 102523. [Google Scholar] [CrossRef]
- D’Silva, K.M.; Wallace, Z.S. COVID-19 and rheumatoid arthritis. Curr. Opin. Rheumatol. 2021, 33, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Disser, N.P.; De Micheli, A.J.; Schonk, M.M.; Konnaris, M.A.; Piacentini, A.N.; Edon, D.L.; Toresdahl, B.G.; Rodeo, S.A.; Casey, E.K.; Mendias, C.L. Musculoskeletal Consequences of COVID-19. J. Bone Jt. Surg. Am. 2020, 102, 1197–1204. [Google Scholar] [CrossRef]
- Ramani, S.L.; Samet, J.; Franz, C.K.; Hsieh, C.; Nguyen, C.V.; Horbinski, C.; Deshmukh, S. Musculoskeletal involvement of COVID-19: Review of imaging. Skelet. Radiol. 2021, 50, 1763–1773. [Google Scholar] [CrossRef]
- Huang, Y.; Tu, M.; Wang, S.; Chen, S.; Zhou, W.; Chen, D.; Zhou, L.; Wang, M.; Zhao, Y.; Zeng, W.; et al. Clinical characteristics of laboratory confirmed positive cases of SARS-CoV-2 infection in Wuhan, China: A retrospective single center analysis. Travel Med. Infect. Dis. 2020, 36, 101606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, S. Corticosteroid-Induced Osteonecrosis in COVID-19: A Call for Caution. J. Bone Miner. Res. 2020, 35, 1828–1829. [Google Scholar] [CrossRef] [PubMed]
- Steinz, M.M.; Santos-Alves, E.; Lanner, J.T. Skeletal muscle redox signaling in rheumatoid arthritis. Clin. Sci. 2020, 134, 2835–2850. [Google Scholar] [CrossRef] [PubMed]
- Vaishya, R.; Jain, V.K.; Iyengar, K.P. Musculoskeletal manifestations of COVID-19. J. Clin. Orthop. Trauma 2021, 17, 280–281. [Google Scholar] [CrossRef]
- Carfì, A.; Bernabei, R.; Landi, F.; Gemelli against COVID-19 Post-Acute Care Study Group. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.T.; Hsu, B.C.; Chen, D.Y. Autoimmune and Rheumatic Manifestations Associated With COVID-19 in Adults: An Updated Systematic Review. Front. Immunol. 2021, 12, 645013. [Google Scholar] [CrossRef]
- Parisi, S.; Borrelli, R.; Bianchi, S.; Fusaro, E. Viral arthritis and COVID-19. Lancet Rheumatol. 2020, 2, e655–e657. [Google Scholar] [CrossRef]
- Derksen, V.F.A.M.; Kissel, T.; Lamers-Karnebeek, F.B.G.; van der Bijl, A.E.; Venhuizen, A.C.; Huizinga, T.W.J.; Toes, R.E.M.; Roukens, A.H.E.; van der Woude, D. Onset of rheumatoid arthritis after COVID-19: Coincidence or connected? Ann. Rheum. Dis. 2021, 80, 1096–1098. [Google Scholar] [CrossRef]
- Perrot, L.; Hemon, M.; Busnel, J.M.; Muis-Pistor, O.; Picard, C.; Zandotti, C.; Pham, T.; Roudier, J.; Desplat-Jego, S.; Balandraud, N. First flare of ACPA-positive rheumatoid arthritis after SARS-CoV-2 infection. Lancet Rheumatol. 2021, 3, e6–e8. [Google Scholar] [CrossRef]
- Roongta, R.; Chattopadhyay, A.; Ghosh, A. Correspondence on Onset of rheumatoid arthritis after COVID-19: Coincidence or connected? Ann. Rheum. Dis. 2021. [Google Scholar] [CrossRef]
- Saadoun, D.; Vieira, M.; Vautier, M.; Baraliakos, X.; Andreica, I.; da Silva, J.A.P.; Sousa, M.; Luis, M.; Khmelinskii, N.; Gracía, J.M.A.; et al. SARS-CoV-2 outbreak in immune-mediated inflammatory diseases: The Euro-COVIMID multicentre cross-sectional study. Lancet Rheumatol. 2021, 3, e481–e488. [Google Scholar] [CrossRef]
- Akiyama, S.; Hamdeh, S.; Micic, D.; Sakuraba, A. Prevalence and clinical outcomes of COVID-19 in patients with autoimmune diseases: A systematic review and meta-analysis. Ann. Rheum. Dis. 2020, 80, 384–391. [Google Scholar] [CrossRef]
- Hyrich, K.L.; Machado, P.M. Rheumatic disease and COVID-19: Epidemiology and outcomes. Nat. Rev. Rheumatol. 2021, 17, 71–72. [Google Scholar] [CrossRef] [PubMed]
- The COVID-19 Global Rheumatology Alliance Global Registry. Available online: https://rheum-covid.org/updates/combined-data.html (accessed on 18 May 2021).
