
Induced Pluripotent Stem Cells as a Tool for Modeling Hematologic Disorders and as a Potential Source for Cell-Based Therapies



Abstract
1. Introduction
2. Discovery and Development of hiPSCs
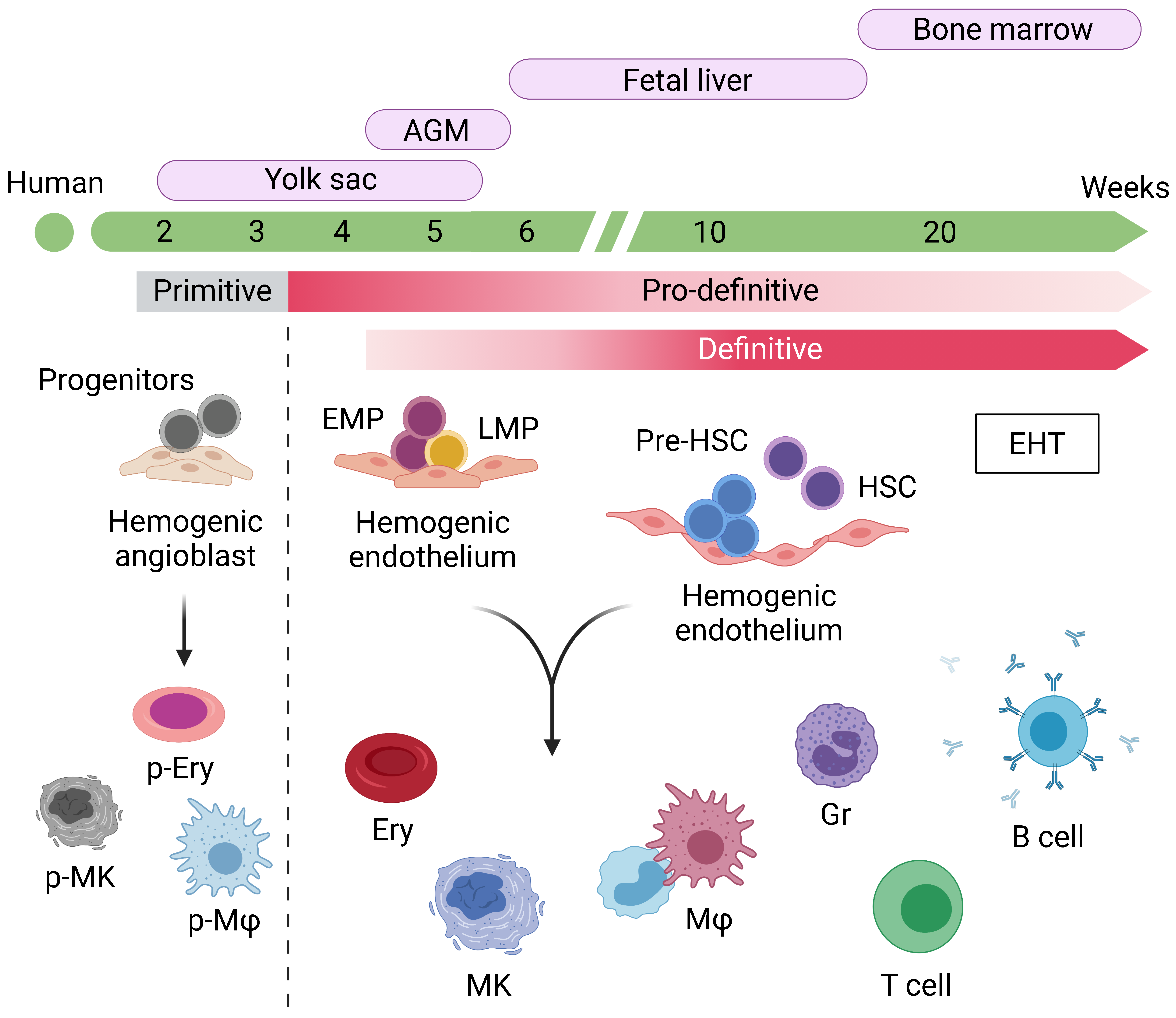
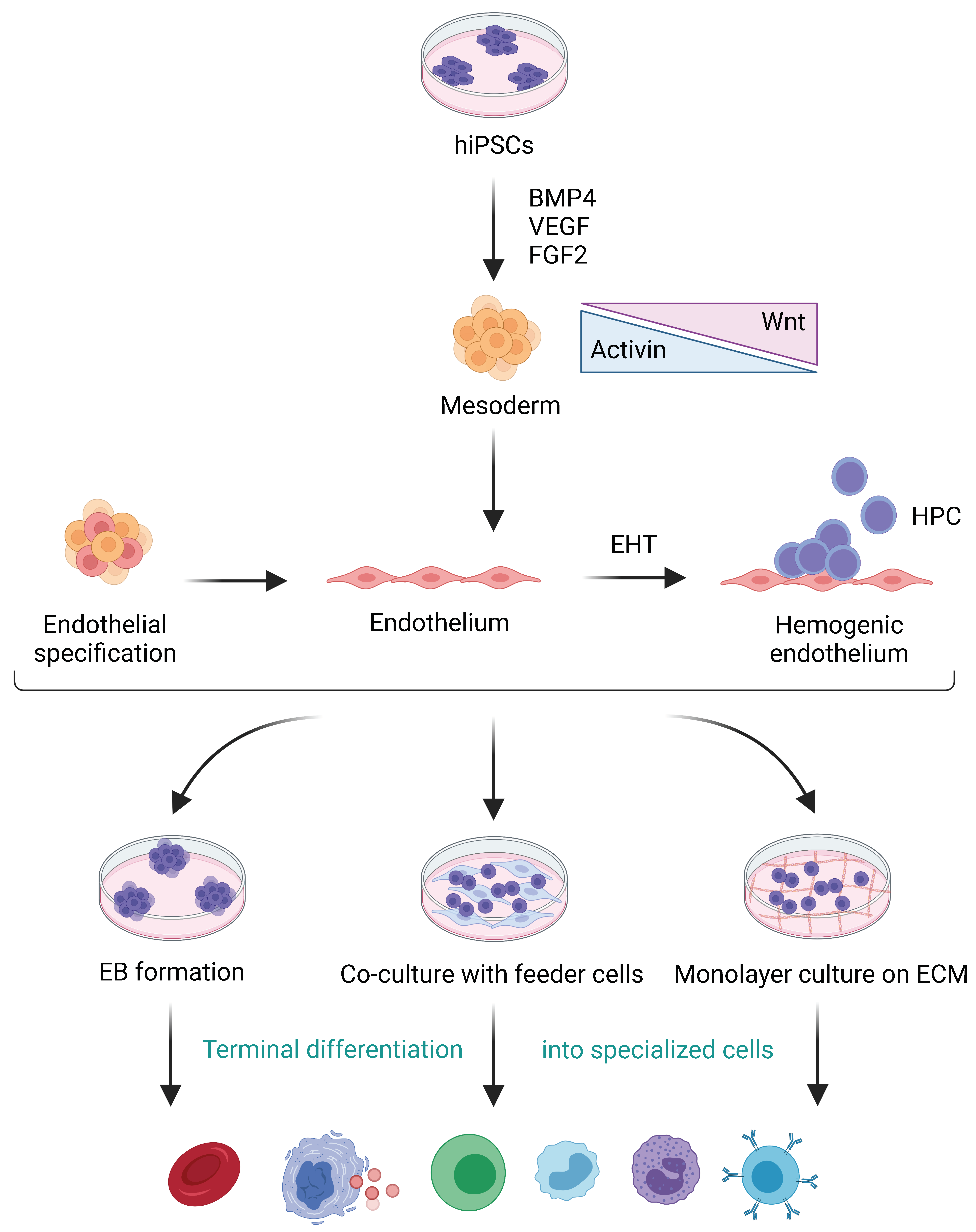
3. Hematopoiesis and Hematopoietic Differentiation from hiPSCs
4. Genetic Disease Modeling with Patient-Specific iPSCs
4.1. Hereditary Hematologic Disorders
4.2. Acquired Hematologic Disorders
5. Patient-Specific iPSCs for Drug Screening
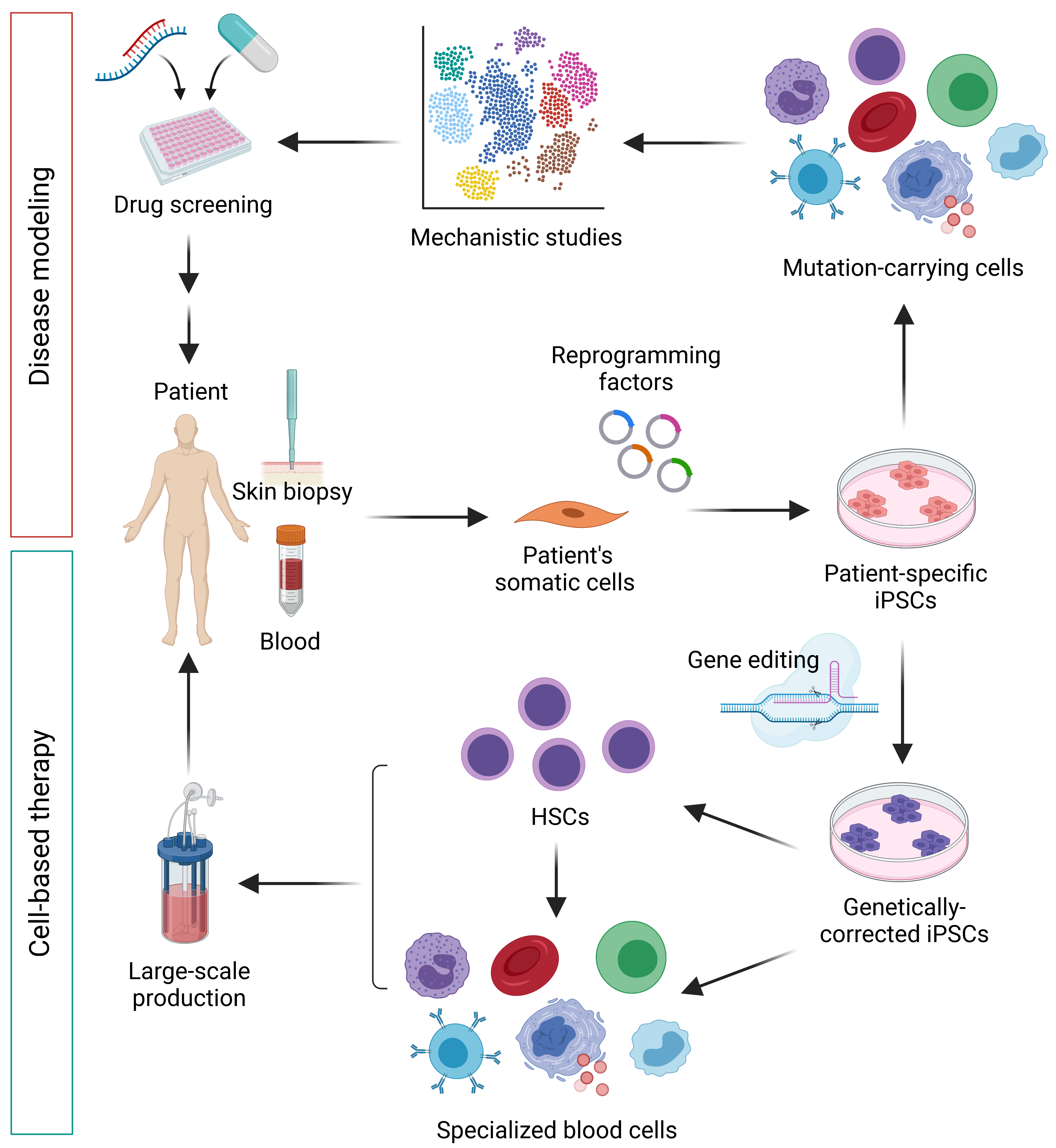
6. iPSCs and Therapeutic Applications
6.1. Cell-Based Therapy
6.2. Cell-Based Cancer Immunotherapy
7. Current Challenges and Future Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Sison, S.L.; Vermilyea, S.C.; Emborg, M.E.; Ebert, A.D. Using patient-derived induced pluripotent stem cells to identify Parkinson’s disease-relevant phenotypes. Curr. Neurol. Neurosci. Rep. 2018, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Seufferlein, T.; Illing, A.; Homann, J. Modelling human channelopathies using induced pluripotent stem cells: A comprehensive review. Stem Cells Int. 2013, 2013, 496501. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xia, G.; Terada, N.; Ashizawa, T. Human iPSC models to study orphan diseases: Muscular dystrophies. Curr. Stem Cell Rep. 2018, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Miyagi-Shiohira, C.; Nakashima, Y. Induced tissue-specific stem cells and epigenetic memory in induced pluripotent stem cells. Int. J. Mol. Sci. 2018, 19, 930. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Papp, B.; Plath, K. Reprogramming to pluripotency: Stepwise resetting of the epigenetic landscape. Cell Res. 2011, 21, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Ohi, Y.; Qin, H.; Hong, C.; Blouin, L.; Polo, J.M.; Guo, T.; Qi, Z.; Downey, S.L.; Manos, P.D.; Rossi, D.J.; et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat. Cell Biol. 2011, 13, 541–549. [Google Scholar] [CrossRef]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef]
- Wang, L.; Su, Y.; Huang, C.; Yin, Y.; Chu, A.; Knupp, A.; Tang, Y. NANOG and LIN28 dramatically improve human cell reprogramming by modulating LIN41 and canonical WNT activities. Biol. Open 2019, 8, 047225. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yin, X.; Qin, H.; Zhu, F.; Liu, H.; Yang, W.; Zhang, Q.; Xiang, C.; Hou, P.; Song, Z.; et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 2008, 3, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Yuan, P.; Yang, H.; Zhang, J.; Soh, B.S.; Li, P.; Lim, S.L.; Cao, S.; Tay, J.; Orlov, Y.L.; et al. Tbx3 improves the germ-line competency of induced pluripotent stem cells. Nature 2010, 463, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Marión, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Belmonte, J.C.I. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Greber, B.; Araúzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Robinton, D.A.; Daley, G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature 2012, 481, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef]
- Wernig, M.; Lengner, C.J.; Hanna, J.; Lodato, M.A.; Steine, E.; Foreman, R.; Staerk, J.; Markoulaki, S.; Jaenisch, R. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat. Biotechnol. 2008, 26, 916–924. [Google Scholar] [CrossRef]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of defective and persistent Sendai virus vector: A unique gene delivery/expression system ideal for cell reprogramming. J. Biol. Chem. 2011, 286, 4760–4771. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef]
- Ye, L.; Muench, M.O.; Fusaki, N.; Beyer, A.I.; Wang, J.; Qi, Z.; Yu, J.; Kan, Y.W. Blood cell-derived induced pluripotent stem cells free of reprogramming factors generated by Sendai viral vectors. Stem Cells Transl. Med. 2013, 2, 558–566. [Google Scholar] [CrossRef]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef]
- Skorik, C.; Mullin, N.K.; Shi, M.; Zhang, Y.; Hunter, P.; Tang, Y.; Hilton, B.; Schlaeger, T.M. Xeno-free reprogramming of peripheral blood mononuclear erythroblasts on laminin-521. Curr. Protoc. Stem Cell Biol. 2020, 52, e103. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Durruthy-Durruthy, J.; Briggs, S.F.; Awe, J.; Ramathal, C.Y.; Karumbayaram, S.; Lee, P.C.; Heidmann, J.D.; Clark, A.; Karakikes, I.; Loh, K.M.; et al. Rapid and efficient conversion of integration-free human induced pluripotent stem cells to GMP-grade culture conditions. PLoS ONE 2014, 9, e94231. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Lin, C. mRNA-based genetic reprogramming. Mol. Ther. 2019, 27, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-H.; Ying, S.-Y. Advances in microRNA-mediated reprogramming technology. Stem Cells Int. 2012, 2012, 823709. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Bao, X.; Liu, L.; Feng, S.; Zovoilis, A.; Liu, W.; Xue, Y.; Cai, J.; Guo, X.; Qin, B.; et al. MicroRNA cluster 302-367 enhances somatic cell reprogramming by accelerating a mesenchymal-to-epithelial transition. J. Biol. Chem. 2011, 286, 17359–17364. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef]
- Eminli, S.; Foudi, A.; Stadtfeld, M.; Maherali, N.; Ahfeldt, T.; Mostoslavsky, G.; Hock, H.; Hochedlinger, K. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat. Genet. 2009, 41, 968–976. [Google Scholar] [CrossRef]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef]
- Ludwig, T.E.; Levenstein, M.E.; Jones, J.M.; Berggren, W.T.; Mitchen, E.R.; Frane, J.L.; Crandall, L.J.; Daigh, C.A.; Conard, K.R.; Piekarczyk, M.S.; et al. Derivation of human embryonic stem cells in defined conditions. Nat. Biotechnol. 2006, 24, 185–187. [Google Scholar] [CrossRef]
- Miyazaki, T.; Futaki, S.; Suemori, H.; Taniguchi, Y.; Yamada, M.; Kawasaki, M.; Hayashi, M.; Kumagai, H.; Nakatsuji, N.; Sekiguchi, K.; et al. Laminin E8 fragments support efficient adhesion and expansion of dissociated human pluripotent stem cells. Nat. Commun. 2012, 3, 1236. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; Robertson, S.; Kennedy, M.; Wall, C.; Keller, G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 1999, 126, 5073–5084. [Google Scholar] [CrossRef]
- Baron, M.H.; Isern, J.; Fraser, S.T. The embryonic origins of erythropoiesis in mammals. Blood 2012, 119, 4828–4837. [Google Scholar] [CrossRef] [PubMed]
