
Transfer of Cellular Content from the Allogeneic Cell-Based Cancer Vaccine DCP-001 to Host Dendritic Cells Hinges on Phosphatidylserine and Is Enhanced by CD47 Blockade


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Generation of DCP-001
2.3. Gamma Irradiation
2.4. Mixed Leukocyte Reaction (MLR)
2.5. Peripheral Blood Mononuclear Cell (PBMC) Stimulation Assay
2.6. Cytokine/Chemokine Analysis Employing the Luminex Platform
2.7. Isolation of PBMCs and Generation of Immature Monocyte-Derived DCs (imoDCs)
2.8. Uptake Assay and Blocking of Uptake
2.9. Confocal Microscopy
2.10. Dynamic Single-Cell Analysis by Droplet Array-Based Microfluidic Platform
2.11. Phenotypic Analysis
2.12. Human Skin Explant Culture and Analysis of Migrating Cells
2.13. Statistics
3. Results
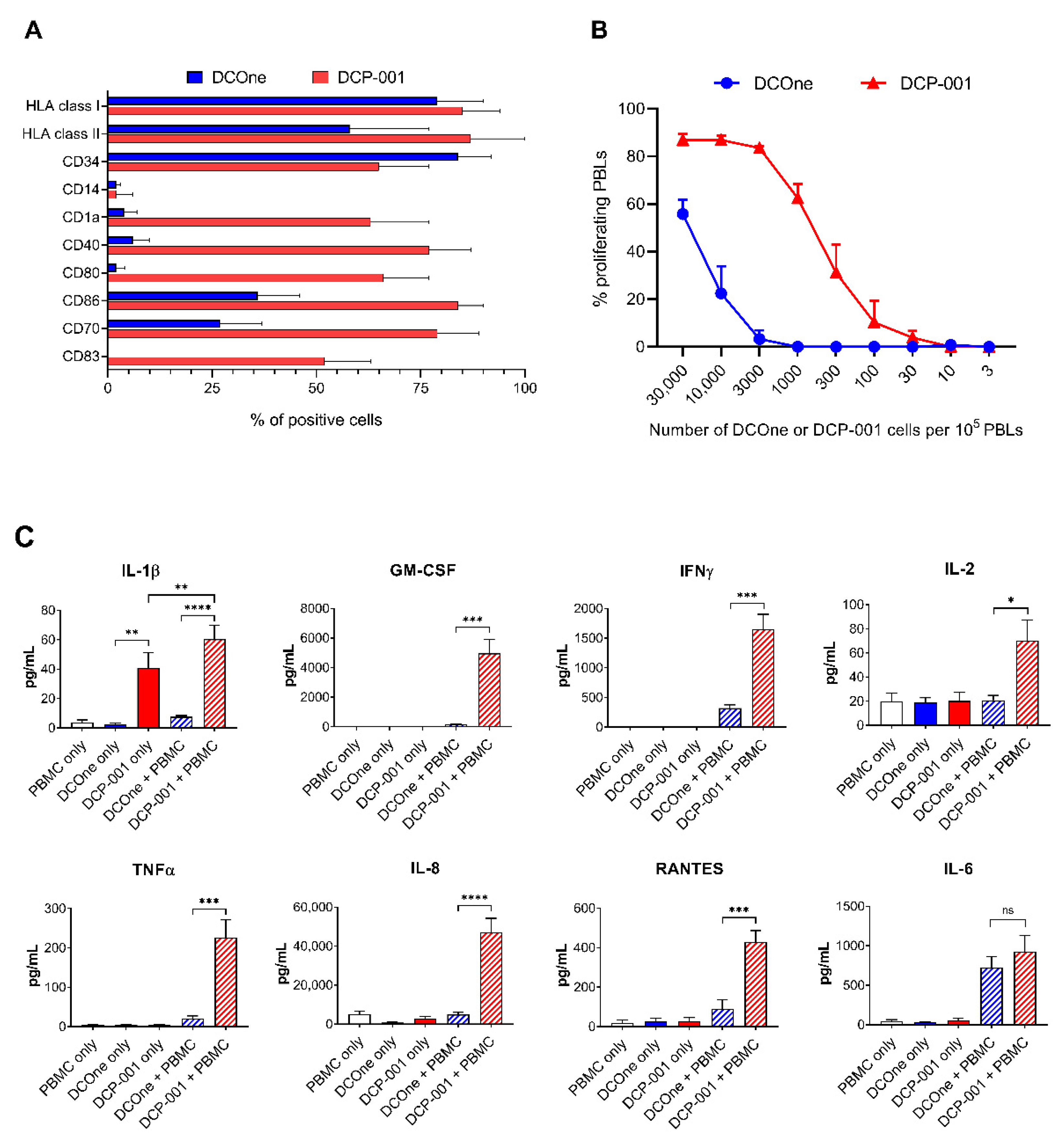
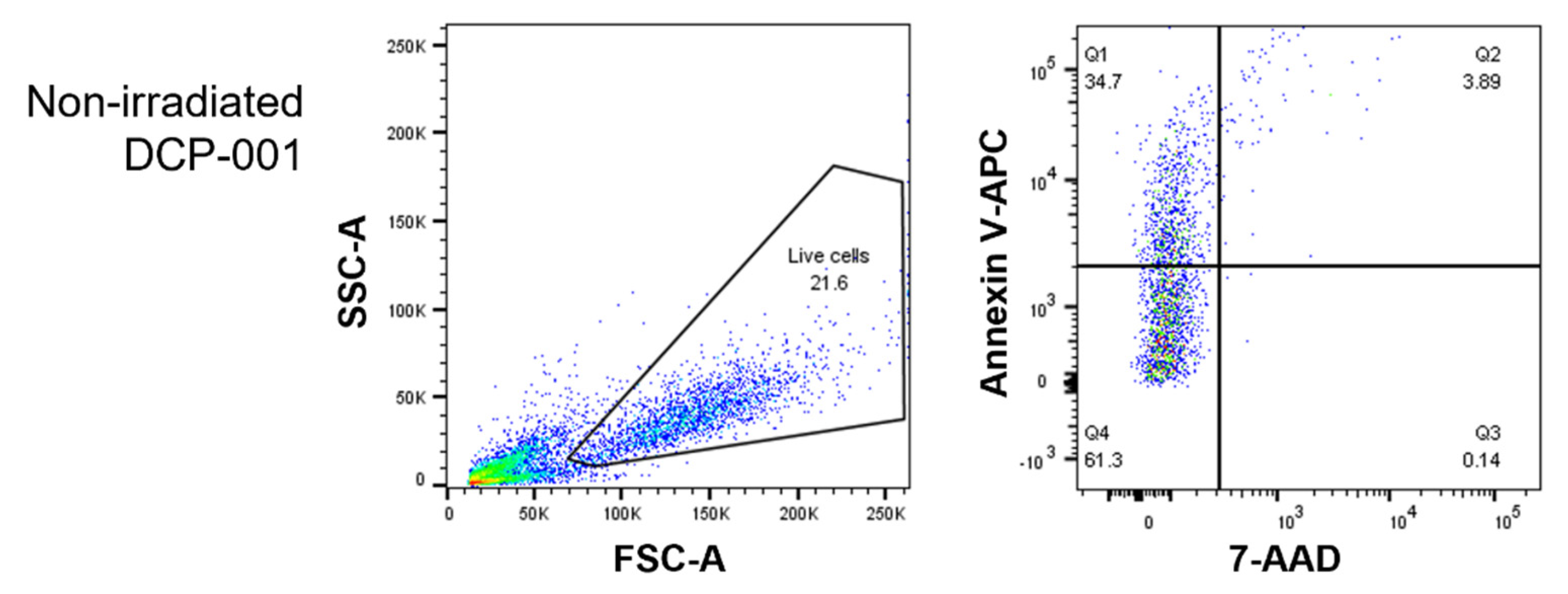
3.1. The Shift towards a Mature DC Phenotype Causes DCP-001 to Induce a Strong Pro-Inflammatory Response When Co-Cultured wth Allogeneic Lymphocytes
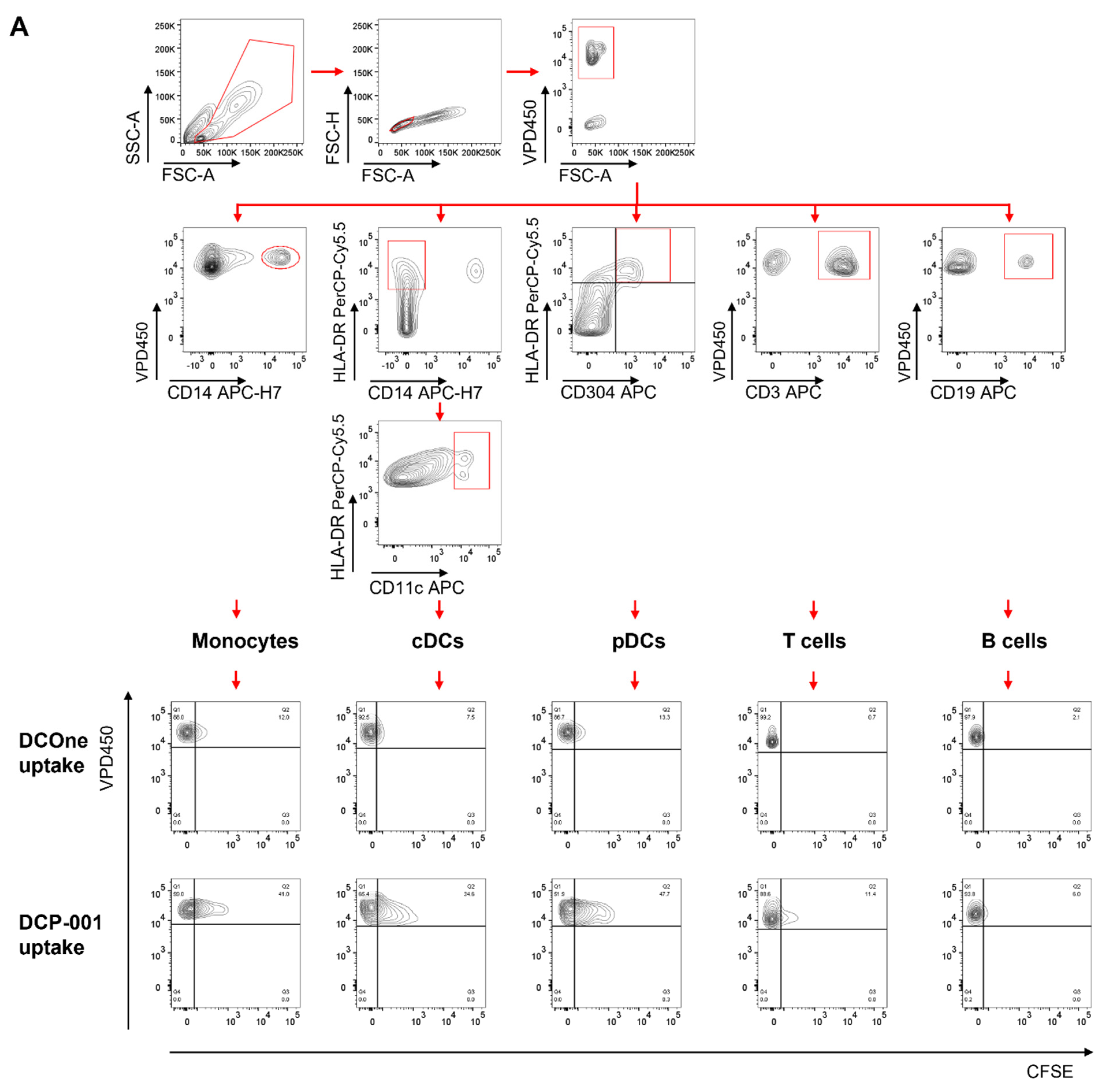
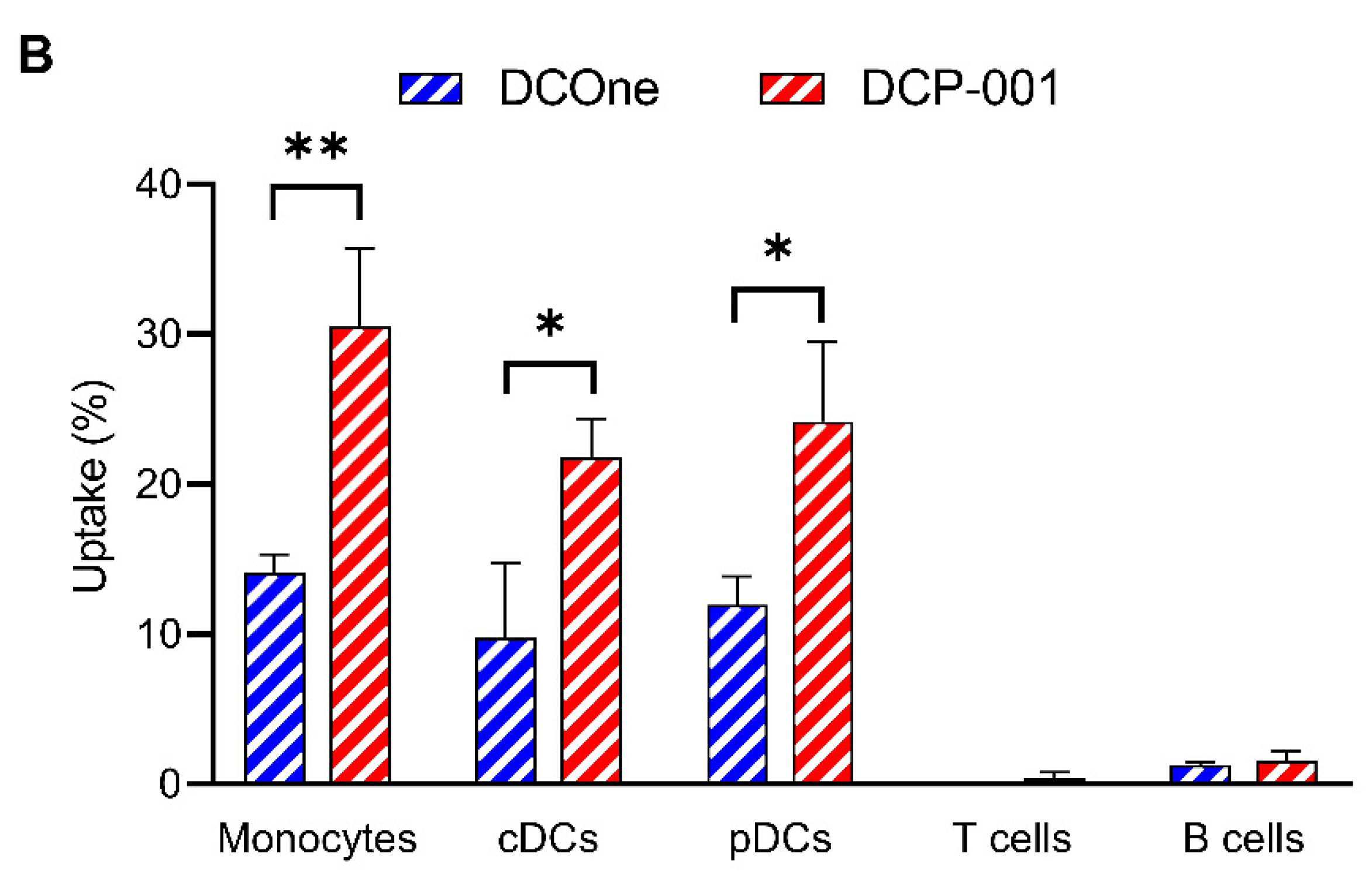
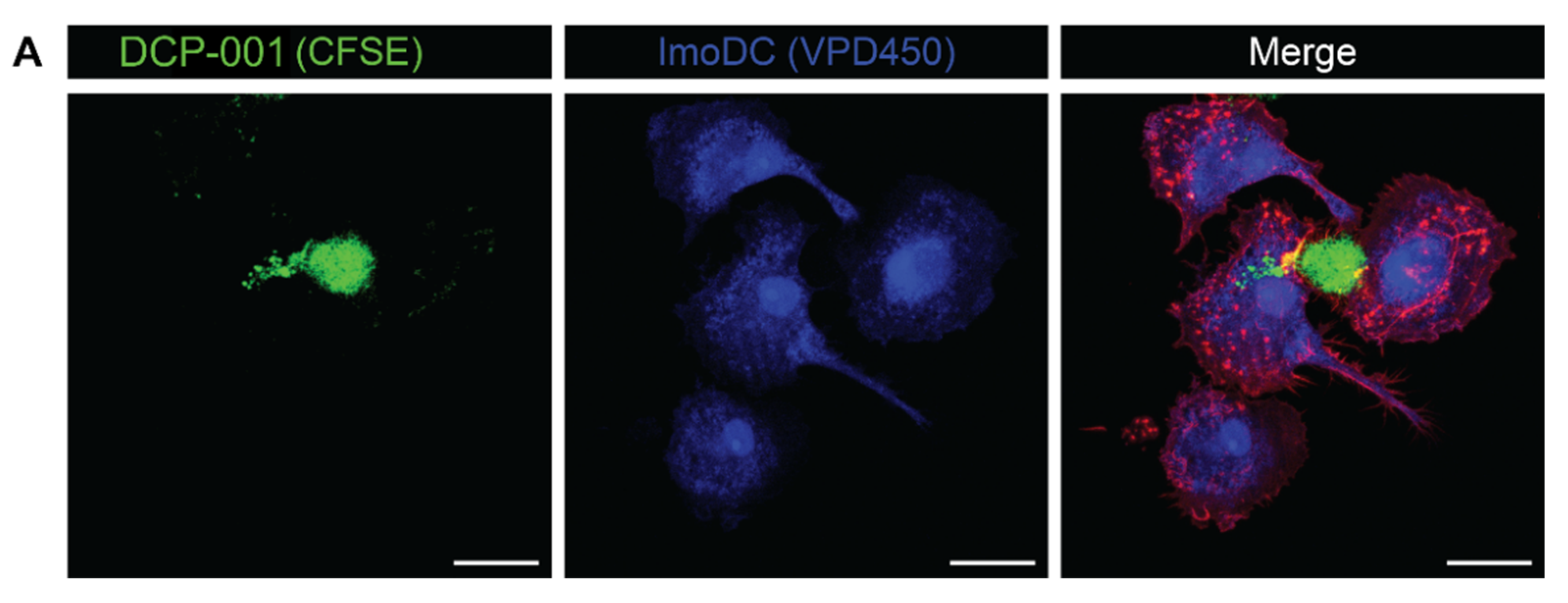
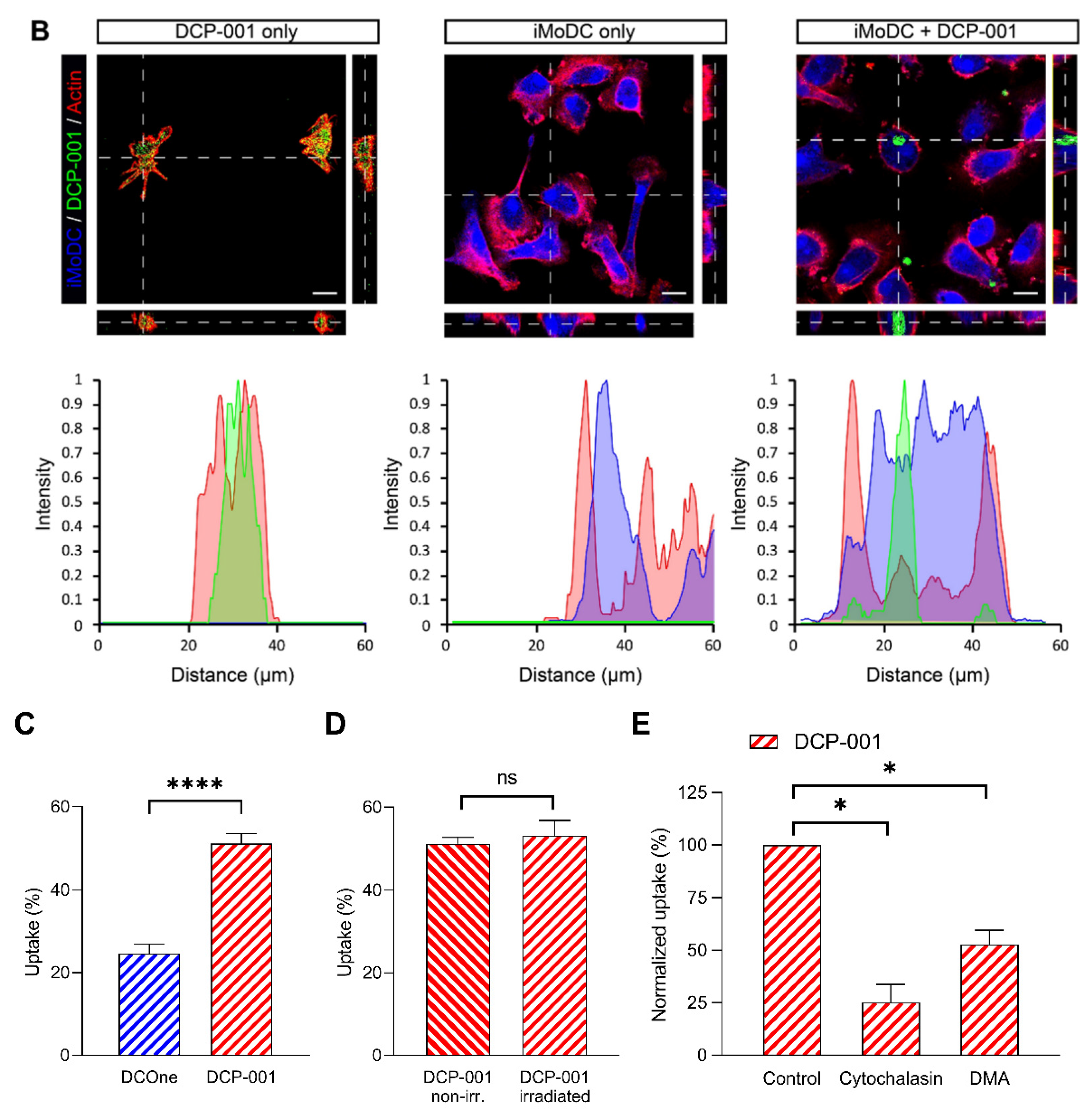
3.2. Cellular Content of DCP-001 Is Efficiently Captured by Host APCs
3.3. Macropinocytosis and/or Phagocytosis Contributes to the Uptake of DCP-001 by APCs
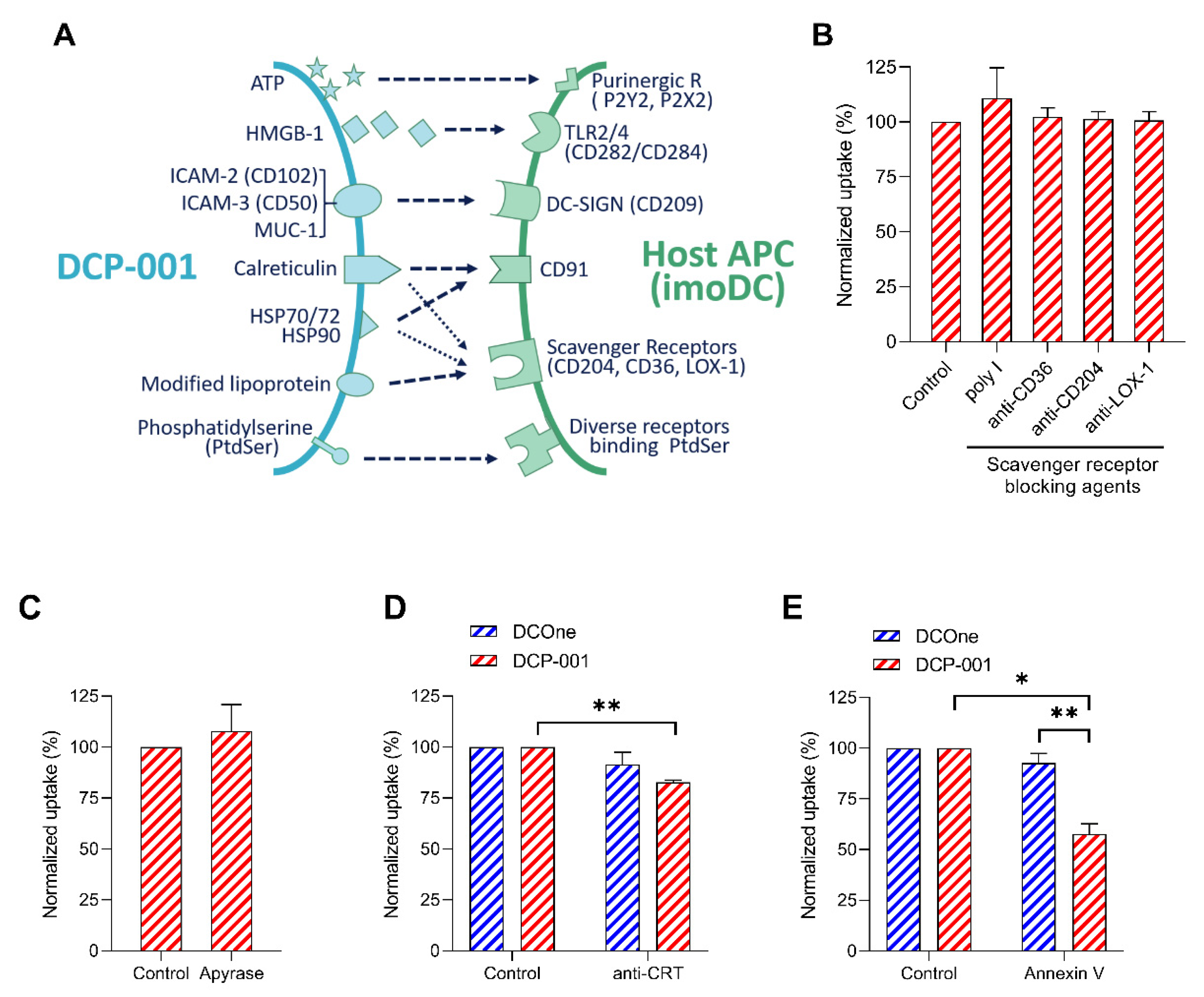
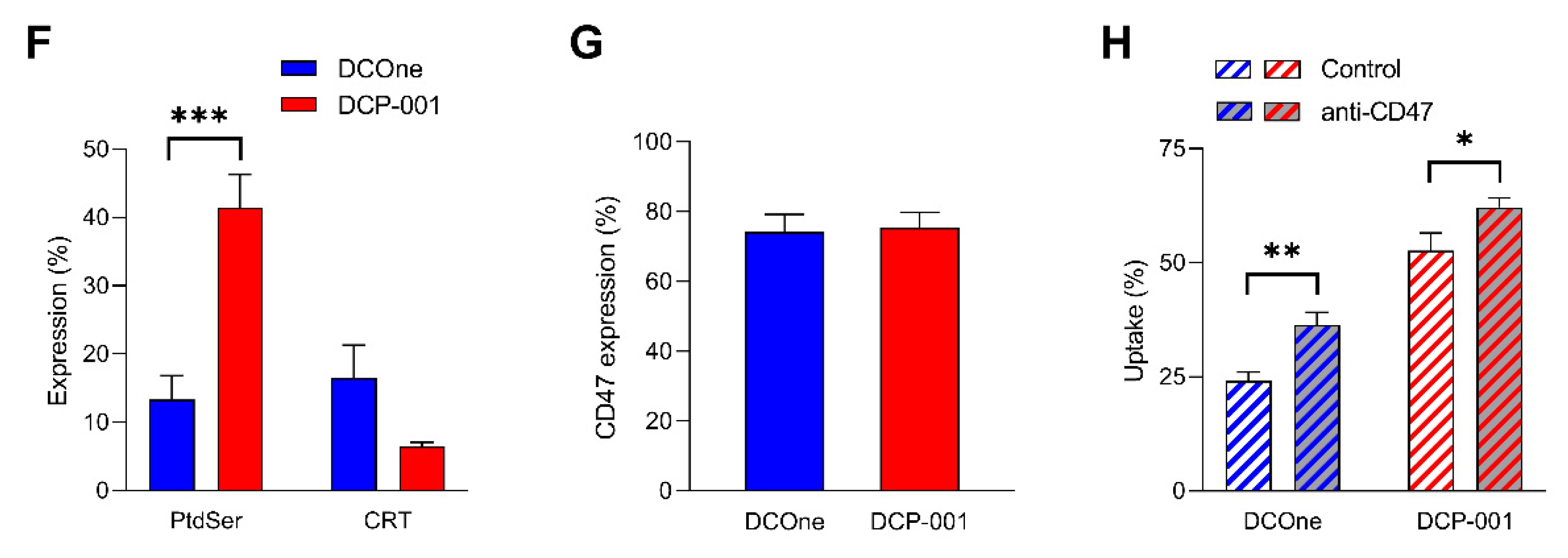
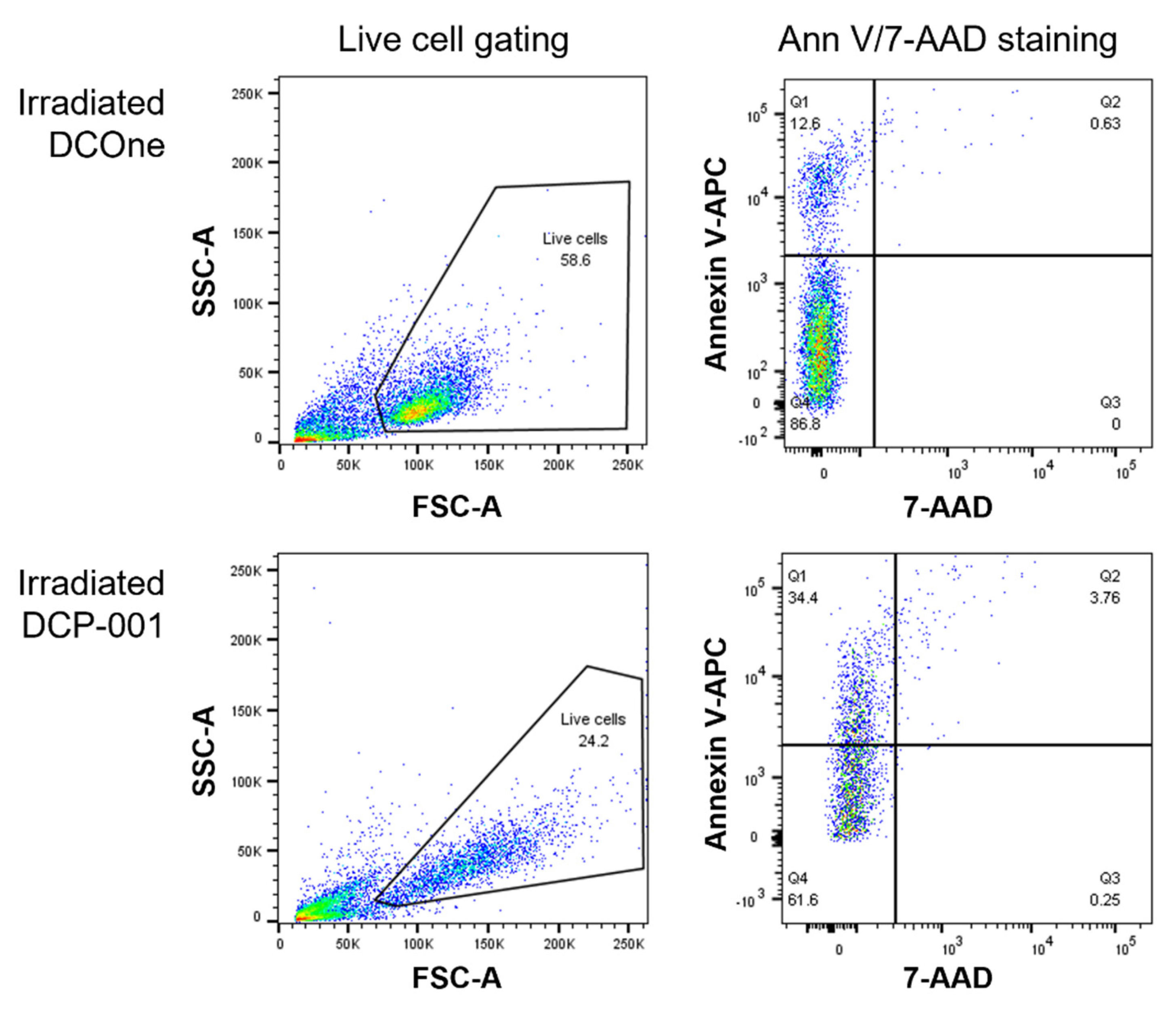
3.4. “Eat-Me” Signals Calreticulin and Phosphatidylserine and “don’t-Eat-Me” Signal CD47 Are Key Factors in the Efficient Endocytosis of DCP-001-Derived Material
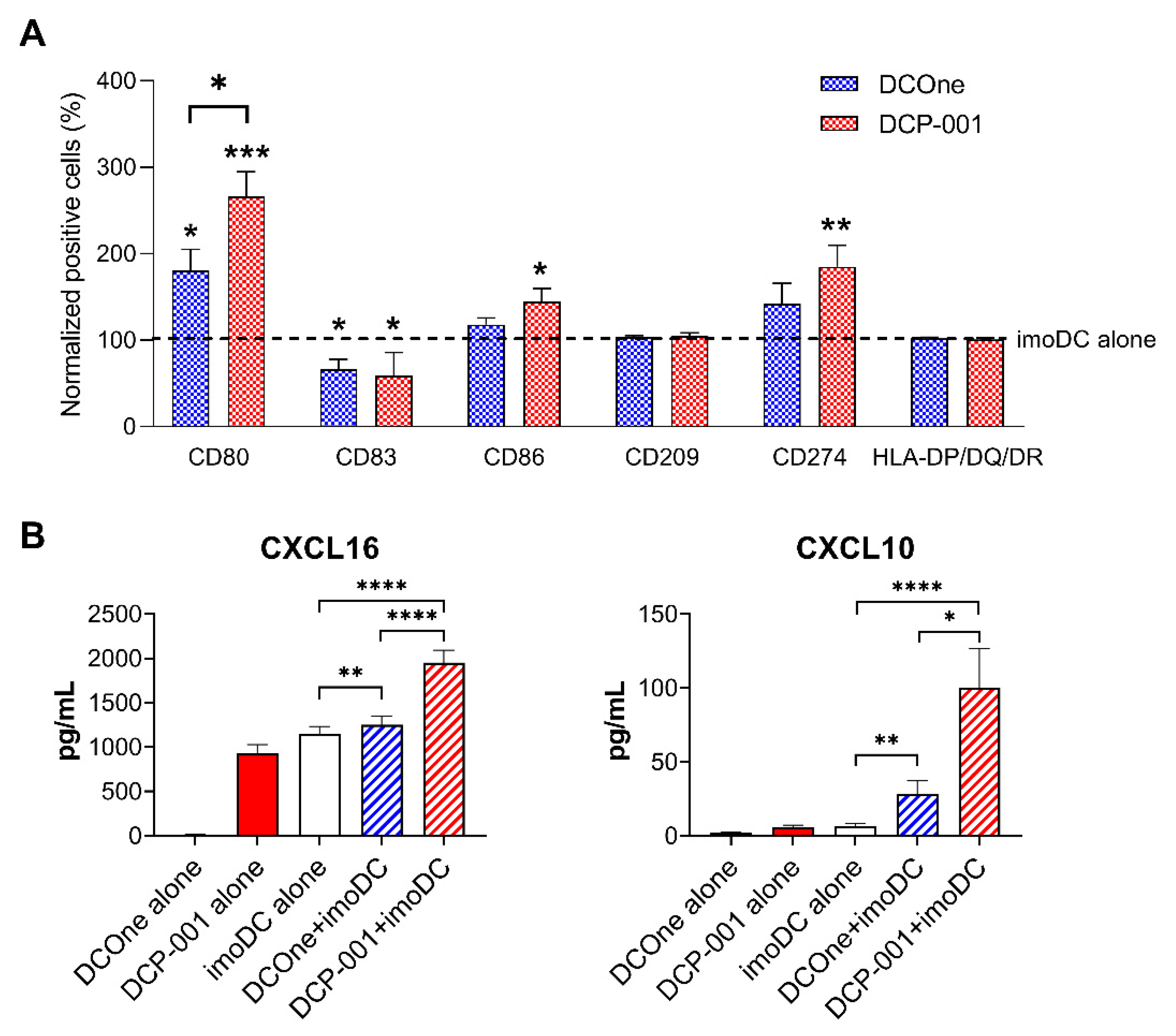
3.5. DCP-001 Induces Phenotypic and Functional Maturation of Co-Cultured Allogeneic DCs
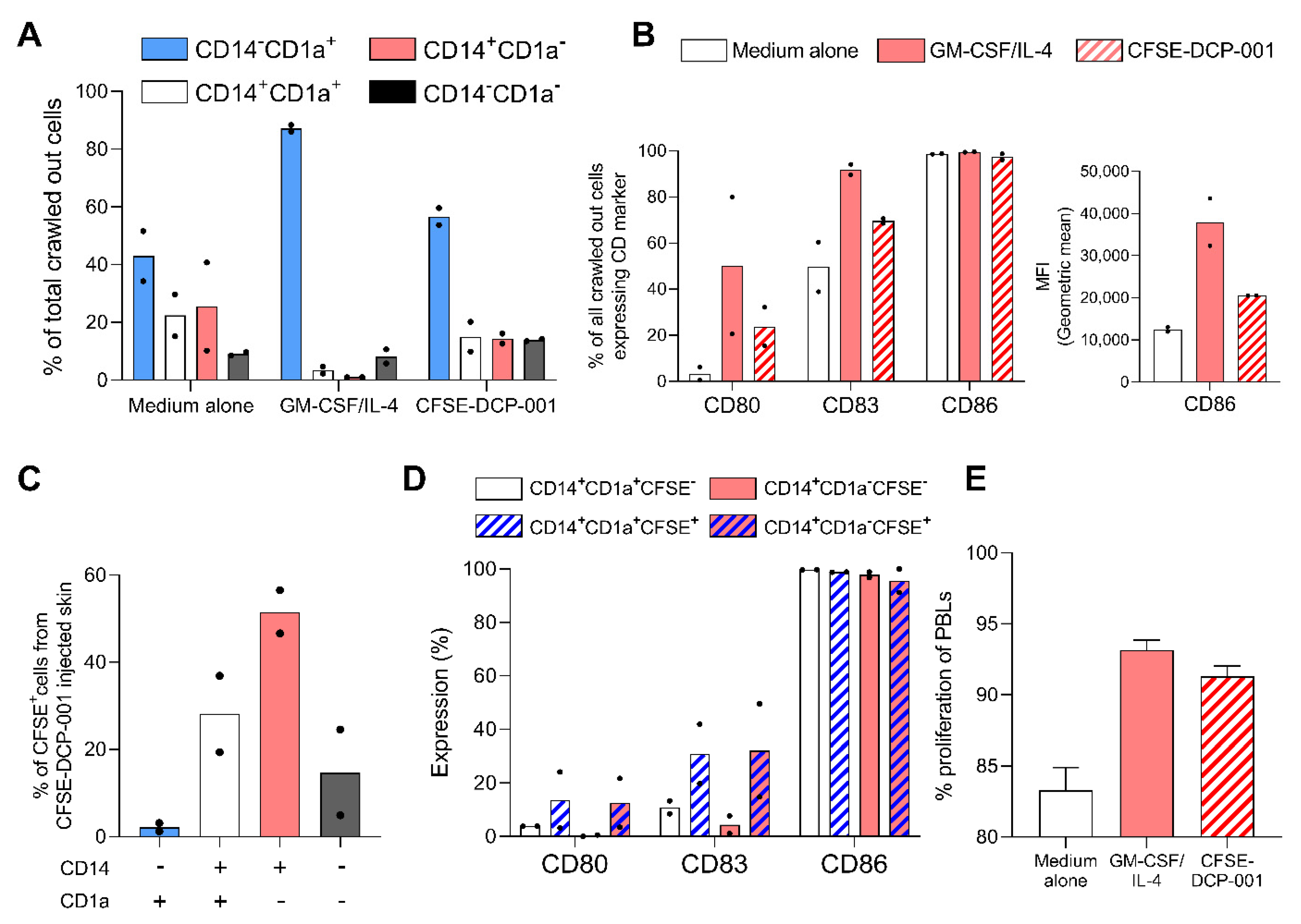