- Favalli, E.G.; Maioli, G.; Biggioggero, M.; Caporali, R. Clinical management of patients with rheumatoid arthritis during the COVID-19 pandemic. Expert Rev. Clin. Immunol. 2021, 17, 561–571. [Google Scholar] [CrossRef]
- Simon, D.; Tascilar, K.; Krönke, G.; Kleyer, A.; Zaiss, M.M.; Hepp, F.; Meder, C.; Atreya, R.; Klenske, E.; Dietrich, P.; et al. Patients with immune-mediated inflammatory diseases receiving cytokine inhibitors have low prevalence of SARS-CoV-2 seroconversion. Nat. Commun. 2020, 11, 3774. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Topless, R.K.; Phipps-Green, A.; Leask, M.; Dalbeth, N.; Stamp, L.K.; Robinson, P.C.; Merriman, T.R. Gout, Rheumatoid Arthritis, and the Risk of Death Related to Coronavirus Disease 2019: An Analysis of the UK Biobank. ACR Open Rheumatol. 2021, 3, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Roongta, R.; Ghosh, A. Managing rheumatoid arthritis during COVID-19. Clin. Rheumatol. 2020, 39, 3237–3244. [Google Scholar] [CrossRef]
- Haberman, R.H.; Castillo, R.; Chen, A.; Yan, D.; Ramirez, D.; Sekar, V.; Lesser, R.; Solomon, G.; Neimann, A.L.; Blank, R.B.; et al. COVID-19 in Patients With Inflammatory Arthritis: A Prospective Study on the Effects of Comorbidities and Disease-Modifying Antirheumatic Drugs on Clinical Outcomes. Arthritis Rheumatol. 2020, 72, 1981–1989. [Google Scholar] [CrossRef]
- Pelaia, C.; Calabrese, C.; Garofalo, E.; Bruni, A.; Vatrella, A.; Pelaia, G. Therapeutic Role of Tocilizumab in SARS-CoV-2-Induced Cytokine Storm: Rationale and Current Evidence. Int. J. Mol. Sci. 2021, 22, 3059. [Google Scholar] [CrossRef] [PubMed]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020, 34, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Orlov, M.; Wander, P.L.; Morrell, E.D.; Mikacenic, C.; Wurfel, M.M. A Case for Targeting Th17 Cells and IL-17A in SARS-CoV-2 Infections. J. Immunol. 2020, 205, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Pum, A.; Ennemoser, M.; Adage, T.; Kungl, A.J. Cytokines and Chemokines in SARS-CoV-2 Infections-Therapeutic Strategies Targeting Cytokine Storm. Biomolecules 2021, 11, 91. [Google Scholar] [CrossRef]
- Yap, H.Y.; Tee, S.Z.; Wong, M.M.; Chow, S.K.; Peh, S.C.; Teow, S.Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [Green Version]
- Araki, Y.; Mimura, T. The Mechanisms Underlying Chronic Inflammation in Rheumatoid Arthritis from the Perspective of the Epigenetic Landscape. J. Immunol. Res. 2016, 2016, 6290682. [Google Scholar] [CrossRef] [Green Version]
- Meednu, N.; Zhang, H.; Owen, T.; Sun, W.; Wang, V.; Cistrone, C.; Rangel-Moreno, J.; Xing, L.; Anolik, J.H. Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 805–816. [Google Scholar] [CrossRef]
- Elemam, N.M.; Hannawi, S.; Maghazachi, A.A. Role of Chemokines and Chemokine Receptors in Rheumatoid Arthritis. Immunotargets Ther. 2020, 9, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Cai, T.; Fan, L.; Lou, K.; Hua, X.; Huang, Z.; Gao, G. The potential role of serum angiotensin-converting enzyme in coronavirus disease 2019. BMC Infect. Dis. 2020, 20, 883. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057. [Google Scholar] [CrossRef]