- Tober, J.; Koniski, A.; McGrath, K.E.; Vemishetti, R.; Emerson, R.; de Mesy-Bentley, K.K.; Waugh, R.; Palis, J. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 2007, 109, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Li, Y.; De Obaldia, M.E.; Yang, Q.; Yzaguirre, A.D.; Yamada-Inagawa, T.; Vink, C.S.; Bhandoola, A.; Dzierzak, E.; Speck, N.A. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell 2011, 9, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Porayette, P.; Glosson, N.L.; Conway, S.J.; Carlesso, N.; Cardoso, A.A.; Kaplan, M.H.; Yoder, M.C. Autonomous murine T-cell progenitor production in the extra-embryonic yolk sac before HSC emergence. Blood 2012, 119, 5706–5714. [Google Scholar] [CrossRef]
- Lin, Y.; Yoder, M.C.; Yoshimoto, M. Lymphoid progenitor emergence in the murine embryo and yolk sac precedes stem cell detection. Stem Cells Dev. 2014, 23, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.E.; Frame, J.M.; Fegan, K.H.; Bowen, J.R.; Conway, S.J.; Catherman, S.C.; Kingsley, P.D.; Koniski, A.D.; Palis, J. Distinct sources of hematopoietic progenitors emerge before HSCs and provide functional blood cells in the mammalian embryo. Cell Rep. 2015, 11, 1892–1904. [Google Scholar] [CrossRef]
- Medvinsky, A.; Dzierzak, E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 1996, 86, 897–906. [Google Scholar] [CrossRef]
- de Bruijn, M.F.; Speck, N.A.; Peeters, M.C.; Dzierzak, E. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. Embo J. 2000, 19, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Ivanovs, A.; Rybtsov, S.; Welch, L.; Anderson, R.A.; Turner, M.L.; Medvinsky, A. Highly potent human hematopoietic stem cells first emerge in the intraembryonic aorta-gonad-mesonephros region. J. Exp. Med. 2011, 208, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.L.; Wright, D.E.; Wagers, A.J.; Weissman, I.L. Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol. 2004, 2, e75. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, K.; Wang, L.; Li, L.; Menendez, P.; Murdoch, B.; Rouleau, A.; Bhatia, M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003, 102, 906–915. [Google Scholar] [CrossRef]
- Kennedy, M.; D’Souza, S.L.; Lynch-Kattman, M.; Schwantz, S.; Keller, G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood 2007, 109, 2679–2687. [Google Scholar] [CrossRef]
- Pearson, S.; Sroczynska, P.; Lacaud, G.; Kouskoff, V. The stepwise specification of embryonic stem cells to hematopoietic fate is driven by sequential exposure to Bmp4, activin A, bFGF and VEGF. Development 2008, 135, 1525–1535. [Google Scholar] [CrossRef]
- Pick, M.; Azzola, L.; Mossman, A.; Stanley, E.G.; Elefanty, A.G. Differentiation of human embryonic stem cells in serum-free medium reveals distinct roles for bone morphogenetic protein 4, vascular endothelial growth factor, stem cell factor, and fibroblast growth factor 2 in hematopoiesis. Stem Cells 2007, 25, 2206–2214. [Google Scholar] [CrossRef]
- Slukvin, I.I. Hematopoietic specification from human pluripotent stem cells: Current advances and challenges toward de novo generation of hematopoietic stem cells. Blood 2013, 122, 4035–4046. [Google Scholar] [CrossRef]
- Sturgeon, C.M.; Ditadi, A.; Awong, G.; Kennedy, M.; Keller, G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 554–561. [Google Scholar] [CrossRef]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zúñiga-Pflücker, J.C.; Keller, G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef]
- Sturgeon, C.M.; Ditadi, A.; Clarke, R.L.; Keller, G. Defining the path to hematopoietic stem cells. Nat. Biotechnol. 2013, 31, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Demirci, S.; Tisdale, J.F. Definitive erythropoiesis from pluripotent stem cells: Recent advances and perspectives. Adv. Exp. Med. Biol. 2018, 1107, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ran, D.; Shia, W.J.; Lo, M.C.; Fan, J.B.; Knorr, D.A.; Ferrell, P.I.; Ye, Z.; Yan, M.; Cheng, L.; Kaufman, D.S.; et al. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood 2013, 121, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Hu, F.; Weng, Q.; Lv, C.; Wu, H.; Liu, L.; Li, Z.; Zeng, Y.; Bai, Z.; Zhang, M.; et al. Guiding T lymphopoiesis from pluripotent stem cells by defined transcription factors. Cell Res. 2020, 30, 21–33. [Google Scholar] [CrossRef]
- Sugimura, R.; Jha, D.K.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef]
- Tan, Y.T.; Ye, L.; Xie, F.; Beyer, A.I.; Muench, M.O.; Wang, J.; Chen, Z.; Liu, H.; Chen, S.J.; Kan, Y.W. Respecifying human iPSC-derived blood cells into highly engraftable hematopoietic stem and progenitor cells with a single factor. Proc. Natl. Acad. Sci. USA 2018, 115, 2180–2185. [Google Scholar] [CrossRef]
- Amabile, G.; Welner, R.S.; Nombela-Arrieta, C.; D’Alise, A.M.; Di Ruscio, A.; Ebralidze, A.K.; Kraytsberg, Y.; Ye, M.; Kocher, O.; Neuberg, D.S.; et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 2013, 121, 1255–1264. [Google Scholar] [CrossRef]
- Suzuki, N.; Yamazaki, S.; Yamaguchi, T.; Okabe, M.; Masaki, H.; Takaki, S.; Otsu, M.; Nakauchi, H. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol. Ther. 2013, 21, 1424–1431. [Google Scholar] [CrossRef]
- Tsukada, M.; Ota, Y.; Wilkinson, A.C.; Becker, H.J.; Osato, M.; Nakauchi, H.; Yamazaki, S. In vivo generation of engraftable murine hematopoietic stem cells by Gfi1b, c-Fos, and Gata2 overexpression within teratoma. Stem Cell Rep. 2017, 9, 1024–1033. [Google Scholar] [CrossRef]
- Olivier, E.N.; Marenah, L.; McCahill, A.; Condie, A.; Cowan, S.; Mountford, J.C. High-efficiency serum-free feeder-free erythroid differentiation of human pluripotent stem cells using small molecules. Stem Cells Transl. Med. 2016, 5, 1394–1405. [Google Scholar] [CrossRef]
- Fujita, A.; Uchida, N.; Haro-Mora, J.J.; Winkler, T.; Tisdale, J. β-globin-expressing definitive erythroid progenitor cells generated from embryonic and induced pluripotent stem cell-derived sacs. Stem Cells 2016, 34, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Ochi, K.; Takayama, N.; Hirose, S.; Nakahata, T.; Nakauchi, H.; Eto, K. Multicolor staining of globin subtypes reveals impaired globin switching during erythropoiesis in human pluripotent stem cells. Stem Cells Transl. Med. 2014, 3, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.T.; Ma, R.; Axton, R.A.; Jackson, M.; Taylor, A.H.; Fidanza, A.; Marenah, L.; Frayne, J.; Mountford, J.C.; Forrester, L.M. Activation of KLF1 enhances the differentiation and maturation of red blood cells from human pluripotent stem cells. Stem Cells 2017, 35, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Trakarnsanga, K.; Wilson, M.C.; Lau, W.; Singleton, B.K.; Parsons, S.F.; Sakuntanaga, P.; Kurita, R.; Nakamura, Y.; Anstee, D.J.; Frayne, J. Induction of adult levels of β-globin in human erythroid cells that intrinsically express embryonic or fetal globin by transduction with KLF1 and BCL11A-XL. Haematologica 2014, 99, 1677–1685. [Google Scholar] [CrossRef]