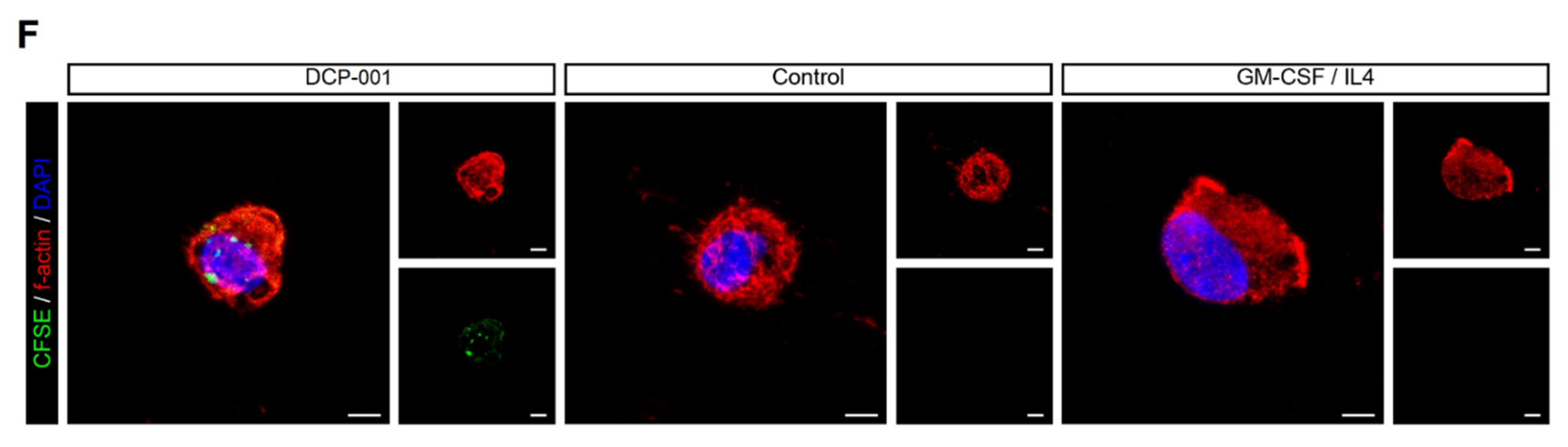
3.6. Uptake of DCP-001 by Skin-Emigrated DC Subsets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Gruijl, T.D.; van den Eertwegh, A.J.; Pinedo, H.M.; Scheper, R.J. Whole-cell cancer vaccination: From autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol. Immunother. 2008, 57, 1569–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, T.; Kamigaki, T.; Kawasaki, K.; Nakamura, T.; Yamamoto, M.; Kanemitsu, K.; Takase, S.; Kuroda, D.; Kim, Y.; Ajiki, T.; et al. Superior anti-tumor protection and therapeutic efficacy of vaccination with allogeneic and semiallogeneic dendritic cell/tumor cell fusion hybrids for murine colon adenocarcinoma. Cancer Immunol. Immunother. 2007, 56, 1025–1036. [Google Scholar] [CrossRef]
- Karlsson-Parra, A.; Kovacka, J.; Heimann, E.; Jorvid, M.; Zeilemaker, S.; Longhurst, S.; Suenaert, P. Ilixadencel—An Allogeneic Cell-Based Anticancer Immune Primer for Intratumoral Administration. Pharm. Res. 2018, 35, 156. [Google Scholar] [CrossRef] [Green Version]
- Anguille, S.; Van Tendeloo, V.F.; Berneman, Z.N. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia 2012, 26, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Leaf, R.K.; Stroopinsky, D.; Pyzer, A.R.; Kruisbeek, A.M.; van Wetering, S.; Washington, A.; Ephraim, A.; Cole, L.; Morin, A.; Jain, S.; et al. DCOne as an Allogeneic Cell-based Vaccine for Multiple Myeloma. J. Immunother. 2017, 40, 315–322. [Google Scholar] [CrossRef]
- Vermeij, R.; Daemen, T.; de Bock, G.H.; de Graeff, P.; Leffers, N.; Lambeck, A.; ten Hoor, K.A.; Hollema, H.; van der Zee, A.G.; Nijman, H.W. Potential target antigens for a universal vaccine in epithelial ovarian cancer. Clin. Dev. Immunol. 2010, 2010, 891505. [Google Scholar] [CrossRef]