- Choudhary, S.; Sharma, K.; Silakari, O. The interplay between inflammatory pathways and COVID-19: A critical review on pathogenesis and therapeutic options. Microb. Pathog. 2021, 150, 104673. [Google Scholar] [CrossRef]
- Kalra, R.S.; Kandimalla, R. Engaging the spikes: Heparan sulfate facilitates SARS-CoV-2 spike protein binding to ACE2 and potentiates viral infection. Signal. Transduct. Target. Ther. 2021, 6, 39. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Ronconi, G. Coronavirus-19 (SARS-CoV-2) induces acute severe lung inflammation via IL-1 causing cytokine storm in COVID-19: A promising inhibitory strategy. J. Biol. Regul. Homeost. Agents 2020, 34, 1971–1975. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Puertas, R. ACE2 activators for the treatment of COVID 19 patients. J. Med. Virol. 2020, 92, 1701–1702. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.C.S.; Wong, S.; Huang, J.; Yan, B. Relating angiotensin-converting enzyme inhibitors or angiotensin receptor blockers with incidence or mortality of COVID-19. ESC Heart Fail. 2020, 7, 3119–3123. [Google Scholar] [CrossRef]
- Chang, Y.; Wei, W. Angiotensin II in inflammation, immunity and rheumatoid arthritis. Clin. Exp. Immunol. 2015, 179, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Najafi, S.; Rajaei, E.; Moallemian, R.; Nokhostin, F. The potential similarities of COVID-19 and autoimmune disease pathogenesis and therapeutic options: New insights approach. Clin. Rheumatol. 2020, 39, 3223–3235. [Google Scholar] [CrossRef]
- Mirastschijski, U.; Dembinski, R.; Maedler, K. Lung Surfactant for Pulmonary Barrier Restoration in Patients with COVID-19 Pneumonia. Front. Med. 2020, 7, 254. [Google Scholar] [CrossRef]
- MacDonald, L.; Alivernini, S.; Tolusso, B.; Elmesmari, A.; Somma, D.; Perniola, S.; Paglionico, A.; Petricca, L.; Bosello, S.L.; Carfì, A.; et al. COVID-19 and RA share an SPP1 myeloid pathway that drives PD-L1+ neutrophils and CD14+ monocytes. JCI Insight 2021, 6, e147413. [Google Scholar] [CrossRef]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 2020, 26, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Dougados, M.; Soubrier, M.; Antunez, A.; Balint, P.; Balsa, A.; Buch, M.H.; Casado, G.; Detert, J.; El-Zorkany, B.; Emery, P.; et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: Results of an international, cross-sectional study (COMORA). Ann. Rheum. Dis. 2014, 73, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Mikuls, T.R.; Johnson, S.R.; Fraenkel, L.; Arasaratnam, R.J.; Baden, L.R.; Bermas, B.L.; Chatham, W.; Cohen, S.; Costenbader, K.; Gravallese, E.M.; et al. American College of Rheumatology Guidance for the Management of Rheumatic Disease in Adult Patients During the COVID-19 Pandemic: Version 1. Arthritis Rheumatol. 2020, 72, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Landewé, R.B.; Machado, P.M.; Kroon, F.; Bijlsma, H.W.; Burmester, G.R.; Carmona, L.; Combe, B.; Galli, M.; Gossec, L.; Iagnocco, A.; et al. EULAR provisional recommendations for the management of rheumatic and musculoskeletal diseases in the context of SARS-CoV-2. Ann. Rheum. Dis. 2020, 79, 851–858. [Google Scholar] [CrossRef]