- Merryweather-Clarke, A.T.; Tipping, A.J.; Lamikanra, A.A.; Fa, R.; Abu-Jamous, B.; Tsang, H.P.; Carpenter, L.; Robson, K.J.; Nandi, A.K.; Roberts, D.J. Distinct gene expression program dynamics during erythropoiesis from human induced pluripotent stem cells compared with adult and cord blood progenitors. BMC Genom. 2016, 17, 817. [Google Scholar] [CrossRef]
- Dorn, I.; Klich, K.; Arauzo-Bravo, M.J.; Radstaak, M.; Santourlidis, S.; Ghanjati, F.; Radke, T.F.; Psathaki, O.E.; Hargus, G.; Kramer, J.; et al. Erythroid differentiation of human induced pluripotent stem cells is independent of donor cell type of origin. Haematologica 2015, 100, 32–41. [Google Scholar] [CrossRef]
- Lapillonne, H.; Kobari, L.; Mazurier, C.; Tropel, P.; Giarratana, M.C.; Zanella-Cleon, I.; Kiger, L.; Wattenhofer-Donzé, M.; Puccio, H.; Hebert, N.; et al. Red blood cell generation from human induced pluripotent stem cells: Perspectives for transfusion medicine. Haematologica 2010, 95, 1651–1659. [Google Scholar] [CrossRef]
- Hansen, M.; Varga, E.; Aarts, C.; Wust, T.; Kuijpers, T.; von Lindern, M.; van den Akker, E. Efficient production of erythroid, megakaryocytic and myeloid cells, using single cell-derived iPSC colony differentiation. Stem Cell Res. 2018, 29, 232–244. [Google Scholar] [CrossRef]
- Hansen, M.; von Lindern, M.; van den Akker, E.; Varga, E. Human-induced pluripotent stem cell-derived blood products: State of the art and future directions. FEBS Lett. 2019, 593, 3288–3303. [Google Scholar] [CrossRef]
- Aihara, A.; Koike, T.; Abe, N.; Nakamura, S.; Sawaguchi, A.; Nakamura, T.; Sugimoto, N.; Nakauchi, H.; Nishino, T.; Eto, K. Novel TPO receptor agonist TA-316 contributes to platelet biogenesis from human iPS cells. Blood Adv. 2017, 1, 468–476. [Google Scholar] [CrossRef]
- Feng, Q.; Shabrani, N.; Thon, J.N.; Huo, H.; Thiel, A.; Machlus, K.R.; Kim, K.; Brooks, J.; Li, F.; Luo, C.; et al. Scalable generation of universal platelets from human induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Moreau, T.; Evans, A.L.; Vasquez, L.; Tijssen, M.R.; Yan, Y.; Trotter, M.W.; Howard, D.; Colzani, M.; Arumugam, M.; Wu, W.H.; et al. Large-scale production of megakaryocytes from human pluripotent stem cells by chemically defined forward programming. Nat. Commun. 2016, 7, 11208. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Nakamura, S.; Sugimoto, N.; Shigemori, T.; Kato, Y.; Ohno, M.; Sakuma, S.; Ito, K.; Kumon, H.; Hirose, H.; et al. Turbulence activates platelet biogenesis to enable clinical scale ex vivo production. Cell 2018, 174, 636–648.e18. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.H.; Howard, D.; Waller, A.K.; Foster, H.R.; Mueller, A.; Moreau, T.; Evans, A.L.; Arumugam, M.; Bouët Chalon, G.; Vriend, E.; et al. Structurally graduated collagen scaffolds applied to the ex vivo generation of platelets from human pluripotent stem cell-derived megakaryocytes: Enhancing production and purity. Biomaterials 2018, 182, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Akabayashi, A.; Nakazawa, E.; Jecker, N.S. The world’s first clinical trial for an aplastic anemia patient with thrombocytopenia administering platelets generated from autologous iPS cells. Int J. Hematol. 2019, 109, 239–240. [Google Scholar] [CrossRef]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity 2017, 47, 183–198.e6. [Google Scholar] [CrossRef]
- Lee, C.Z.W.; Kozaki, T.; Ginhoux, F. Studying tissue macrophages in vitro: Are iPSC-derived cells the answer? Nat. Rev. Immunol. 2018, 18, 716–725. [Google Scholar] [CrossRef]
- Lachmann, N.; Ackermann, M.; Frenzel, E.; Liebhaber, S.; Brennig, S.; Happle, C.; Hoffmann, D.; Klimenkova, O.; Lüttge, D.; Buchegger, T.; et al. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Rep. 2015, 4, 282–296. [Google Scholar] [CrossRef]
- Brauer, P.M.; Pessach, I.M.; Clarke, E.; Rowe, J.H.; Ott de Bruin, L.; Lee, Y.N.; Dominguez-Brauer, C.; Comeau, A.M.; Awong, G.; Felgentreff, K.; et al. Modeling altered T-cell development with induced pluripotent stem cells from patients with RAG1-dependent immune deficiencies. Blood 2016, 128, 783–793. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakauchi, H. Generation of antigen-specific T cells from human induced pluripotent stem cells. Methods Mol. Biol. 2019, 1899, 25–40. [Google Scholar] [CrossRef]
- Shukla, S.; Langley, M.A.; Singh, J.; Edgar, J.M.; Mohtashami, M.; Zúñiga-Pflücker, J.C.; Zandstra, P.W. Progenitor T-cell differentiation from hematopoietic stem cells using Delta-like-4 and VCAM-1. Nat. Methods 2017, 14, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Wattanapanitch, M.; Damkham, N.; Potirat, P.; Trakarnsanga, K.; Janan, M.; U-Pratya, Y.; Kheolamai, P.; Klincumhom, N.; Issaragrisil, S. One-step genetic correction of hemoglobin E/beta-thalassemia patient-derived iPSCs by the CRISPR/Cas9 system. Stem Cell Res. Ther. 2018, 9, 46. [Google Scholar] [CrossRef]
- Cai, L.; Bai, H.; Mahairaki, V.; Gao, Y.; He, C.; Wen, Y.; Jin, Y.C.; Wang, Y.; Pan, R.L.; Qasba, A.; et al. A universal approach to correct various HBB gene mutations in human stem cells for gene therapy of beta-thalassemia and sickle cell disease. Stem Cells Transl. Med. 2018, 7, 87–97. [Google Scholar] [CrossRef]
- Niu, X.; He, W.; Song, B.; Ou, Z.; Fan, D.; Chen, Y.; Fan, Y.; Sun, X. Combining single strand oligodeoxynucleotides and CRISPR/Cas9 to correct gene mutations in β-thalassemia-induced pluripotent stem cells. J. Biol. Chem. 2016, 291, 16576–16585. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, C.G.; Jiang, Y.; Zhang, J.; Chen, J.; Yao, C.; Zhao, Q.; Liu, S.; Chen, K.; Du, J.; et al. Genetic correction of β-thalassemia patient-specific iPS cells and its use in improving hemoglobin production in irradiated SCID mice. Cell Res. 2012, 22, 637–648. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Kang, X.; Lin, B.; Yu, Q.; Song, B.; Gao, G.; Chen, Y.; Sun, X.; Li, X.; et al. One-step biallelic and scarless correction of a β-thalassemia mutation in patient-specific iPSCs without drug selection. Mol. Ther. Nucleic Acids 2017, 6, 57–67. [Google Scholar] [CrossRef]
- Xiong, Z.; Xie, Y.; Yang, Y.; Xue, Y.; Wang, D.; Lin, S.; Chen, D.; Lu, D.; He, L.; Song, B.; et al. Efficient gene correction of an aberrant splice site in β-thalassaemia iPSCs by CRISPR/Cas9 and single-strand oligodeoxynucleotides. J. Cell. Mol. Med. 2019, 23, 8046–8057. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Liao, B.; Zhang, H.; Wang, L.; Shan, Y.; Xue, Y.; Huang, K.; Chen, S.; Zhou, X.; Chen, Y.; et al. Transcription activator-like effector nuclease (TALEN)-mediated gene correction in integration-free β-thalassemia induced pluripotent stem cells. J. Biol. Chem. 2013, 288, 34671–34679. [Google Scholar] [CrossRef]