- van de Loosdrecht, A.A.; van Wetering, S.; Santegoets, S.; Singh, S.K.; Eeltink, C.M.; den Hartog, Y.; Koppes, M.; Kaspers, J.; Ossenkoppele, G.J.; Kruisbeek, A.M.; et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol. Immunother. 2018, 67, 1505–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, L.L.G.; Westers, T.M.; Rovers, J.; Valk, P.; Cloos, J.; de Gruijl, T.D.; Van de Loosdrecht, A.A. Durable Responses and Survival in High Risk AML and MDS Patients Treated with an Allogeneic Leukemia-Derived Dendritic Cell Vaccine. Blood 2019, 134, 1381. [Google Scholar] [CrossRef]
- Verdijk, P.; Aarntzen, E.H.; Lesterhuis, W.J.; Boullart, A.C.; Kok, E.; van Rossum, M.M.; Strijk, S.; Eijckeler, F.; Bonenkamp, J.J.; Jacobs, J.F.; et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin. Cancer Res. 2009, 15, 2531–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yewdall, A.W.; Drutman, S.B.; Jinwala, F.; Bahjat, K.S.; Bhardwaj, N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS ONE 2010, 5, e11144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, L.M.; Ezekowitz, R.A. Phagocytosis: Elegant complexity. Immunity 2005, 22, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Roche, P.A. Macropinocytosis in phagocytes: Regulation of MHC class-II-restricted antigen presentation in dendritic cells. Front. Physiol. 2015, 6, 1. [Google Scholar] [CrossRef]
- Frydrychowicz, M.; Kolecka-Bednarczyk, A.; Madejczyk, M.; Yasar, S.; Dworacki, G. Exosomes-structure, biogenesis and biological role in non-small-cell lung cancer. Scand. J. Immunol. 2015, 81, 2–10. [Google Scholar] [CrossRef]
- Théry, C.; Duban, L.; Segura, E.; Véron, P.; Lantz, O.; Amigorena, S. Indirect activation of naïve CD4+ T cells by dendritic cell-derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef]