- Russell, C.D.; Millar, J.E.; Baillie, J.K. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 2020, 395, 473–475. [Google Scholar] [CrossRef] [Green Version]
- Strangfeld, A.; Schäfer, M.; Gianfrancesco, M.A.; Lawson-Tovey, S.; Liew, J.W.; Ljung, L.; Mateus, E.F.; Richez, C.; Santos, M.J.; Schmajuk, G.; et al. Factors associated with COVID-19-related death in people with rheumatic diseases: Results from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann. Rheum. Dis. 2021, 80, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Hyrich, K.L.; Gianfrancesco, M.; Machado, P.M.; Yazdany, J.; Robinson, P.C. Rheumatic disease activity, glucocorticoid use and COVID-19. Response to: ’Comment on ’Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease: Data from the COVID-19 Global Rheumatology Alliance physician-reported registry’ by Gianfrancesco et al. Disease activity, rather than glucocorticoid therapy, may be associated with COVID-19 severity in patients with rheumatic musculoskeletal diseases’ by Giollo et al. Ann. Rheum. Dis. 2020, 79, 859–866. [Google Scholar] [CrossRef]
- Abu Esba, L.C.; Alqahtani, R.A.; Thomas, A.; Shamas, N.; Alswaidan, L.; Mardawi, G. Ibuprofen and NSAID Use in COVID-19 Infected Patients Is Not Associated with Worse Outcomes: A Prospective Cohort Study. Infect. Dis. Ther. 2021, 10, 253–268. [Google Scholar] [CrossRef]
- Chen, J.S.; Alfajaro, M.M.; Chow, R.D.; Wei, J.; Filler, R.B.; Eisenbarth, S.C.; Wilen, C.B. Non-steroidal anti-inflammatory drugs dampen the cytokine and antibody response to SARS-CoV-2 infection. J. Virol. 2021, 95, e00014–e00021. [Google Scholar] [CrossRef]
- Micallef, J.; Soeiro, T.; Jonville-Béra, A.P.; French Society of Pharmacology, Therapeutics (SFPT). Non-steroidal anti-inflammatory drugs, pharmacology, and COVID-19 infection. Therapie 2020, 75, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.; Pedrosa, M.A.; Garrido-Gil, P.; Labandeira, C.M.; Navarro, G.; Franco, R.; Rodriguez-Perez, A.I.; Labandeira-Garcia, J.L. Interactions between ibuprofen, ACE2, renin-angiotensin system, and spike protein in the lung. Implications for COVID-19. Clin. Transl. Med. 2021, 11, e371. [Google Scholar] [CrossRef] [PubMed]
- Zolk, O.; Hafner, S.; Schmidt, C.Q.; German Society for Experimental and Clinical Pharmacology and Toxicology (DGPT). COVID-19 pandemic and therapy with ibuprofen or renin-angiotensin system blockers: No need for interruptions or changes in ongoing chronic treatments. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 1131–1135. [Google Scholar] [CrossRef]
- Reese, J.T.; Coleman, B.; Chan, L.; Blau, H.; Callahan, T.J.; Cappelletti, L.; Fontana, T.; Bradwell, K.R.; Harris, N.L.; Casiraghi, E.; et al. Cyclooxygenase inhibitor use is associated with increased COVID-19 severity. medRxiv 2021. [Google Scholar] [CrossRef]
- Fantini, J.; Di Scala, C.; Chahinian, H.; Yahi, N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int. J. Antimicrob. Agents 2020, 55, 105960. [Google Scholar] [CrossRef] [PubMed]