- Ma, N.; Shan, Y.; Liao, B.; Kong, G.; Wang, C.; Huang, K.; Zhang, H.; Cai, X.; Chen, S.; Pei, D.; et al. Factor-induced reprogramming and zinc finger nuclease-aided gene targeting cause different genome instability in β-thalassemia induced pluripotent stem cells (iPSCs). J. Biol. Chem. 2015, 290, 12079–12089. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Tong, Y.; Liu, X.Z.; Wang, T.T.; Cheng, L.; Wang, B.Y.; Lv, X.; Huang, Y.; Liu, D.P. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci. Rep. 2015, 5, 12065. [Google Scholar] [CrossRef]
- Chang, C.J.; Bouhassira, E.E. Zinc-finger nuclease-mediated correction of α-thalassemia in iPS cells. Blood 2012, 120, 3906–3914. [Google Scholar] [CrossRef] [PubMed]
- Garate, Z.; Quintana-Bustamante, O.; Crane, A.M.; Olivier, E.; Poirot, L.; Galetto, R.; Kosinski, P.; Hill, C.; Kung, C.; Agirre, X.; et al. Generation of a high number of healthy erythroid cells from gene-edited pyruvate kinase deficiency patient-specific induced pluripotent stem cells. Stem Cell Rep. 2015, 5, 1053–1066. [Google Scholar] [CrossRef]
- Kohara, H.; Utsugisawa, T.; Sakamoto, C.; Hirose, L.; Ogawa, Y.; Ogura, H.; Sugawara, A.; Liao, J.; Aoki, T.; Iwasaki, T.; et al. KLF1 mutation E325K induces cell cycle arrest in erythroid cells differentiated from congenital dyserythropoietic anemia patient-specific induced pluripotent stem cells. Exp. Hematol. 2019, 73, 25–37.e8. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Mali, P.; Huang, X.; Dowey, S.N.; Cheng, L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 2011, 118, 4599–4608. [Google Scholar] [CrossRef]
- Ramalingam, S.; Annaluru, N.; Kandavelou, K.; Chandrasegaran, S. TALEN-mediated generation and genetic correction of disease-specific human induced pluripotent stem cells. Curr. Gene Ther. 2014, 14, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol. Bioeng. 2014, 111, 1048–1053. [Google Scholar] [CrossRef]
- Huang, X.; Wang, Y.; Yan, W.; Smith, C.; Ye, Z.; Wang, J.; Gao, Y.; Mendelsohn, L.; Cheng, L. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells 2015, 33, 1470–1479. [Google Scholar] [CrossRef]
- Park, S.; Gianotti-Sommer, A.; Molina-Estevez, F.J.; Vanuytsel, K.; Skvir, N.; Leung, A.; Rozelle, S.S.; Shaikho, E.M.; Weir, I.; Jiang, Z.; et al. A comprehensive, ethnically diverse library of sickle cell disease-specific induced pluripotent stem cells. Stem Cell Rep. 2017, 8, 1076–1085. [Google Scholar] [CrossRef]
- Zou, J.; Sweeney, C.L.; Chou, B.K.; Choi, U.; Pan, J.; Wang, H.; Dowey, S.N.; Cheng, L.; Malech, H.L. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: Functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood 2011, 117, 5561–5572. [Google Scholar] [CrossRef]
- Jiang, Y.; Cowley, S.A.; Siler, U.; Melguizo, D.; Tilgner, K.; Browne, C.; Dewilton, A.; Przyborski, S.; Saretzki, G.; James, W.S.; et al. Derivation and functional analysis of patient-specific induced pluripotent stem cells as an in vitro model of chronic granulomatous disease. Stem Cells 2012, 30, 599–611. [Google Scholar] [CrossRef]
- Merling, R.K.; Sweeney, C.L.; Chu, J.; Bodansky, A.; Choi, U.; Priel, D.L.; Kuhns, D.B.; Wang, H.; Vasilevsky, S.; De Ravin, S.S.; et al. An AAVS1-targeted minigene platform for correction of iPSCs from all five types of chronic granulomatous disease. Mol. Ther. 2015, 23, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.; Grundmann, A.; Renz, P.; Hänseler, W.; James, W.S.; Cowley, S.A.; Moore, M.D. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp. Hematol. 2015, 43, 838–848.e3. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, A.K.; Hoffmann, D.; Lachmann, N.; Ackermann, M.; Steinemann, D.; Timm, B.; Siler, U.; Reichenbach, J.; Grez, M.; Moritz, T.; et al. TALEN-mediated functional correction of X-linked chronic granulomatous disease in patient-derived induced pluripotent stem cells. Biomaterials 2015, 69, 191–200. [Google Scholar] [CrossRef]
- Laugsch, M.; Rostovskaya, M.; Velychko, S.; Richter, C.; Zimmer, A.; Klink, B.; Schröck, E.; Haase, M.; Neumann, K.; Thieme, S.; et al. Functional restoration of gp91phox-oxidase activity by BAC transgenesis and gene targeting in X-linked chronic granulomatous disease iPSCs. Mol. Ther. 2016, 24, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Merling, R.K.; Kuhns, D.B.; Sweeney, C.L.; Wu, X.; Burkett, S.; Chu, J.; Lee, J.; Koontz, S.; Di Pasquale, G.; Afione, S.A.; et al. Gene-edited pseudogene resurrection corrects p47(phox)-deficient chronic granulomatous disease. Blood Adv. 2017, 1, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Klatt, D.; Cheng, E.; Philipp, F.; Selich, A.; Dahlke, J.; Schmidt, R.E.; Schott, J.W.; Büning, H.; Hoffmann, D.; Thrasher, A.J.; et al. Targeted repair of p47-CGD in iPSCs by CRISPR/Cas9: Functional correction without cleavage in the highly homologous pseudogenes. Stem Cell Rep. 2019, 13, 590–598. [Google Scholar] [CrossRef]
- Menon, T.; Firth, A.L.; Scripture-Adams, D.D.; Galic, Z.; Qualls, S.J.; Gilmore, W.B.; Ke, E.; Singer, O.; Anderson, L.S.; Bornzin, A.R.; et al. Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell 2015, 16, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Lai, Y.S.; Westin, E.; Khodadadi-Jamayran, A.; Pawlik, K.M.; Lamb, L.S., Jr.; Goldman, F.D.; Townes, T.M. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-enhanced gene targeting. Cell Rep. 2015, 12, 1668–1677. [Google Scholar] [CrossRef]
- Themeli, M.; Chhatta, A.; Boersma, H.; Prins, H.J.; Cordes, M.; de Wilt, E.; Farahani, A.S.; Vandekerckhove, B.; van der Burg, M.; Hoeben, R.C.; et al. iPSC-based modeling of RAG2 severe combined immunodeficiency reveals multiple T cell developmental arrests. Stem Cell Rep. 2020, 14, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Ingrungruanglert, P.; Amarinthnukrowh, P.; Rungsiwiwut, R.; Maneesri-le Grand, S.; Sosothikul, D.; Suphapeetiporn, K.; Israsena, N.; Shotelersuk, V. Wiskott-Aldrich syndrome iPS cells produce megakaryocytes with defects in cytoskeletal rearrangement and proplatelet formation. Thromb. Haemost. 2015, 113, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, T.J.; Van Caeneghem, Y.; Pourebrahim, R.; Ma, C.; Ni, Z.; Garate, Z.; Crane, A.M.; Li, X.S.; Liao, W.; Gonzalez-Garay, M.; et al. Gene correction of iPSCs from a Wiskott-Aldrich syndrome patient normalizes the lymphoid developmental and functional defects. Stem Cell Rep. 2016, 7, 139–148. [Google Scholar] [CrossRef]