- Sarkar, S.; Sabhachandani, P.; Stroopinsky, D.; Palmer, K.; Cohen, N.; Rosenblatt, J.; Avigan, D.; Konry, T. Dynamic analysis of immune and cancer cell interactions at single cell level in microfluidic droplets. Biomicrofluidics 2016, 10, 054115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruben, J.M.; Bontkes, H.J.; Westers, T.M.; Hooijberg, E.; Ossenkoppele, G.J.; van de Loosdrecht, A.A.; de Gruijl, T.D. In situ loading of skin dendritic cells with apoptotic bleb-derived antigens for the induction of tumor-directed immunity. Oncoimmunology 2014, 3, e946360. [Google Scholar] [CrossRef] [Green Version]
- de Gruijl, T.D.; Sombroek, C.C.; Lougheed, S.M.; Oosterhoff, D.; Buter, J.; van den Eertwegh, A.J.; Scheper, R.J.; Pinedo, H.M. A postmigrational switch among skin-derived dendritic cells to a macrophage-like phenotype is predetermined by the intracutaneous cytokine balance. J. Immunol. 2006, 176, 7232–7242. [Google Scholar] [CrossRef]
- Garg, A.D.; Romano, E.; Rufo, N.; Agostinis, P. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: Mechanisms and clinical translation. Cell Death Differ. 2016, 23, 938–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hourigan, C.S.; Levitsky, H.I. Evaluation of current cancer immunotherapy: Hemato-oncology. Cancer J. (Sudbury Mass.) 2011, 17, 309–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloudova, K.; Hromadkova, H.; Partlova, S.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Hensler, M.; Halaska, M.J.; Spisek, R.; Fialova, A. Expression of tumor antigens on primary ovarian cancer cells compared to established ovarian cancer cell lines. Oncotarget 2016, 7, 46120–46126. [Google Scholar] [CrossRef] [Green Version]