- Rentsch, C.T.; DeVito, N.J.; MacKenna, B.; Morton, C.E.; Bhaskaran, K.; Brown, J.P.; Schultze, A.; Hulme, W.J.; Croker, R.; Walker, A.J.; et al. Effect of pre-exposure use of hydroxychloroquine on COVID-19 mortality: A population-based cohort study in patients with rheumatoid arthritis or systemic lupus erythematosus using the OpenSAFELY platform. Lancet Rheumatol. 2021, 3, e19–e27. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Rochwerg, B.; Agarwal, A.; Siemieniuk, R.A.; Agoritsas, T.; Lamontagne, F.; Askie, L.; Lytvyn, L.; Leo, Y.S.; Macdonald, H.; Zeng, L.; et al. A living WHO guideline on drugs for covid-19. BMJ 2020, 370, m3379. [Google Scholar] [CrossRef]
- Robinson, P.C.; Liew, D.F.L.; Liew, J.W.; Monaco, C.; Richards, D.; Shivakumar, S.; Tanner, H.L.; Feldmann, M. The Potential for Repurposing Anti-TNF as a Therapy for the Treatment of COVID-19. Med 2020, 1, 90–102. [Google Scholar] [CrossRef]
- Rosas, I.O.; Bräu, N.; Waters, M.; Go, R.C.; Hunter, B.D.; Bhagani, S.; Skiest, D.; Aziz, M.S.; Cooper, N.; Douglas, I.S.; et al. Tocilizumab in Hospitalized Patients with Severe Covid-19 Pneumonia. N. Engl. J. Med. 2021, 384, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.A.; Paiva, J.G.; Pimentel, F.N.; Guimarães, R.S.; Moreira, M.R. COVID-19 in patients with rheumatological diseases treated with anti-TNF. Ann. Rheum. Dis. 2021, 80, e62. [Google Scholar] [CrossRef] [PubMed]
- Favalli, E.G.; De Lucia, O.; Biggioggero, M.; Del Papa, N.; Caporali, R. Role of antimalarials in COVID-19: Observational data from a cohort of rheumatic patients. Ann. Rheum. Dis. 2021, 80, e75. [Google Scholar] [CrossRef]
- Sepriano, A.; Kerschbaumer, A.; Smolen, J.S.; van der Heijde, D.; Dougados, M.; van Vollenhoven, R.; McInnes, I.B.; Bijlsma, J.W.; Burmester, G.R.; de Wit, M.; et al. Safety of synthetic and biological DMARDs: A systematic literature review informing the 2019 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann. Rheum. Dis. 2020, 79, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Monti, S.; Balduzzi, S.; Delvino, P.; Bellis, E.; Quadrelli, V.S.; Montecucco, C. Clinical course of COVID-19 in a series of patients with chronic arthritis treated with immunosuppressive targeted therapies. Ann. Rheum. Dis. 2020, 79, 667–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO; Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group; Sterne, J.A.C.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar] [CrossRef]
- Ranjbar, K.; Moghadami, M.; Mirahmadizadeh, A.; Fallahi, M.J.; Khaloo, V.; Shahriarirad, R.; Erfani, A.; Khodamoradi, Z.; Gholampoor Saadi, M.H. Methylprednisolone or dexamethasone, which one is superior corticosteroid in the treatment of hospitalized COVID-19 patients: A triple-blinded randomized controlled trial. BMC Infect. Dis. 2021, 21, 337. [Google Scholar] [CrossRef]
- Gianfrancesco, M.; Hyrich, K.L.; Al-Adely, S.; Carmona, L.; Danila, M.I.; Gossec, L.; Izadi, Z.; Jacobsohn, L.; Katz, P.; Lawson-Tovey, S.; et al. Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease: Data from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann. Rheum. Dis. 2020, 79, 859–866. [Google Scholar] [CrossRef]
- Khuroo, M.S. Chloroquine and hydroxychloroquine in coronavirus disease 2019 (COVID-19). Facts, fiction and the hype: A critical appraisal. Int. J. Antimicrob. Agents 2020, 56, 106101. [Google Scholar] [CrossRef]