- Park, C.Y.; Kim, J.; Kweon, J.; Son, J.S.; Lee, J.S.; Yoo, J.E.; Cho, S.R.; Kim, J.H.; Kim, J.S.; Kim, D.W. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. USA 2014, 111, 9253–9258. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hu, Z.; Li, Z.; Pang, J.; Feng, M.; Hu, X.; Wang, X.; Lin-Peng, S.; Liu, B.; Chen, F.; et al. In situ genetic correction of F8 intron 22 inversion in hemophilia A patient-specific iPSCs. Sci. Rep. 2016, 6, 18865. [Google Scholar] [CrossRef]
- Park, C.Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.H.; Kim, D.W.; Kim, J.S. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Wu, Y.; Li, Z.; Hu, Z.; Wang, X.; Hu, X.; Wang, X.; Liu, X.; Zhou, M.; Liu, B.; et al. Targeting of the human F8 at the multicopy rDNA locus in hemophilia A patient-derived iPSCs using TALENickases. Biochem. Biophys. Res. Commun. 2016, 472, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Olgasi, C.; Talmon, M.; Merlin, S.; Cucci, A.; Richaud-Patin, Y.; Ranaldo, G.; Colangelo, D.; Di Scipio, F.; Berta, G.N.; Borsotti, C.; et al. Patient-specific iPSC-derived endothelial cells provide long-term phenotypic correction of hemophilia A. Stem Cell Rep. 2018, 11, 1391–1406. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tonnu, N.; Menon, T.; Lewis, B.M.; Green, K.T.; Wampler, D.; Monahan, P.E.; Verma, I.M. Autologous and heterologous cell therapy for hemophilia B toward functional restoration of factor IX. Cell Rep. 2018, 23, 1565–1580. [Google Scholar] [CrossRef]
- He, Q.; Wang, H.H.; Cheng, T.; Yuan, W.P.; Ma, Y.P.; Jiang, Y.P.; Ren, Z.H. Genetic correction and hepatic differentiation of hemophilia B-specific human induced pluripotent stem cells. Chin. Med. Sci. J. 2017, 32, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Lyu, C.; Shen, J.; Wang, R.; Gu, H.; Zhang, J.; Xue, F.; Liu, X.; Liu, W.; Fu, R.; Zhang, L.; et al. Targeted genome engineering in human induced pluripotent stem cells from patients with hemophilia B using the CRISPR-Cas9 system. Stem Cell Res. Ther. 2018, 9, 92. [Google Scholar] [CrossRef]
- Garçon, L.; Ge, J.; Manjunath, S.H.; Mills, J.A.; Apicella, M.; Parikh, S.; Sullivan, L.M.; Podsakoff, G.M.; Gadue, P.; French, D.L.; et al. Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood 2013, 122, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Doulatov, S.; Vo, L.T.; Macari, E.R.; Wahlster, L.; Kinney, M.A.; Taylor, A.M.; Barragan, J.; Gupta, M.; McGrath, K.; Lee, H.Y.; et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci Transl Med. 2017, 9, aah5645. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Apicella, M.; Mills, J.A.; Garçon, L.; French, D.L.; Weiss, M.J.; Bessler, M.; Mason, P.J. Dysregulation of the transforming growth factor β pathway in induced pluripotent stem cells generated from patients with Diamond Blackfan anemia. PLoS ONE 2015, 10, e0134878. [Google Scholar] [CrossRef]
- Kotini, A.G.; Chang, C.J.; Boussaad, I.; Delrow, J.J.; Dolezal, E.K.; Nagulapally, A.B.; Perna, F.; Fishbein, G.A.; Klimek, V.M.; Hawkins, R.D.; et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 646–655. [Google Scholar] [CrossRef]
- Kotini, A.G.; Chang, C.J.; Chow, A.; Yuan, H.; Ho, T.C.; Wang, T.; Vora, S.; Solovyov, A.; Husser, C.; Olszewska, M.; et al. Stage-specific human induced pluripotent stem cells map the progression of myeloid transformation to transplantable leukemia. Cell Stem Cell 2017, 20, 315–328.e7. [Google Scholar] [CrossRef]
- Chao, M.P.; Gentles, A.J.; Chatterjee, S.; Lan, F.; Reinisch, A.; Corces, M.R.; Xavy, S.; Shen, J.; Haag, D.; Chanda, S.; et al. Human AML-iPSCs reacquire leukemic properties after differentiation and model clonal variation of disease. Cell Stem Cell 2017, 20, 329–344.e7. [Google Scholar] [CrossRef] [PubMed]
- Bedel, A.; Pasquet, J.M.; Lippert, E.; Taillepierre, M.; Lagarde, V.; Dabernat, S.; Dubus, P.; Charaf, L.; Beliveau, F.; de Verneuil, H.; et al. Variable behavior of iPSCs derived from CML patients for response to TKI and hematopoietic differentiation. PLoS ONE 2013, 8, e71596. [Google Scholar] [CrossRef]
- Suknuntha, K.; Ishii, Y.; Tao, L.; Hu, K.; McIntosh, B.E.; Yang, D.; Swanson, S.; Stewart, R.; Wang, J.Y.J.; Thomson, J.; et al. Discovery of survival factor for primitive chronic myeloid leukemia cells using induced pluripotent stem cells. Stem Cell Res. 2015, 15, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Charaf, L.; Mahon, F.X.; Lamrissi-Garcia, I.; Moranvillier, I.; Beliveau, F.; Cardinaud, B.; Dabernat, S.; de Verneuil, H.; Moreau-Gaudry, F.; Bedel, A. Effect of tyrosine kinase inhibitors on stemness in normal and chronic myeloid leukemia cells. Leukemia 2017, 31, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Toofan, P.; Busch, C.; Morrison, H.; O’Brien, S.; Jørgensen, H.; Copland, M.; Wheadon, H. Chronic myeloid leukaemia cells require the bone morphogenic protein pathway for cell cycle progression and self-renewal. Cell Death Dis. 2018, 9, 927. [Google Scholar] [CrossRef]
- Miyauchi, M.; Koya, J.; Arai, S.; Yamazaki, S.; Honda, A.; Kataoka, K.; Yoshimi, A.; Taoka, K.; Kumano, K.; Kurokawa, M. ADAM8 is an antigen of tyrosine kinase inhibitor-resistant chronic myeloid leukemia cells identified by patient-derived induced pluripotent stem cells. Stem Cell Rep. 2018, 10, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Braunstein, E.M.; Ye, Z.; Liu, C.F.; Chen, G.; Zou, J.; Cheng, L.; Brodsky, R.A. Generation of glycosylphosphatidylinositol anchor protein-deficient blood cells from human induced pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.; Yi, L.; Hou, Z.; Chen, J.; Kou, X.; Zhao, Y.; Wang, H.; Sun, X.F.; Jiang, C.; et al. Naïve induced pluripotent stem cells generated from β-thalassemia fibroblasts allow efficient gene correction with CRISPR/Cas9. Stem Cells Transl. Med. 2016, 5, 8–19. [Google Scholar] [CrossRef]
- Potirat, P.; Wattanapanitch, M.; Viprakasit, V.; Kheolamai, P.; Issaragrisil, S. An integration-free iPSC line (MUSIi008-A) derived from a patient with severe hemolytic anemia carrying compound heterozygote mutations in KLF1 gene for disease modeling. Stem Cell Res. 2019, 34, 101344. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Reilly, A.; Hayes, B.J.; Clough, C.A.; Konnick, E.Q.; Torok-Storb, B.; Gulsuner, S.; Wu, D.; Becker, P.S.; Keel, S.B.; et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood 2019, 134, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Pine, A.R.; Kotini, A.G.; Yuan, H.; Zamparo, L.; Starczynowski, D.T.; Leslie, C.; Papapetrou, E.P. Sequential CRISPR gene editing in human iPSCs charts the clonal evolution of myeloid leukemia and identifies early disease targets. Cell Stem Cell 2021, 28, 1074–1089.e7. [Google Scholar] [CrossRef]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009, 114, 5473–5480. [Google Scholar] [CrossRef]