- Brode, S.; Macary, P.A. Cross-presentation: Dendritic cells and macrophages bite off more than they can chew! Immunology 2004, 112, 345–351. [Google Scholar] [CrossRef]
- Nagata, S.; Tanaka, M. Programmed cell death and the immune system. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Harms-Ringdahl, M.; Nicotera, P.; Radford, I.R. Radiation induced apoptosis. Mutat. Res. Rev. Genet. Toxicol. 1996, 366, 171–179. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, N.; Riol-Blanco, L.; de la Rosa, G.; Puig-Kroger, A.; Garcia-Bordas, J.; Martin, D.; Longo, N.; Cuadrado, A.; Cabanas, C.; Corbi, A.L.; et al. Chemokine receptor CCR7 induces intracellular signaling that inhibits apoptosis of mature dendritic cells. Blood 2004, 104, 619–625. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Voelkl, S.; Berger, J.; Andreesen, R.; Pomorski, T.; Mackensen, A. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood 2006, 108, 4094–4101. [Google Scholar] [CrossRef] [PubMed]
- Sauter, B.; Albert, M.L.; Francisco, L.; Larsson, M.; Somersan, S.; Bhardwaj, N. Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000, 191, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.A.; Wu, D.-C.; Cheung, J.; Navarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 2020, 1, 681–691. [Google Scholar] [CrossRef]
- D’Orsogna, L.J.; Roelen, D.L.; Doxiadis, I.I.N.; Claas, F.H. Alloreactivity from human viral specific memory T-cells. Transpl. Immunol. 2010, 23, 149–155. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8⁺ T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Menares, E.; Galvez-Cancino, F.; Caceres-Morgado, P.; Ghorani, E.; Lopez, E.; Diaz, X.; Saavedra-Almarza, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A.; et al. Tissue-resident memory CD8(+) T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 4401. [Google Scholar] [CrossRef] [Green Version]