- Jankelson, L.; Karam, G.; Becker, M.L.; Chinitz, L.A.; Tsai, M.C. QT prolongation, torsades de pointes, and sudden death with short courses of chloroquine or hydroxychloroquine as used in COVID-19: A systematic review. Heart Rhythm. 2020, 17, 1472–1479. [Google Scholar] [CrossRef]
- White, N.J.; Watson, J.A.; Hoglund, R.M.; Chan, X.H.S.; Cheah, P.Y.; Tarning, J. COVID-19 prevention and treatment: A critical analysis of chloroquine and hydroxychloroquine clinical pharmacology. PLoS Med. 2020, 17, e1003252. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atzeni, F.; Masala, I.F.; Rodríguez-Carrio, J.; Ríos-Garcés, R.; Gerratana, E.; La Corte, L.; Giallanza, M.; Nucera, V.; Riva, A.; Espinosa, G.; et al. The Rheumatology Drugs for COVID-19 Management: Which and When? J. Clin. Med. 2021, 10, 783. [Google Scholar] [CrossRef]
- Mahil, S.K.; Dand, N.; Mason, K.J.; Yiu, Z.Z.N.; Tsakok, T.; Meynell, F.; Coker, B.; McAteer, H.; Moorhead, L.; Mackenzie, T.; et al. Factors associated with adverse COVID-19 outcomes in patients with psoriasis-insights from a global registry-based study. J. Allergy Clin. Immunol. 2021, 147, 60–71. [Google Scholar] [CrossRef]
- Landi, L.; Ravaglia, C.; Russo, E.; Cataleta, P.; Fusari, M.; Boschi, A.; Giannarelli, D.; Facondini, F.; Valentini, I.; Panzini, I.; et al. Blockage of interleukin-1β with canakinumab in patients with Covid-19. Sci. Rep. 2020, 10, 21775. [Google Scholar] [CrossRef]
- Geng, J.; Wang, F.; Huang, Z.; Chen, X.; Wang, Y. Perspectives on anti-IL-1 inhibitors as potential therapeutic interventions for severe COVID-19. Cytokine 2021, 143, 155544. [Google Scholar] [CrossRef] [PubMed]
- Casillo, G.M.; Mansour, A.A.; Raucci, F.; Saviano, A.; Mascolo, N.; Iqbal, A.J.; Maione, F. Could IL-17 represent a new therapeutic target for the treatment and/or management of COVID-19-related respiratory syndrome? Pharmacol. Res. 2020, 156, 104791. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef]
- Calabrese, C.; Rajendram, P.; Sacha, G.L.; Calabrese, L. Practical aspects of targeting IL-6 in COVID-19 disease. Cleve. Clin. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Castelnovo, L.; Tamburello, A.; Lurati, A.; Zaccara, E.; Marrazza, M.G.; Olivetti, M.; Mumoli, N.; Mastroiacovo, D.; Colombo, D.; Ricchiuti, E.; et al. Anti-IL6 treatment of serious COVID-19 disease: A monocentric retrospective experience. Medicine 2021, 100, e23582. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, K.M.; Wallace, Z.S. COVID-19 and Disease-Modifying Anti-rheumatic Drugs. Curr. Rheumatol. Rep. 2021, 23, 28. [Google Scholar] [CrossRef]
- Pacha, O.; Sallman, M.A.; Evans, S.E. COVID-19: A case for inhibiting IL-17? Nat. Rev. Immunol. 2020, 20, 345–346. [Google Scholar] [CrossRef]
- Seif, F.; Aazami, H.; Khoshmirsafa, M.; Kamali, M.; Mohsenzadegan, M.; Pornour, M.; Mansouri, D. JAK Inhibition as a New Treatment Strategy for Patients with COVID-19. Int. Arch. Allergy Immunol. 2020, 181, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, G.E.; Maguire, S.; Haroon, N. COVID-19: What Do Rheumatologists Need to Know? Curr. Rheumatol. Rep. 2021, 23, 5. [Google Scholar] [CrossRef]