- Nilsri, N.; Jangprasert, P.; Pawinwongchai, J.; Israsena, N.; Rojnuckarin, P. Distinct effects of V617F and exon12-mutated JAK2 expressions on erythropoiesis in a human induced pluripotent stem cell (iPSC)-based model. Sci. Rep. 2021, 11, 5255. [Google Scholar] [CrossRef]
- Saliba, J.; Hamidi, S.; Lenglet, G.; Langlois, T.; Yin, J.; Cabagnols, X.; Secardin, L.; Legrand, C.; Galy, A.; Opolon, P.; et al. Heterozygous and homozygous JAK2(V617F) states modeled by induced pluripotent stem cells from myeloproliferative neoplasm patients. PLoS ONE 2013, 8, e74257. [Google Scholar] [CrossRef]
- Takei, H.; Edahiro, Y.; Mano, S.; Masubuchi, N.; Mizukami, Y.; Imai, M.; Morishita, S.; Misawa, K.; Ochiai, T.; Tsuneda, S.; et al. Skewed megakaryopoiesis in human induced pluripotent stem cell-derived haematopoietic progenitor cells harbouring calreticulin mutations. Br. J. Haematol. 2018, 181, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Secardin, L.; Gomez Limia, C.; da Silva-Benedito, S.; Lordier, L.; El-Khoury, M.; Marty, C.; Ianotto, J.C.; Raslova, H.; Constantinescu, S.N.; Bonamino, M.H.; et al. Induced pluripotent stem cells enable disease modeling and drug screening in calreticulin del52 and ins5 myeloproliferative Neoplasms. Hemasphere 2021, 5, e593. [Google Scholar] [CrossRef]
- Brault, J.; Vaganay, G.; Le Roy, A.; Lenormand, J.L.; Cortes, S.; Stasia, M.J. Therapeutic effects of proteoliposomes on X-linked chronic granulomatous disease: Proof of concept using macrophages differentiated from patient-specific induced pluripotent stem cells. Int. J. Nanomed. 2017, 12, 2161–2177. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Suzuki, K.; Li, M.; Qu, J.; Montserrat, N.; Tarantino, C.; Gu, Y.; Yi, F.; Xu, X.; Zhang, W.; et al. Modelling Fanconi anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs. Nat. Commun. 2014, 5, 4330. [Google Scholar] [CrossRef]
- Ruiz-Gutierrez, M.; Bölükbaşı, Ö.V.; Alexe, G.; Kotini, A.G.; Ballotti, K.; Joyce, C.E.; Russell, D.W.; Stegmaier, K.; Myers, K.; Novina, C.D.; et al. Therapeutic discovery for marrow failure with MDS predisposition using pluripotent stem cells. JCI Insight 2019, 5, 125157. [Google Scholar] [CrossRef] [PubMed]
- Qanash, H.; Li, Y.; Smith, R.H.; Linask, K.; Young-Baird, S.; Hakami, W.; Keyvanfar, K.; Choy, J.S.; Zou, J.; Larochelle, A. Eltrombopag improves erythroid differentiation in a human induced pluripotent stem cell model of Diamond Blackfan anemia. Cells 2021, 10, 734. [Google Scholar] [CrossRef] [PubMed]
- Taoka, K.; Arai, S.; Kataoka, K.; Hosoi, M.; Miyauchi, M.; Yamazaki, S.; Honda, A.; Aixinjueluo, W.; Kobayashi, T.; Kumano, K.; et al. Using patient-derived iPSCs to develop humanized mouse models for chronic myelomonocytic leukemia and therapeutic drug identification, including liposomal clodronate. Sci. Rep. 2018, 8, 15855. [Google Scholar] [CrossRef]
- Chang, C.J.; Kotini, A.G.; Olszewska, M.; Georgomanoli, M.; Teruya-Feldstein, J.; Sperber, H.; Sanchez, R.; DeVita, R.; Martins, T.J.; Abdel-Wahab, O.; et al. Dissecting the contributions of cooperating gene mutations to cancer phenotypes and drug responses with patient-derived iPSCs. Stem Cell Rep. 2018, 10, 1610–1624. [Google Scholar] [CrossRef]
- Sullivan, S.; Stacey, G.N.; Akazawa, C.; Aoyama, N.; Baptista, R.; Bedford, P.; Bennaceur Griscelli, A.; Chandra, A.; Elwood, N.; Girard, M.; et al. Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen. Med. 2018, 13, 859–866. [Google Scholar] [CrossRef]
- Barker, J.N.; Kurtzberg, J.; Ballen, K.; Boo, M.; Brunstein, C.; Cutler, C.; Horwitz, M.; Milano, F.; Olson, A.; Spellman, S.; et al. Optimal practices in unrelated donor cord blood transplantation for hematologic malignancies. Biol. Blood Marrow Transpl. 2017, 23, 882–896. [Google Scholar] [CrossRef]
- Phondeechareon, T.; Wattanapanitch, M.; U-Pratya, Y.; Damkham, C.; Klincumhom, N.; Lorthongpanich, C.; Kheolamai, P.; Laowtammathron, C.; Issaragrisil, S. Generation of induced pluripotent stem cells as a potential source of hematopoietic stem cells for transplant in PNH patients. Ann. Hematol. 2016, 95, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takayama, N.; Hirata, S.; Seo, H.; Endo, H.; Ochi, K.; Fujita, K.; Koike, T.; Harimoto, K.; Dohda, T.; et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell 2014, 14, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Umekage, M.; Sato, Y.; Takasu, N. Overview: An iPS cell stock at CiRA. Inflamm. Regen. 2019, 39, 17. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Leslie, S.; Martin, N.G.; Peschanski, M.; Rao, M.; Taylor, C.J.; Trounson, A.; Turner, D.; Yamanaka, S.; Wilmut, I. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell 2013, 13, 382–384. [Google Scholar] [CrossRef]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted disruption of HLA genes via CRISPR-Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell 2019, 24, 566–578.e7. [Google Scholar] [CrossRef] [PubMed]
- Börger, A.K.; Eicke, D.; Wolf, C.; Gras, C.; Aufderbeck, S.; Schulze, K.; Engels, L.; Eiz-Vesper, B.; Schambach, A.; Guzman, C.A.; et al. Generation of HLA-universal iPSC-derived megakaryocytes and platelets for survival under refractoriness conditions. Mol. Med. 2016, 22, 274–285. [Google Scholar] [CrossRef]
- Mattapally, S.; Pawlik, K.M.; Fast, V.G.; Zumaquero, E.; Lund, F.E.; Randall, T.D.; Townes, T.M.; Zhang, J. Human leukocyte antigen class I and II knockout human induced pluripotent stem cell-derived cells: Universal donor for cell therapy. J. Am. Heart Assoc. 2018, 7, e010239. [Google Scholar] [CrossRef]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Suzuki, D.; Flahou, C.; Yoshikawa, N.; Stirblyte, I.; Hayashi, Y.; Sawaguchi, A.; Akasaka, M.; Nakamura, S.; Higashi, N.; Xu, H.; et al. iPSC-derived platelets depleted of HLA class I are inert to anti-HLA class I and natural killer cell immunity. Stem Cell Rep. 2020, 14, 49–59. [Google Scholar] [CrossRef]
- Norbnop, P.; Ingrungruanglert, P.; Israsena, N.; Suphapeetiporn, K.; Shotelersuk, V. Generation and characterization of HLA-universal platelets derived from induced pluripotent stem cells. Sci. Rep. 2020, 10, 8472. [Google Scholar] [CrossRef]