- Naeini, M.B.; Bianconi, V.; Pirro, M.; Sahebkar, A. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell Mol. Biol. Lett. 2020, 25, 23. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Roberts, E.W.; Cai, E.; Mujal, A.M.; Marchuk, K.; Beppler, C.; Nam, D.; Serwas, N.K.; Binnewies, M.; Krummel, M.F. Visualizing Synaptic Transfer of Tumor Antigens among Dendritic Cells. Cancer Cell 2020, 37, 786–799.e785. [Google Scholar] [CrossRef]
- Caronni, N.; Piperno, G.M.; Simoncello, F.; Romano, O.; Vodret, S.; Yanagihashi, Y.; Dress, R.; Dutertre, C.A.; Bugatti, M.; Bourdeley, P.; et al. TIM4 expression by dendritic cells mediates uptake of tumor-associated antigens and anti-tumor responses. Nat. Commun. 2021, 12, 2237. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.M.G.; Bianconi, V.; Pirro, M.; Jaafari, M.R.; Hatamipour, M.; Sahebkar, A. CD47: Role in the immune system and application to cancer therapy. Cell. Oncol. 2020, 43, 19–30. [Google Scholar] [CrossRef]
- Lian, S.; Xie, R.; Ye, Y.; Xie, X.; Li, S.; Lu, Y.; Li, B.; Cheng, Y.; Katanaev, V.L.; Jia, L. Simultaneous blocking of CD47 and PD-L1 increases innate and adaptive cancer immune responses and cytokine release. EBioMedicine 2019, 42, 281–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashem, S.W.; Haniffa, M.; Kaplan, D.H. Antigen-Presenting Cells in the Skin. Annu. Rev. Immunol. 2017, 35, 469–499. [Google Scholar] [CrossRef]
- Sporri, R.; Reis e Sousa, C. Newly activated T cells promote maturation of bystander dendritic cells but not IL-12 production. J. Immunol. 2003, 171, 6406–6413. [Google Scholar] [CrossRef] [Green Version]