- Gupta, L.; Misra, D.P.; Agarwal, V.; Balan, S.; Agarwal, V. Management of rheumatic diseases in the time of covid-19 pandemic: Perspectives of rheumatology practitioners from India. Ann. Rheum. Dis. 2021, 80, e1. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.; Tasso, M.; Scotti, N.; Mostacciuolo, E.; Girolimetto, N.; Foglia, F.; Del Puente, A.; Scarpa, R.; Caso, F. Telerheumatology in COVID-19 era: A study from a psoriatic arthritis cohort. Ann. Rheum. Dis. 2020, 80, e46. [Google Scholar] [CrossRef] [PubMed]
- Monaghesh, E.; Hajizadeh, A. The role of telehealth during COVID-19 outbreak: A systematic review based on current evidence. BMC Public Health 2020, 20, 1193. [Google Scholar] [CrossRef]
- Mistry, J.; Sharif, M.; Prideaux, A.; Smith, C.; Sumbwanyambe, M.; Sibley, M.; Carpenter, L.; Sweeney, M.; Kiely, P. Use of rheumatoid arthritis impact of disease (RAID) in routine care; identification of DAS28 remission and unmet patient-reported outcomes. Rheumatol. Adv. Pract. 2020, 4, rkaa013. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.R.; de Thurah, A.; Lomborg, K. Experiences with Telehealth Followup in Patients with Rheumatoid Arthritis: A Qualitative Interview Study. Arthritis Care Res. 2018, 70, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, P.; Ahmed, S.; Paul, A.; Skaria, T.G.; Joby, J.; Alias, B. Switching to teleconsultation for rheumatology in the wake of the COVID-19 pandemic: Feasibility and patient response in India. Clin. Rheumatol. 2020, 39, 2757–2762. [Google Scholar] [CrossRef]
- Seo, M.R.; Kim, J.W.; Park, E.J.; Jung, S.M.; Sung, Y.K.; Kim, H.; Kim, G.; Kim, H.S.; Lee, M.S.; Lee, J.; et al. Recommendations for the management of patients with systemic rheumatic diseases during the coronavirus disease pandemic. Korean J. Intern. Med. 2020, 35, 1317–1332. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dewanjee, S.; Kandimalla, R.; Kalra, R.S.; Valupadas, C.; Vallamkondu, J.; Kolli, V.; Dey Ray, S.; Reddy, A.P.; Reddy, P.H. COVID-19 and Rheumatoid Arthritis Crosstalk: Emerging Association, Therapeutic Options and Challenges. Cells 2021, 10, 3291. https://doi.org/10.3390/cells10123291
Dewanjee S, Kandimalla R, Kalra RS, Valupadas C, Vallamkondu J, Kolli V, Dey Ray S, Reddy AP, Reddy PH. COVID-19 and Rheumatoid Arthritis Crosstalk: Emerging Association, Therapeutic Options and Challenges. Cells. 2021; 10(12):3291. https://doi.org/10.3390/cells10123291
Chicago/Turabian StyleDewanjee, Saikat, Ramesh Kandimalla, Rajkumar Singh Kalra, Chandrasekhar Valupadas, Jayalakshmi Vallamkondu, Viswakalyan Kolli, Sarbani Dey Ray, Arubala P. Reddy, and P. Hemachandra Reddy. 2021. "COVID-19 and Rheumatoid Arthritis Crosstalk: Emerging Association, Therapeutic Options and Challenges" Cells 10, no. 12: 3291. https://doi.org/10.3390/cells10123291
APA StyleDewanjee, S., Kandimalla, R., Kalra, R. S., Valupadas, C., Vallamkondu, J., Kolli, V., Dey Ray, S., Reddy, A. P., & Reddy, P. H. (2021). COVID-19 and Rheumatoid Arthritis Crosstalk: Emerging Association, Therapeutic Options and Challenges. Cells, 10(12), 3291. https://doi.org/10.3390/cells10123291