- Hanna, J.; Wernig, M.; Markoulaki, S.; Sun, C.W.; Meissner, A.; Cassady, J.P.; Beard, C.; Brambrink, T.; Wu, L.C.; Townes, T.M.; et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 2007, 318, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Ma, C.; Awong, G.; Kennedy, M.; Gornalusse, G.; Keller, G.; Kaufman, D.S.; Russell, D.W. Silent IL2RG gene editing in human pluripotent stem cells. Mol. Ther. 2016, 24, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves fourth CAR-T cell therapy. Nat. Rev. Drug Discov. 2021, 20, 166. [Google Scholar] [CrossRef] [PubMed]
- Luanpitpong, S.; Poohadsuan, J.; Klaihmon, P.; Issaragrisil, S. Selective cytotoxicity of single and dual anti-CD19 and anti-CD138 chimeric antigen receptor-natural killer cells against hematologic malignancies. J. Immunol. Res. 2021, 2021, 5562630. [Google Scholar] [CrossRef]
- Graham, C.; Jozwik, A.; Pepper, A.; Benjamin, R. Allogeneic CAR-T cells: More than ease of access? Cells 2018, 7, 155. [Google Scholar] [CrossRef]
- Pfefferle, A.; Huntington, N.D. You have got a fast CAR: Chimeric antigen receptor NK cells in cancer therapy. Cancers 2020, 12, 706. [Google Scholar] [CrossRef]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, H.; Diao, Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef]
- Arias, J.; Yu, J.; Varshney, M.; Inzunza, J.; Nalvarte, I. Hematopoietic stem cell- and induced pluripotent stem cell-derived CAR-NK cells as reliable cell-based therapy solutions. Stem Cells Transl. Med. 2021, 10, 987–995. [Google Scholar] [CrossRef]
- Mehta, R.S.; Rezvani, K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric antigen receptors T cell therapy in solid tumor: Challenges and clinical applications. Front. Immunol. 2017, 8, 1850. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Z.; Tan, X.; Jiang, H.; Xu, Z.; Fang, Y.; Han, D.; Hong, W.; Wei, W.; Tu, J. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 2021, 139, 111605. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Ghobadi, A.; Pennella, E.J.; Vanas, J.; Powell, C.; Pavelova, M.; Wagner, C.; Kuo, M.; Ullmann, C.D.; Hassan, R.; et al. Feasibility and preliminary safety and efficacy of first-in-human intraperitoneal delivery of MCY-M11, anti-human-mesothelin CAR mRNA transfected into peripheral blood mononuclear cells, for ovarian cancer and malignant peritoneal mesothelioma. J. Clin. Oncol. 2020, 38, 3014. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 153. [Google Scholar] [CrossRef]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 2017, 546, 370–375. [Google Scholar] [CrossRef]
- DeBoever, C.; Li, H.; Jakubosky, D.; Benaglio, P.; Reyna, J.; Olson, K.M.; Huang, H.; Biggs, W.; Sandoval, E.; D’Antonio, M.; et al. Large-scale profiling reveals the influence of genetic variation on gene expression in human induced pluripotent stem cells. Cell Stem Cell 2017, 20, 533–546.e7. [Google Scholar] [CrossRef]
- Nishizawa, M.; Chonabayashi, K.; Nomura, M.; Tanaka, A.; Nakamura, M.; Inagaki, A.; Nishikawa, M.; Takei, I.; Oishi, A.; Tanabe, K.; et al. Epigenetic variation between human induced pluripotent stem cell lines is an indicator of differentiation capacity. Cell Stem Cell 2016, 19, 341–354. [Google Scholar] [CrossRef]
- Kyttälä, A.; Moraghebi, R.; Valensisi, C.; Kettunen, J.; Andrus, C.; Pasumarthy, K.K.; Nakanishi, M.; Nishimura, K.; Ohtaka, M.; Weltner, J.; et al. Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Rep. 2016, 6, 200–212. [Google Scholar] [CrossRef]
- Kajiwara, M.; Aoi, T.; Okita, K.; Takahashi, R.; Inoue, H.; Takayama, N.; Endo, H.; Eto, K.; Toguchida, J.; Uemoto, S.; et al. Donor-dependent variations in hepatic differentiation from human-induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12538–12543. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.W.; Powell, B.E.; Wang, H.; Mitalipova, M.; Faddah, D.A.; Reddy, J.; Fan, Z.P.; Maetzel, D.; Ganz, K.; Shi, L.; et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell 2014, 15, 471–487. [Google Scholar] [CrossRef]
- Takashima, Y.; Guo, G.; Loos, R.; Nichols, J.; Ficz, G.; Krueger, F.; Oxley, D.; Santos, F.; Clarke, J.; Mansfield, W.; et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 2014, 158, 1254–1269. [Google Scholar] [CrossRef] [PubMed]
- Canu, G.; Athanasiadis, E.; Grandy, R.A.; Garcia-Bernardo, J.; Strzelecka, P.M.; Vallier, L.; Ortmann, D.; Cvejic, A. Analysis of endothelial-to-haematopoietic transition at the single cell level identifies cell cycle regulation as a driver of differentiation. Genome Biol. 2020, 21, 157. [Google Scholar] [CrossRef]
- Rostovskaya, M.; Bredenkamp, N.; Smith, A. Towards consistent generation of pancreatic lineage progenitors from human pluripotent stem cells. Philos. Trans. R Soc. Lond. B Biol. Sci. 2015, 370, 20140365. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Tang, C.; Cao, F.; Xie, X.; van der Bogt, K.; Hwang, A.; Connolly, A.J.; Robbins, R.C.; Wu, J.C. Effects of cell number on teratoma formation by human embryonic stem cells. Cell Cycle 2009, 8, 2608–2612. [Google Scholar] [CrossRef] [PubMed]
- Hentze, H.; Soong, P.L.; Wang, S.T.; Phillips, B.W.; Putti, T.C.; Dunn, N.R. Teratoma formation by human embryonic stem cells: Evaluation of essential parameters for future safety studies. Stem Cell Res. 2009, 2, 198–210. [Google Scholar] [CrossRef]
- Haridass, D.; Yuan, Q.; Becker, P.D.; Cantz, T.; Iken, M.; Rothe, M.; Narain, N.; Bock, M.; Nörder, M.; Legrand, N.; et al. Repopulation efficiencies of adult hepatocytes, fetal liver progenitor cells, and embryonic stem cell-derived hepatic cells in albumin-promoter-enhancer urokinase-type plasminogen activator mice. Am. J. Pathol. 2009, 175, 1483–1492. [Google Scholar] [CrossRef]
- Martin, R.M.; Fowler, J.L.; Cromer, M.K.; Lesch, B.J.; Ponce, E.; Uchida, N.; Nishimura, T.; Porteus, M.H.; Loh, K.M. Improving the safety of human pluripotent stem cell therapies using genome-edited orthogonal safeguards. Nat. Commun. 2020, 11, 2713. [Google Scholar] [CrossRef]
- Kojima, K.; Miyoshi, H.; Nagoshi, N.; Kohyama, J.; Itakura, G.; Kawabata, S.; Ozaki, M.; Iida, T.; Sugai, K.; Ito, S.; et al. Selective ablation of tumorigenic cells following human induced pluripotent stem cell-derived neural stem/progenitor cell transplantation in spinal cord injury. Stem Cells Transl. Med. 2019, 8, 260–270. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disorder | Genotype | Cell Source | Phenotypes (vs. Normal hiPSCs) | Genetic Modifications | Phenotypic Rescue (vs. Disease-Specific hiPSCs) | References |
|---|---|---|---|---|---|---|
| Hereditary Hematologic Disorders | ||||||