- Lindenbergh, M.F.S.; Koerhuis, D.G.J.; Borg, E.G.F.; van’t Veld, E.M.; Driedonks, T.A.P.; Wubbolts, R.; Stoorvogel, W.; Boes, M. Bystander T-Cells Support Clonal T-Cell Activation by Controlling the Release of Dendritic Cell-Derived Immune-Stimulatory Extracellular Vesicles. Front. Immunol. 2019, 10, 448. [Google Scholar] [CrossRef] [Green Version]
- Lapteva, N.; Huang, X.F. CCL5 as an adjuvant for cancer immunotherapy. Expert Opin. Biol. Ther. 2010, 10, 725–733. [Google Scholar] [CrossRef]
- Lebre, M.C.; Burwell, T.; Vieira, P.L.; Lora, J.; Coyle, A.J.; Kapsenberg, M.L.; Clausen, B.E.; De Jong, E.C. Differential expression of inflammatory chemokines by Th1- and Th2-cell promoting dendritic cells: A role for different mature dendritic cell populations in attracting appropriate effector cells to peripheral sites of inflammation. Immunol. Cell Biol. 2005, 83, 525–535. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Haniffa, M.; Shin, A.; Bigley, V.; McGovern, N.; Teo, P.; See, P.; Wasan, P.S.; Wang, X.N.; Malinarich, F.; Malleret, B.; et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 2012, 37, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Granot, T.; Senda, T.; Carpenter, D.J.; Matsuoka, N.; Weiner, J.; Gordon, C.L.; Miron, M.; Kumar, B.V.; Griesemer, A.; Ho, S.H.; et al. Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics over Human Life. Immunity 2017, 46, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, R.S.; Waithman, J.; Bedoui, S.; Jones, C.M.; Villadangos, J.A.; Zhan, Y.; Lew, A.M.; Shortman, K.; Heath, W.R.; Carbone, F.R. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 2006, 25, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, I.; Feferman, T.; Milo, I.; Tal, O.; Golani, O.; Drexler, I.; Shakhar, G. Active dissemination of cellular antigens by DCs facilitates CD8(+) T-cell priming in lymph nodes. Eur. J. Immunol. 