| β-thalassemia major | HbE/β-thalassemia (βE/β0 (β41/42)) | Fibroblasts | HbE/β-thalassemia iPSCs produced lower hematopoietic progenitor cells and erythroid cells | CRISPR/Cas9-mediated HbE correction | Restored the number of hematopoietic progenitor cells and erythroid cells | [93] |
| Homozygous β-thalassemia (β0/β0 (β41/42)) | PBMCs, fibroblasts | β41/42-thalassemia iPSCs displayed lower differentiation efficiency and produced erythrocytes with absence of HBB gene and protein expression | CRISPR/Cas9-, HR-mediated HBB correction, | Restored HBB gene and protein expression in the corrected iPSC-derived erythrocytes | [94,95,96,97] | |
| Homozygous β-thalassemia (β+/β+ (IVS2-654)) | Fibroblasts, amniotic fluid | IVS2-654 thalassemia iPSC-derived erythrocytes lacked HBB gene and protein expression | TALEN-, ZFN-, or CRISPR/Cas9-mediated HBB correction | Restored HBB gene and protein expression in the corrected iPSC-derived erythrocytes | [98,99,100,101] | |
| α-thalassemia | Homozygous α-thalassemia major (− −/− −) | Fibroblasts | Homozygous α-thalassemia iPSC-derived erythrocytes expressed no α-globin chains | ZFN-mediated HBA1 correction | Improved globin chain imbalance in the corrected iPSC-derived erythrocytes | [102] |
| Hemolytic anemia | Heterozygous PKLR (359C > T) and (1168G > A) and homozygous PKLR (IVS9(+1)G > C) | PB | PKD-iPSC-derived erythroid cells displayed the energetic imbalance | TALEN-mediated PKLR correction | Recovered energetic balance in the corrected iPSC-derived erythroid cells | [103] |
| Heterozygous KLF1 (c.973G > A, p.E325K) | PBMCs | CDA-iPSC-derived erythroid cells displayed multinucleated morphology, absence of CD44, dysregulation of target gene and cell cycle regulator genes | N/A | N/A | [104] | |
| SCD | Homozygous (βS/βS) | BM, PBMCs, Fibroblasts | N/A | ZFN-, TALEN-, CRISPR/Cas9-mediated HBB correction | Restored HBB gene and protein expression in corrected iPSC-derived erythrocytes | [105,106,107,108,109] |
| CGD | Homozygous and heterozygous CYBB mutations | BM, PB CD34+ cells, fibroblasts, keratinocytes | CGD-iPSC-derived neutrophils and macrophages lacked ROS production | ZFN-, CRISPR/Cas9-, HR-, TALEN-mediated CYBB correction | Restored CYBB gene expression, functional NADPH oxidase activity, and antimicrobial activity in corrected iPSC-derived neutrophils or macrophages | [110,111,112,113,114,115,116,117] |
| SCID | X-SCID (IL-2Rg 468 + 3A > C) | BM | SCID-X1-iPSCs could not differentiate into functional lymphocytes | TALEN-mediated IL2RG correction | Recovered the production of mature NK cells and T cell precursors differentiated from corrected SCID-X1-iPSCs | [118] |
| JAK3-SCID Homozygous (JAK3 613C > T) | Keratinocytes | JAK3 mutant iPSCs exhibited blockage in early T cell development | TALEN-mediated JAK3 correction | Restored normal T cell development in corrected JAK3 mutant iPSCs | [119] | |
| RAG1-SCID Homozygous and compound heterozygous RAG1 mutations | Fibroblasts | RAG1 mutant iPSCs displayed blockage in early T cell development and TCR re-arrangements | N/A | N/A | [89] | |
| RAG2-SCID Homozygous RAG2 (p.R148X) | Fibroblasts | RAG2 mutant iPSCs displayed blockage in early T cell development and TCR re-arrangements | HR-mediated RAG2 correction | Restored normal T cell development and TCR rearrangements in corrected RAG2 mutant iPSCs | [120] | |
| WAS | WASP (c.1507T > A) and (c.55C > T) | Fibroblasts | WAS-iPSCs exhibited defects in platelet production | Overexpression of WASP using lentiviral vector | Improved proplatelet structure and increased the platelet size in overexpressed WAS-iPSCs | [121] |
| WASP 1305 insG | Fibroblasts | WAS-iPSCs exhibited deficient T lymphopoiesis and NK cell differentiation and function | ZFN-mediated WASP correction | Restored T and NK cell differentiation and function in corrected WAS-iPSCs | [122] | |
| Hemophilia A | F8 mutations | Fibroblasts, epithelial cells, PB CD34+ cells | HA-iPSCs-derived endothelial cells lacked F8 gene expression, secretory protein, and activity | TALEN-, CRISPR/Cas9-, lentiviral vector-mediated F8 correction | Restored F8 transcript, protein secretion, and activity in corrected HA-iPSCs both in vitro and in vivo | [123,124,125,126,127] |
| Hemophilia B | F9 mutations | PBMCs | HB-iPSCs-derived hepatocyte-like cells could not secrete coagulation factor FIX | CRISPR/Cas9-mediated F9 correction | Restored F9 transcript, protein secretion, and activity in corrected HB-iPSCs both in vitro and in vivo | [128,129,130] |
| DBA | RPS19 and RPL5 mutations | Fibroblasts | DBA-iPSCs exhibited ribosomal defects, impaired erythropoiesis | ZFN-, CRISPR/Cas9-mediated RPS19 or RPL5 correction | Rescue of ribosomal defects and erythropoiesis in corrected DBA-iPSCs | [131,132,133] |
| Acquired Hematologic Disorders | ||||||
| MDS | del(7q) | BM, PBMCs | MDS-iPSCs exhibited impaired hematopoietic differentiation, clonogenic capacity, cell growth, and viability | Spontaneous dosage chr7q correction, CRISPR/Cas9-mediated gene correction | Restored hematopoietic differentiation in corrected MDS-iPSCs | [134,135] |
| AML | MLL rearrangement | Primary AML cells | AML-iPSCs exhibited leukemic behavior and methylation patterns upon hematopoietic differentiation | N/A | N/A | [136] |
| CML | BCR/ABL | PBMCs, BM | CML-iPSCs resistant to tyrosine kinase inhibitor (TKI) and reduced hematopoietic differentiation | N/A | N/A | [137,138,139,140,141] |
| PNH | PIGA mutations | Fibroblasts | PIGA-iPSCs were unable to produce hematopoietic cells or mesodermal cells expressing KDR/VEGFR2 and CD56 markers | N/A | N/A | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratumkaew, P.; Issaragrisil, S.; Luanpitpong, S. Induced Pluripotent Stem Cells as a Tool for Modeling Hematologic Disorders and as a Potential Source for Cell-Based Therapies. Cells 2021, 10, 3250. https://doi.org/10.3390/cells10113250
Pratumkaew P, Issaragrisil S, Luanpitpong S. Induced Pluripotent Stem Cells as a Tool for Modeling Hematologic Disorders and as a Potential Source for Cell-Based Therapies. Cells. 2021; 10(11):3250. https://doi.org/10.3390/cells10113250
Chicago/Turabian StylePratumkaew, Ponthip, Surapol Issaragrisil, and Sudjit Luanpitpong. 2021. "Induced Pluripotent Stem Cells as a Tool for Modeling Hematologic Disorders and as a Potential Source for Cell-Based Therapies" Cells 10, no. 11: 3250. https://doi.org/10.3390/cells10113250
APA StylePratumkaew, P., Issaragrisil, S., & Luanpitpong, S. (2021). Induced Pluripotent Stem Cells as a Tool for Modeling Hematologic Disorders and as a Potential Source for Cell-Based Therapies. Cells, 10(11), 3250. https://doi.org/10.3390/cells10113250