2017, 47, 1802–1818. [Google Scholar] [CrossRef] [Green Version]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, H.; van Lierop, M.-J.C.; Kaspers, J.; Bos, R.; Reurs, A.; Sarkar, S.; Konry, T.; Kamermans, A.; Kooij, G.; de Vries, H.E.; et al. Transfer of Cellular Content from the Allogeneic Cell-Based Cancer Vaccine DCP-001 to Host Dendritic Cells Hinges on Phosphatidylserine and Is Enhanced by CD47 Blockade. Cells 2021, 10, 3233. https://doi.org/10.3390/cells10113233
Zuo H, van Lierop M-JC, Kaspers J, Bos R, Reurs A, Sarkar S, Konry T, Kamermans A, Kooij G, de Vries HE, et al. Transfer of Cellular Content from the Allogeneic Cell-Based Cancer Vaccine DCP-001 to Host Dendritic Cells Hinges on Phosphatidylserine and Is Enhanced by CD47 Blockade. Cells. 2021; 10(11):3233. https://doi.org/10.3390/cells10113233
Chicago/Turabian StyleZuo, Haoxiao, Marie-José C. van Lierop, Jorn Kaspers, Remco Bos, Anneke Reurs, Saheli Sarkar, Tania Konry, Alwin Kamermans, Gijs Kooij, Helga E. de Vries, and et al. 2021. "Transfer of Cellular Content from the Allogeneic Cell-Based Cancer Vaccine DCP-001 to Host Dendritic Cells Hinges on Phosphatidylserine and Is Enhanced by CD47 Blockade" Cells 10, no. 11: 3233. https://doi.org/10.3390/cells10113233
APA StyleZuo, H., van Lierop, M.-J. C., Kaspers, J., Bos, R., Reurs, A., Sarkar, S., Konry, T., Kamermans, A., Kooij, G., de Vries, H. E., de Gruijl, T. D., Karlsson-Parra, A., Manting, E. H., Kruisbeek, A. M., & Singh, S. K. (2021). Transfer of Cellular Content from the Allogeneic Cell-Based Cancer Vaccine DCP-001 to Host Dendritic Cells Hinges on Phosphatidylserine and Is Enhanced by CD47 Blockade. Cells, 10(11), 3233. https://doi.org/10.3390/cells10113233